
Use of the Molecular Dynamics Method to Investigate the Stability of α-α-Corner Structural Motifs in Proteins


Abstract
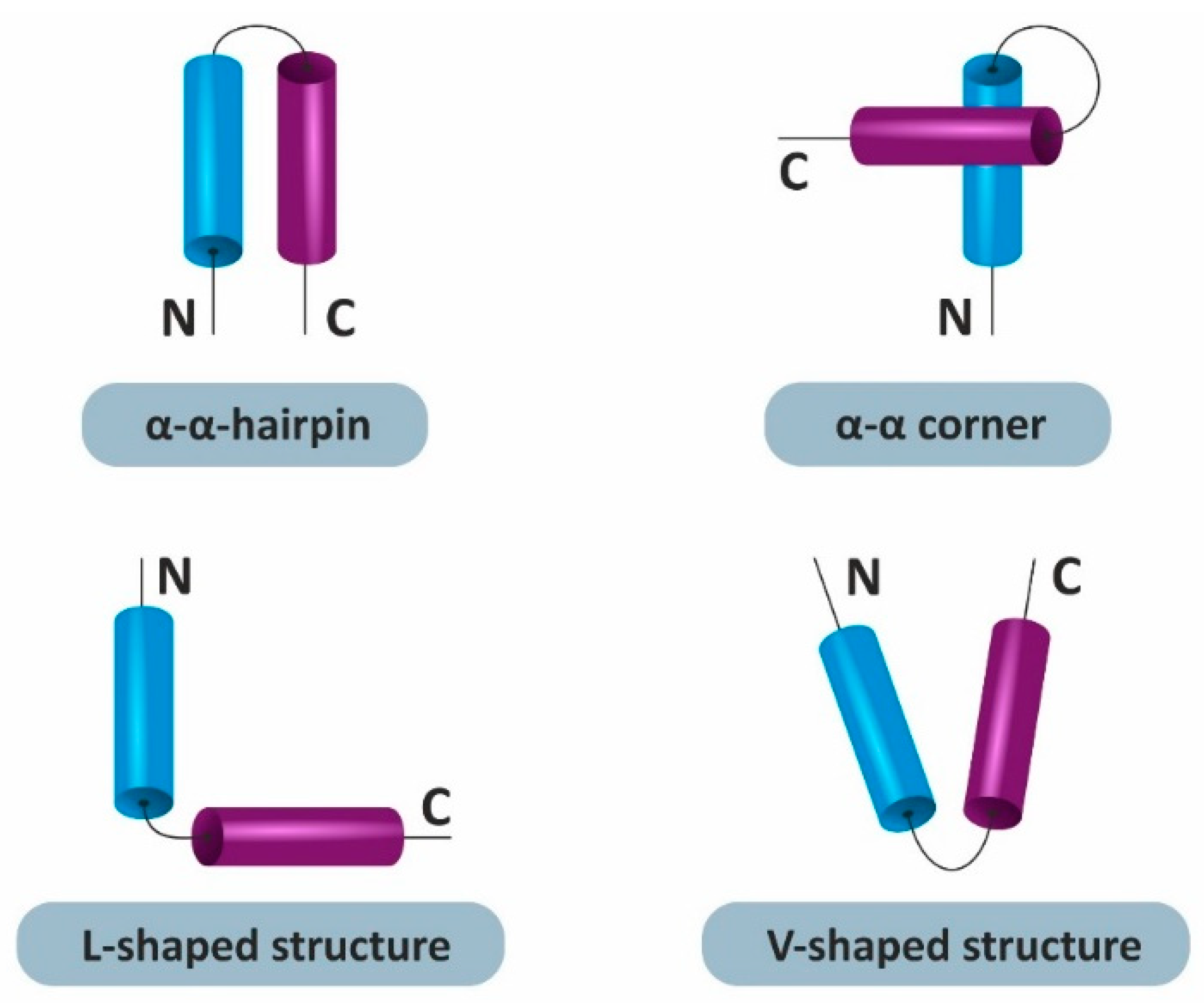
:1. Introduction
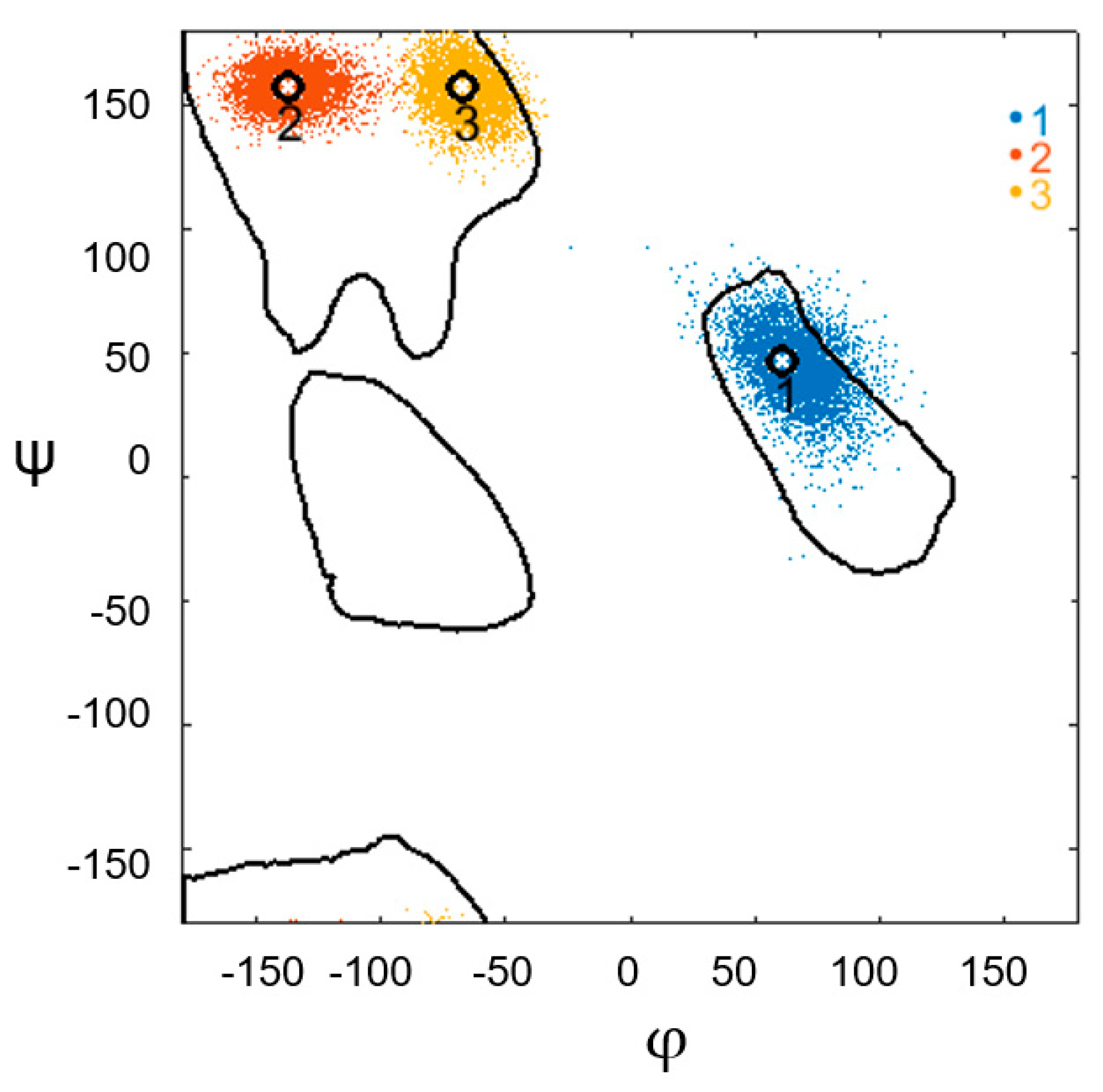
2. Materials and Methods
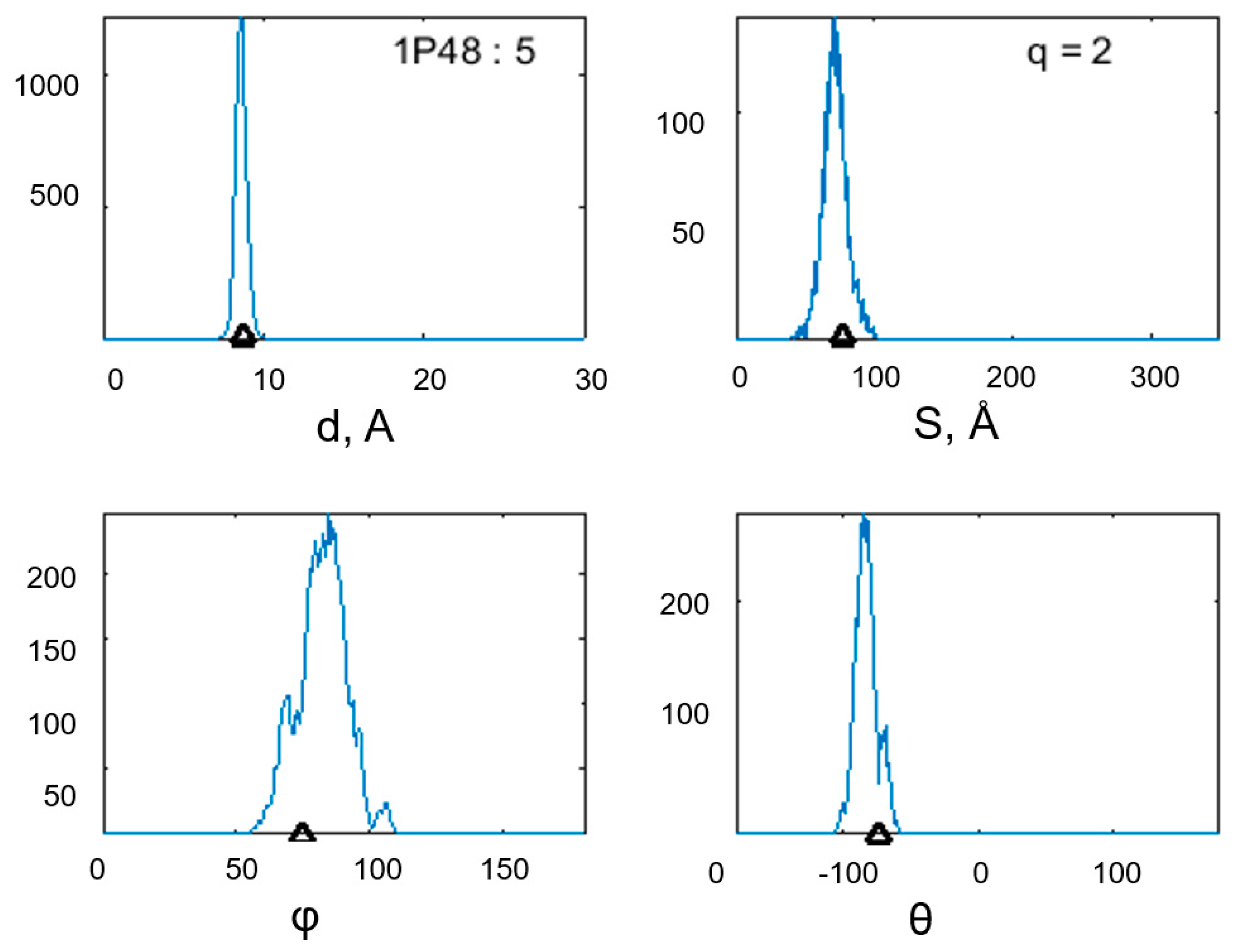
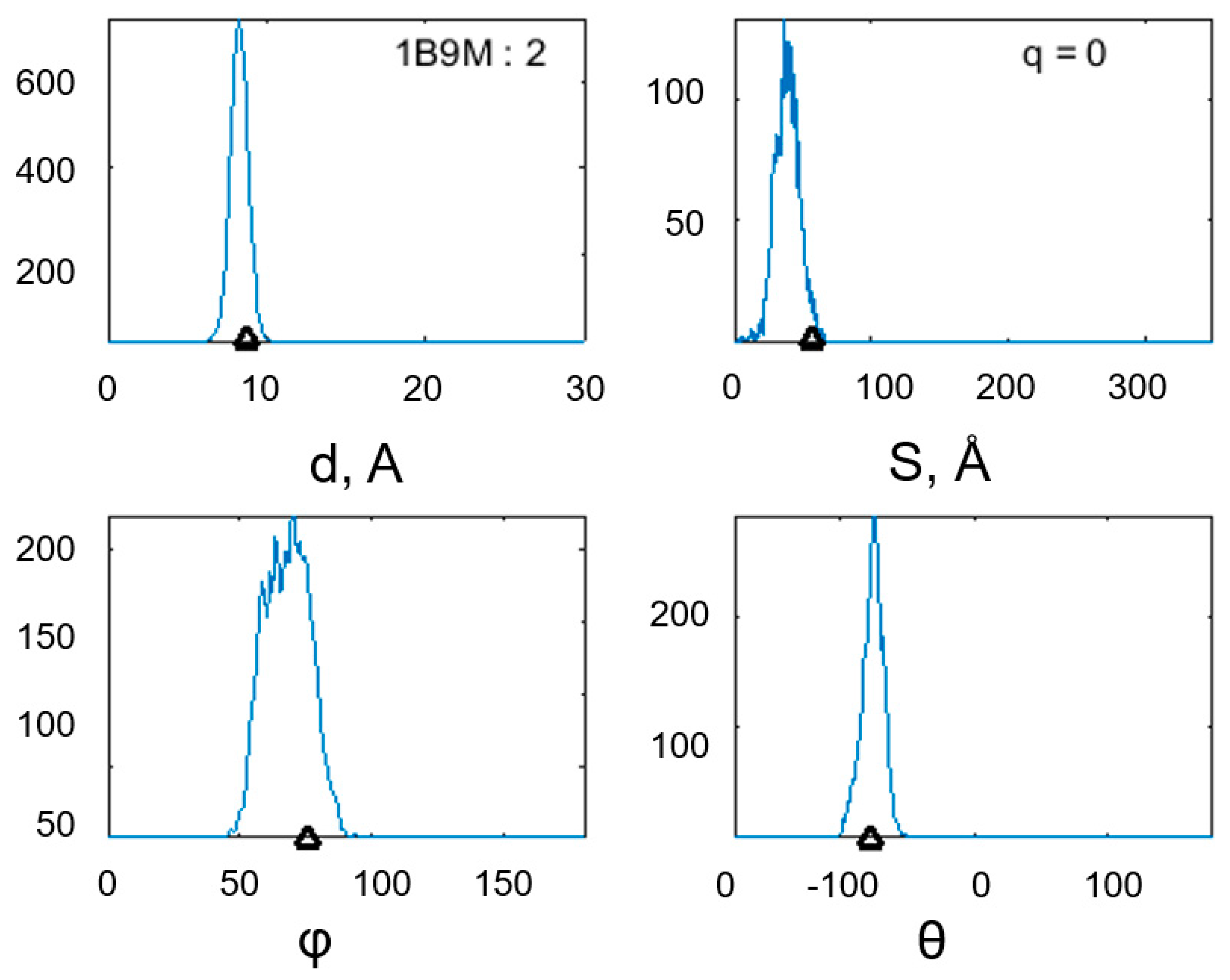
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Efimov, A.V. Standard structures in proteins. Prog. Biophys. Mol. Biol. 1993, 60, 201–239. [Google Scholar] [CrossRef]
- Brazhnikov, E.V.; Efimov, A.V. Structure of α-α-hairpins with short connections in globular proteins. Mol. Biol. 2001, 35, 89–97. [Google Scholar] [CrossRef]
- Efimov, A.V. L-shaped structure of two α-helices with a proline residue between them. Mol. Biol. 1992, 26, 1370–1376. (In Russian) [Google Scholar]
- Efimov, A.V. New supersecondary structure of proteins: α-α-corner. Mol. Biol. 1984, 18, 1524–1537. (In Russian) [Google Scholar]
- Tikhonov, D.A.; Kulikova, L.I.; Efimov, A.V. Statistical analysis of the internal distances of helical pairs in protein molecules. Math. Biol. Bioinf. 2016, 11, 170–190. (In Russian) [Google Scholar] [CrossRef]
- Tikhonov, D.A.; Kulikova, L.I.; Efimov, A.V. The Study of interhelical angles in the structural motifs formed by two helices. Math. Biol. Bioinf. 2017, 12, 83–101. (In Russian) [Google Scholar] [CrossRef] [Green Version]
- Tikhonov, D.A.; Kulikova, L.I.; Efimov, A.V. Analysis of the Torsion Angles between Helical Axes inPairs of 745 Helices in Protein Molecules. Math. Biol. Bioinf. 2018, 13, t17–t28. [Google Scholar] [CrossRef]
- Tikhonov, D.A.; Kulikova, L.I.; Efimov, A.V. Relationship between the interhelical packing angles and the length of α-helices in proteins. Mol. Biol. 2020, 54, 292–298. [Google Scholar] [CrossRef]
- Tikhonov, D.; Kulikova, L.; Kopylov, A.; Malsagova, K.; Stepanov, A.; Rudnev, V.; Kaysheva, A. super secondary structures of proteins with post-translational modifications in colon cancer. Molecules 2020, 25, 3144. [Google Scholar] [CrossRef] [PubMed]
- Tsai, F.C.; Sherman, J.C. Circular dichroism analysis of a synthetic peptide corresponding to the α-α-corner motif of hemoglobin. Biochem. Biophys. Res. Commun. 1993, 196, 435–439. [Google Scholar] [CrossRef]
- Rudnev, V.R.; Pankratov, A.N.; Kulikova, L.I.; Dedus, F.F.; Tikhonov, D.A.; Efimov, A.V. Recognition and stability analysis of structural motifs of α-α-corner type in globular proteins. Math. Biol. Bioinf. 2013, 8, 398–406. [Google Scholar] [CrossRef]
- Rudnev, V.R.; Pankratov, A.N.; Kulikova, L.I.; Dedus, F.F.; Tikhonov, D.A.; Efimov, A.V. Conformational analysis of structural motifs of α-α-corner in the computational experiment of molecular dynamics. Math. Biol. Bioinf. 2014, 9, 575–584. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein databank. Nucleic Acids Res. 2020, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Lee, M.C.; Duan, Y. Distinguish protein decoys by using a scoring function based on a new Amber force field, short molecular dynamics simulations, and the generalized Born solvent model. Proteins 2004, 55, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, N.L. A cell-based approach to the human proteome project. J. Am. Soc. Mass Spectrom 2012, 23, 1617–1624. [Google Scholar] [CrossRef] [Green Version]
- Indeykina, M.I.; Popov, I.A.; Kozin, S.A.; Kononikhin, A.S.; Kharybin, O.N.; Tsvetkov, P.O.; Makarov, A.A.; Nikolaev, E.N. Capabilities of MS for analytical quantitative determination of the ratio of α- and βAsp7 isoforms of the amyloid-β peptide in binary mixtures. Anal. Chem. 2011, 8, 3205–3210. [Google Scholar] [CrossRef]
- Tilli, T.M.; Mello, K.D.; Ferreira, L.B.; Matos, A.R.; Accioly, M.T.S.; Faria, P.A.; Bellahcène, A.; Castronovo, V.; Gimba, E.R. Both osteopontin-c and osteopontin-b splicing isoforms exert pro-tumorigenic roles in prostate cancer cells. Prostate 2012, 72, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.D.; Sun, L.; Yu, D.X.; Li, R.X.; Li, H.X.; Yu, Z.J.; Sheng, Q.H.; Lin, X.; Zeng, R.; Wu, J.R. Quantitative detection of single amino acid polymorphisms by targeted proteomics. Mol. Cell. Biol. 2011, 3, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chaerkady, R.; Wu, J.; Hwang, H.J.; Papadopoulos, N.; Kopelovich, L.; Maitra, A.; Matthaei, H.; Eshleman, J.R.; Hruban, R.H.; et al. Mutant proteins as cancer-specific biomarkers. Proc. Natl. Acad. Sci. USA 2011, 108, 2444–2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | PDB ID | Protein | Mw, kDa | Taxon | Number of a.a. | Locus of α-α-Corner | Number of Helical Pairs | Aliphatic Index |
|---|---|---|---|---|---|---|---|---|
| 1 | 1QAY | 3-hydroxy-3-methylglutaryl-coenzyme A reductase | 45, 6 | Pseudomonas mevalonii | 428 | 14–38 | 1 | 106, 1 |
| 2 | 1P48 | Enolase 1 | 46, 8 | Saccharomyces cerevisiae | 437 | 106–137 | 5 | 90, 7 |
| 3 | 1ONE | Enolase 1 | 46, 8 | Saccharomyces cerevisiae | 437 | 106–137 | 5 | 90, 7 |
| 4 | 1ELS | Enolase 1 | 46, 8 | Saccharomyces cerevisiae | 437 | 107–136 | 5 | 90, 7 |
| 5 | 2ANI | Ribonucleoside-diphosphate reductase subunit beta | 40, 5 | Chlamydia trachomatis | 346 | 106–143 | 8 | 97, 8 |
| 6 | 1LJ9 | Transcriptional regulator | 17, 4 | Enterococcus faecalis | 150 | 47–71 | 4 | 98, 8 |
| 7 | 1KCZ | Methyl aspartate ammonia-lyase | 45, 5 | Clostridium tetanomorphum | 413 | 127–159 | 5 | 87, 4 |
| 8 | 3DTK | Radiation response metalloprotease IrrE | 30 | Deinococcus deserti | 281 | 115–169 | 7 | 83, 3 |
| 9 | 1B9M | DNA-binding transcriptional dual regulator ModE | 28, 3 | Escherichia coli | 262 | 34–59 | 2 | 108, 8 |
| 10 | 3C07 | Putative tetR-family transcriptional regulator | 28, 3 | Streptomyces coeli color | 251 | 41–60 | 2 | 85, 2 |
| 11 | 3C4I | DNA-binding protein HU homolog | 22, 2 | Mycobacterium tuberculosis | 214 | 2–38 | 1 | 67, 3 |
| 12 | 2H1K | Pancreas/duodenum home box protein 1 | 30, 8 | Mesocricetus auratus | 283 | 27–56 | 2 | 59, 1 |
| 13 | 3F6W | DNA-binding protein | 8, 9 | Pseudomonas syringae | 80 | 26–46 | 2 | 92, 9 |
| 14 | 1E2X | Fatty acid metabolism regulator protein | 27 | Escherichia coli | 239 | 33–58 | 2 | 93, 1 |
| 15 | 1DU0 | Segmentation polarity homeobox protein engrailed | 59, 4 | Drosophila melanogaster | 552 | 27–57 | 2 | 58, 4 |
| 16 | 2G7U (A) | Transcriptional regulator | 27 | Rhodococcus jostii | 256 | 30–55 | 2 | 99, 2 |
| 17 | 2G7U (B) | Transcriptional regulator | 27 | Rhodococcus jostii | 256 | 30–55 | 2 | 99, 2 |
| 18 | 1F36 | DNA-binding protein Fis | 11, 2 | Escherichia coli | 98 | 74–94 | 3 | 86, 4 |
| 19 | 2AEK | Trichodiene synthase | 44 | Fusarium sporotrichioides | 374 | 242–278 | 13 | 75, 6 |
| 20 | 1PFU | Methionine-tRNA ligase | 76, 3 | Escherichia coli | 677 | 55–87 | 2 | 80, 3 |
| 21 | 2OER | Probable transcriptional regulator | 23, 3 | Pseudomonas aeruginosa | 210 | 44–63 | 2 | 92, 2 |
| 22 | 2F93 | Sensory rhodopsin-2 | 25, 4 | Natronomonas pharaonis | 239 | 70–117 | 3 | 126, 5 |
| 23 | 1B9N | DNA-binding transcriptional dual regulator ModE | 28, 3 | Escherichia coli | 262 | 34–59 | 2 | 108, 8 |
| 24 | 2CSF | DNA-bindingprotein SATB2 | 82, 6 | Homo sapiens | 733 | 20–44 | 1 | 81, 3 |
| 25 | 1C1D | Phenyl alanine dehydrogenase | 36, 6 | Rhodococcus | 356 | 314–345 | 16 | 91, 4 |
| 26 | 2PS6 | Trichodiene synthase | 44 | Fusarium sporotrichioides | 374 | 242–278 | 13 | 75, 6 |
| 27 | 1J6O | Uncharacterized protein | 29, 2 | Thermotoga maritima | 256 | 219–252 | 13 | 94 |
| 28 | 1PFW | Methionine-tRNA ligase | 76, 3 | Escherichia coli | 677 | 55–87 | 2 | 80, 3 |
| 29 | 1A76 | Flap endonuclease 1 | 37, 5 | Methanocaldococcus jannaschii | 326 | 200–221 | 10 | 90 |
| 30 | 3DBJ | Allophycocyanin alpha subunit | 17, 5 | Thermosynechococcus vulcanus | 161 | 114–145 | 6 | 93, 3 |
| 31 | 1F4L | Methionine-tRNAligase | 76, 3 | Escherichia coli | 677 | 55–87 | 2 | 80, 3 |
| 32 | 1A77 | Flapendonuclease 1 | 37, 5 | Methanocaldococcus jannaschii | 326 | 200–221 | 9 | 90 |
| 33 | 1PFY | Methionine-tRNA ligase | 76, 3 | Escherichia coli | 766 | 55–87 | 2 | 80, 3 |
| 34 | 2NYX | Probable transcriptional regulatory protein | 17, 5 | Mycobacterium tuberculosis | 160 | 62–86 | 3 | 106, 8 |
| 35 | 2OPX | Lactaldehyde dehydrogenase | 52, 3 | Escherichia coli | 479 | 82–117 | 3 | 92, 7 |
| 36 | 2PIJ | Protein Cro | 7, 2 | Pseudomonassp. GM80 | 67 | 15–36 | 2 | 96, 4 |
| 37 | 1PFV | Methionine-tRNA ligase | 76, 3 | Escherichia coli | 766 | 55–87 | 2 | 80, 3 |
| 38 | 3B97 | Alpha-enolase | 47, 2 | Homo sapiens | 434 | 106–137 | 5 | 88, 6 |
| 39 | 1ETO | DNA-binding protein Fis | 11, 2 | Escherichia coli | 98 | 49–82 | 2 | 86, 4 |
| 40 | 2DXI | Glutamate-tRNA ligase | 53, 9 | Thermus thermophilus | 468 | 252–272 | 11 | 88, 4 |
| 41 | 3BGW | DNA helicase | 49, 8 | Bacillus phage SPP1 | 442 | 234–256 | 13 | 83, 9 |
| 42 | 2CV1 | Glutamate-tRNA ligase | 53, 9 | Thermus thermophilus | 468 | 252–273 | 11 | 88, 4 |
| 43 | 2CV2 | Glutamate—tRNA ligase | 53, 9 | Thermus thermophilus | 468 | 252–272 | 10 | 88, 4 |
| 44 | 1B28 | Transcriptional repressor arc | 6, 2 | Salmonella phage P22 | 53 | 16–43 | 1 | 67, 9 |
| 45 | 2PSN | Alpha-enolase | 47, 2 | Homo sapiens | 434 | 106–138 | 5 | 88, 6 |
| 46 | 1PDZ | Enolase | 47 | Homarus gammarus | 433 | 107–136 | 5 | 84, 7 |
| 47 | 1Q3Q | Thermosome subunit alpha | 59, 2 | Thermococcus sp | 548 | 96–143 | 4 | 104, 5 |
| 48 | 1D1L | Regulatory protein cro | 7, 4 | Escherichi aphage lambda | 66 | 15–36 | 2 | 72, 6 |
| 49 | 2OEM | 2,3-diketo-5-methylthiopentyl-1-phosphate enolase | 44, 9 | Geobacillus kaustophilus | 413 | 360–385 | 19 | 103, 5 |
| 50 | 2NX4 | Transcriptional regulator | 21, 7 | Rhodococcus jostii | 193 | 8–40 | 1 | 92, 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rudnev, V.R.; Kulikova, L.I.; Kaysheva, A.L.; Efimov, A.V.; Tikhonov, D.A. Use of the Molecular Dynamics Method to Investigate the Stability of α-α-Corner Structural Motifs in Proteins. Symmetry 2021, 13, 1193. https://doi.org/10.3390/sym13071193
Rudnev VR, Kulikova LI, Kaysheva AL, Efimov AV, Tikhonov DA. Use of the Molecular Dynamics Method to Investigate the Stability of α-α-Corner Structural Motifs in Proteins. Symmetry. 2021; 13(7):1193. https://doi.org/10.3390/sym13071193
Chicago/Turabian StyleRudnev, Vladimir R., Liudmila I. Kulikova, Anna L. Kaysheva, Alexander V. Efimov, and Dmitry A. Tikhonov. 2021. "Use of the Molecular Dynamics Method to Investigate the Stability of α-α-Corner Structural Motifs in Proteins" Symmetry 13, no. 7: 1193. https://doi.org/10.3390/sym13071193
APA StyleRudnev, V. R., Kulikova, L. I., Kaysheva, A. L., Efimov, A. V., & Tikhonov, D. A. (2021). Use of the Molecular Dynamics Method to Investigate the Stability of α-α-Corner Structural Motifs in Proteins. Symmetry, 13(7), 1193. https://doi.org/10.3390/sym13071193