The Status of Edge Strands in Ferredoxin-Like Fold

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Data
2.2. Fuzzy Oil Drop Model
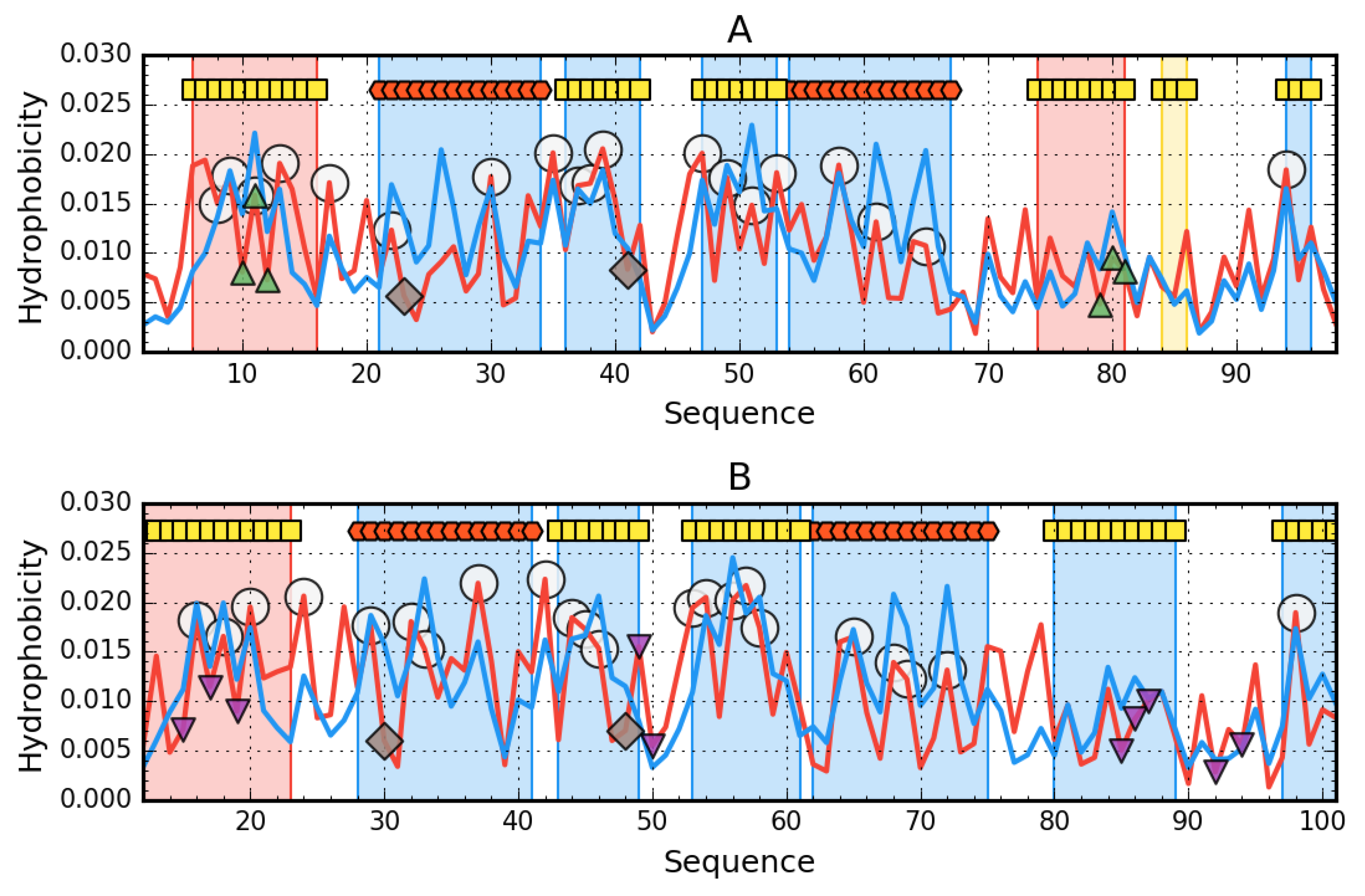
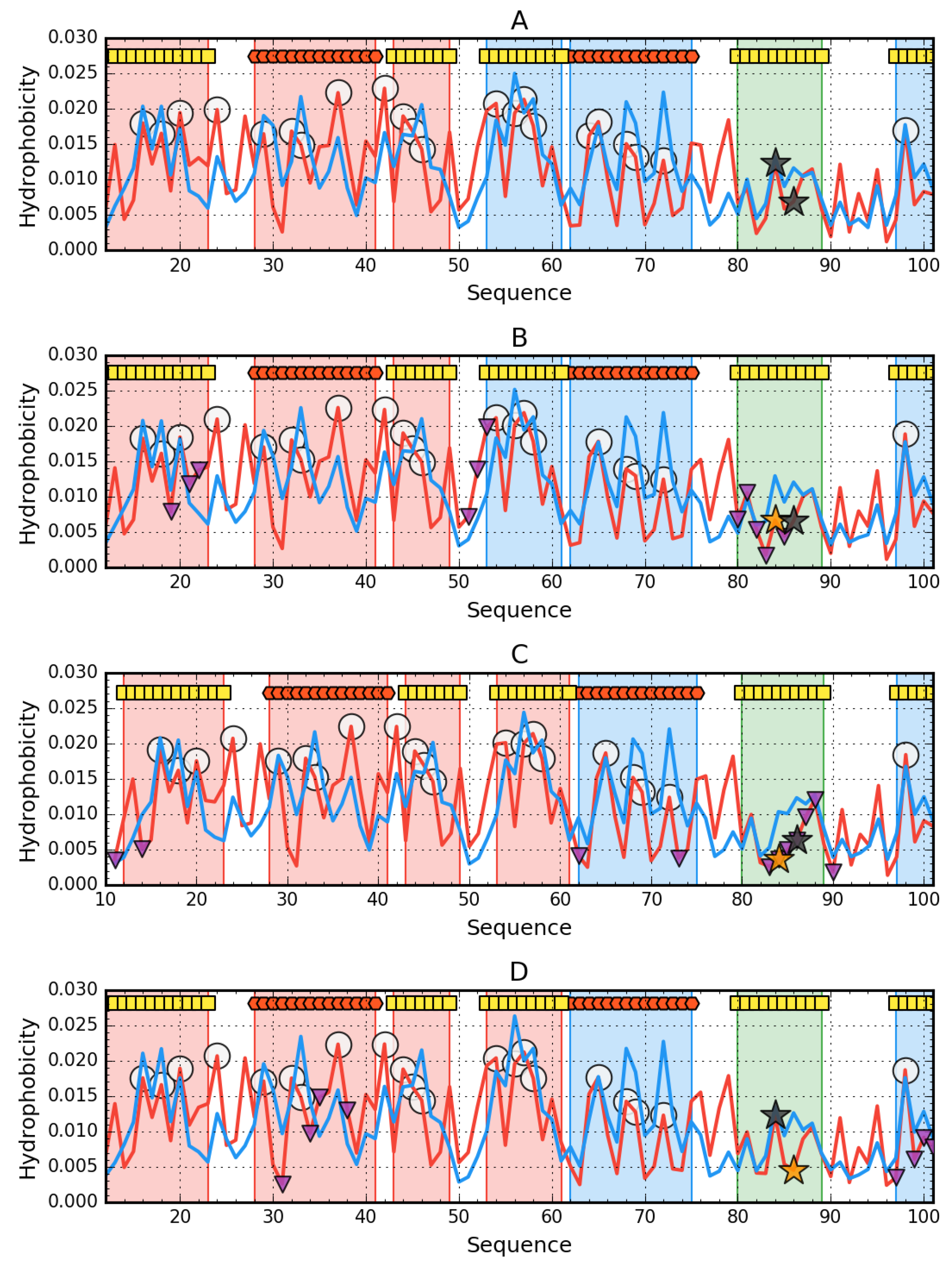
3. Results
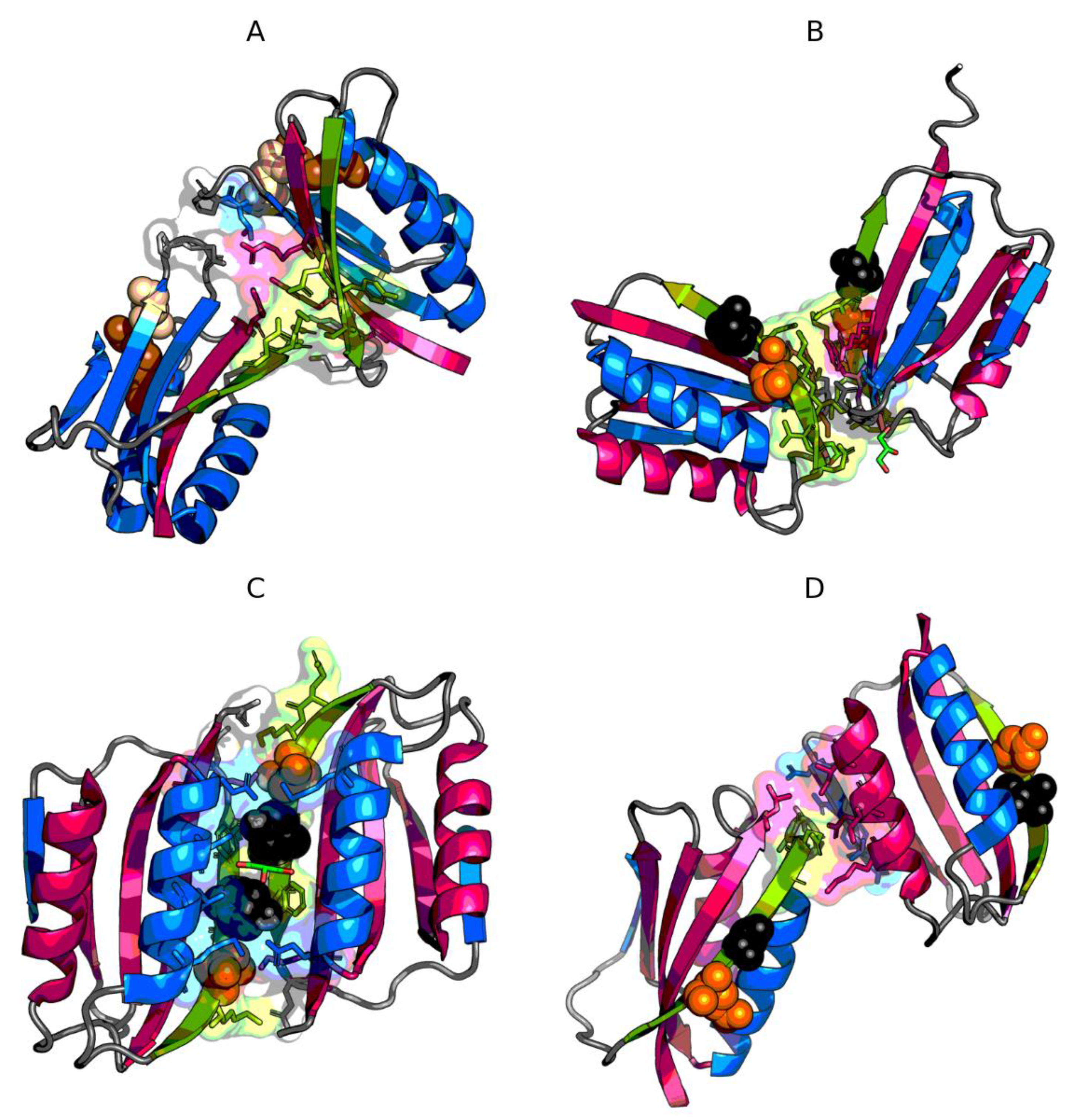
3.1. The Presence of Mutations Limiting the Predisposition to Aggregation of AclSS
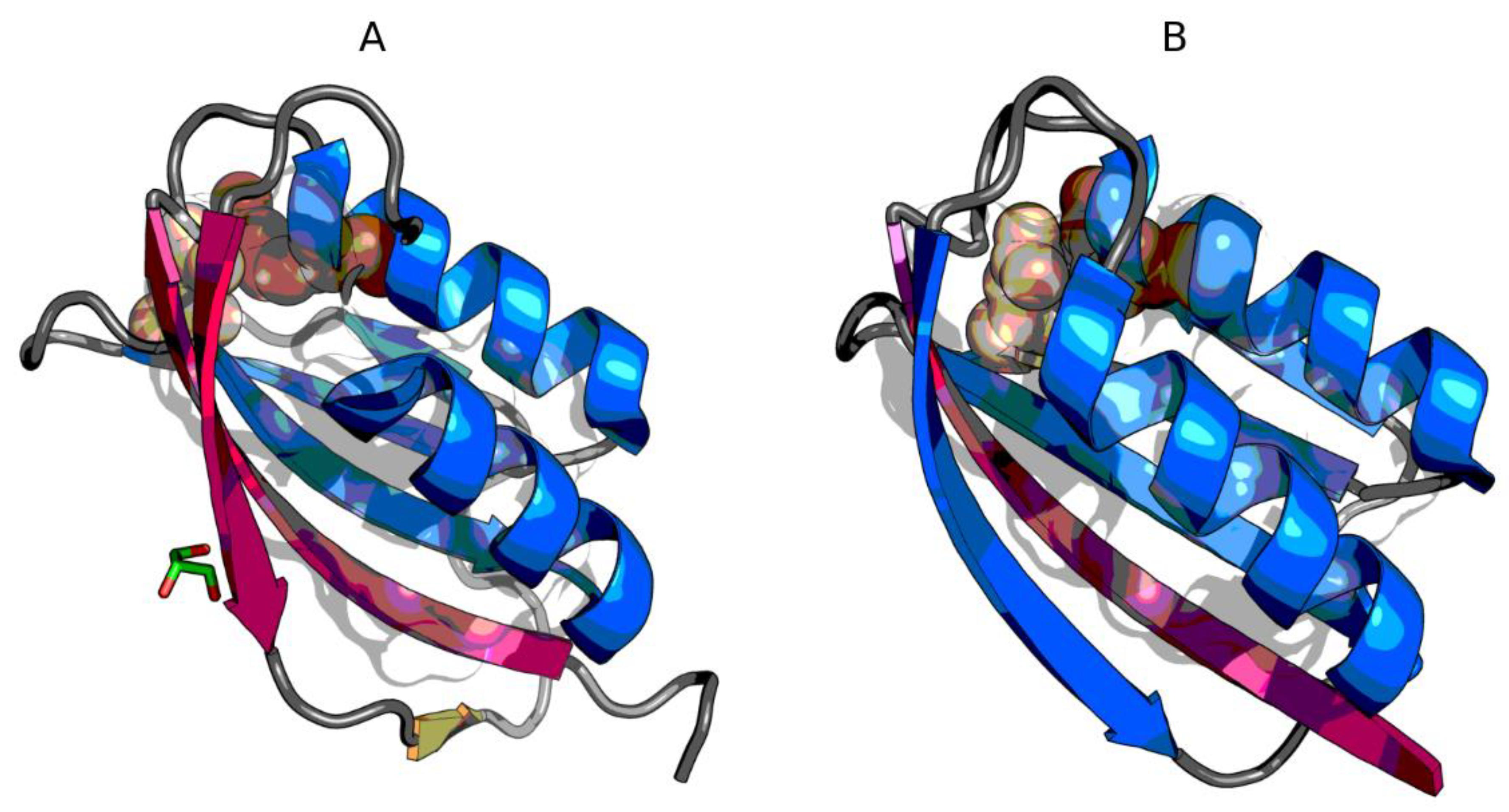
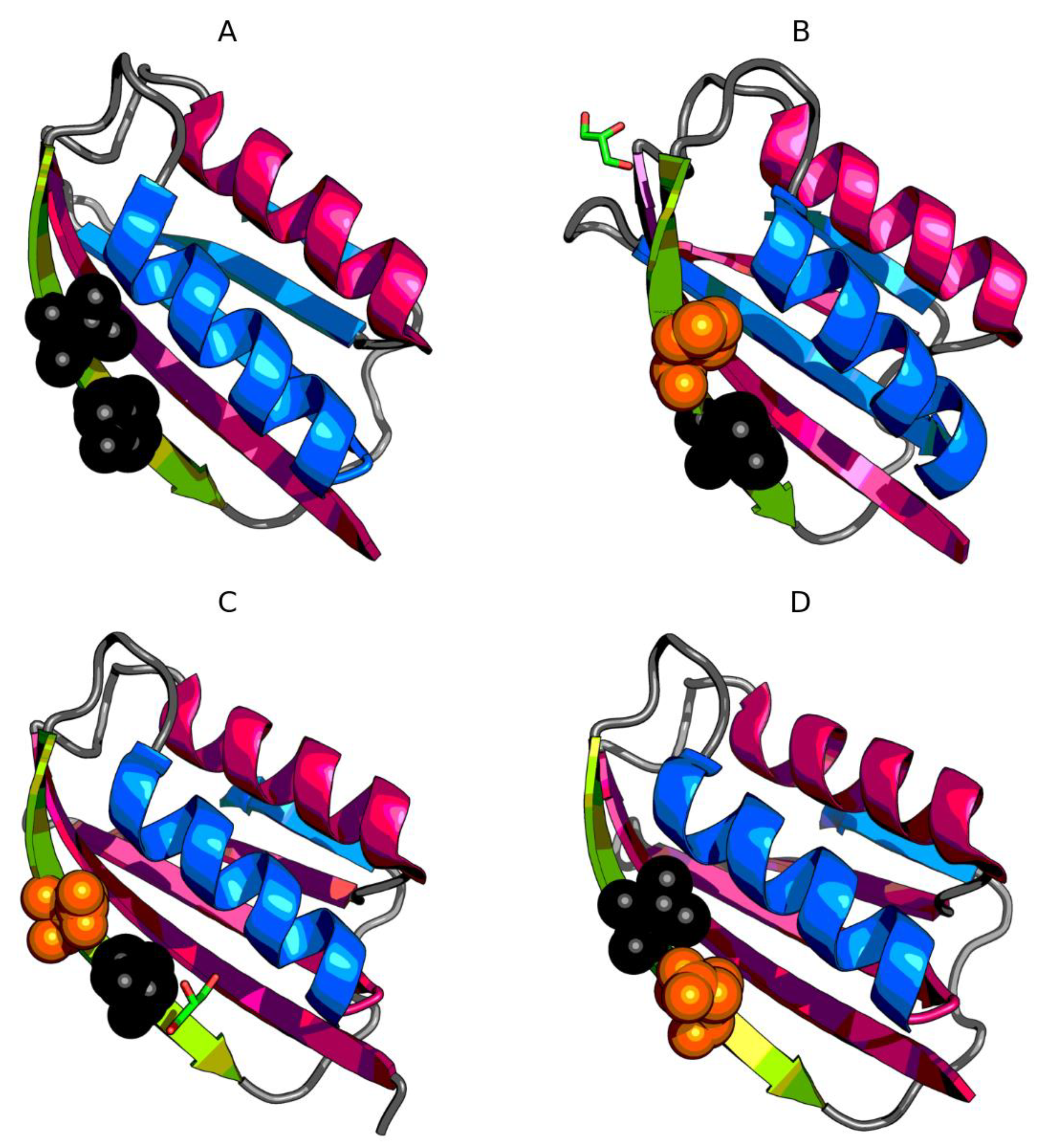
3.2. The Status of Edge β-Strands in Complexes
3.3. Presence of Trifluoroethanol (TFE)
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Selkoe, D. Folding proteins in fatal ways. Nature 2003, 426, 900–904. [Google Scholar] [CrossRef]
- Richardson, J.S.; Richardson, D.C. Natural β-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. USA 2002, 99, 2754–2759. [Google Scholar] [CrossRef]
- Bemporad, F.; Calloni, G.; Campioni, S.; Plakoutsi, G.; Taddei, N.; Chiti, F. Sequence and Structural Determinants of Amyloid Fibril Formation. Accounts Chem. Res. 2006, 39, 620–627. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Amyloid formation by globular proteins under native conditions. Nat. Methods 2008, 5, 15–22. [Google Scholar] [CrossRef]
- Bahar, I.; Cheng, M.H.; Lee, J.Y.; Kaya, C.; Zhang, S. Structure-Encoded Global Motions and Their Role in Mediating Protein-Substrate Interactions. Biophys. J. 2015, 109, 1101–1109. [Google Scholar] [CrossRef]
- Ferrolino, M.C.; Zhuravleva, A.; Budyak, I.L.; Krishnan, B.; Gierasch, L.M. Delicate Balance between Functionally Required Flexibility and Aggregation Risk in a β-Rich Protein. Biochemistry 2013, 52, 8843–8854. [Google Scholar] [CrossRef] [PubMed]
- De Simone, A.; Dhulesia, A.; Soldi, G.; Vendruscolo, M.; Hsu, S.-T.D.; Chiti, F.; Dobson, C.M. Experimental free energy surfaces reveal the mechanisms of maintenance of protein solubility. Proc. Natl. Acad. Sci. USA 2011, 108, 21057–21062. [Google Scholar] [CrossRef]
- Corazza, A.; Rosano, C.; Pagano, K.; Alverdi, V.; Esposito, G.; Capanni, C.; Bemporad, F.; Plakoutsi, G.; Stefani, M.; Chiti, F.; et al. Structure, conformational stability, and enzymatic properties of acylphosphatase from the hyperthermophile Sulfolobus solfataricus. Proteins: Struct. Funct. Bioinform. 2005, 62, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Plakoutsi, G.; Bemporad, F.; Monti, M.; Pagnozzi, D.; Pucci, P.; Chiti, F.; Pucci, P. Exploring the Mechanism of Formation of Native-like and Precursor Amyloid Oligomers for the Native Acylphosphatase from Sulfolobus solfataricus. Structure 2006, 14, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, M.; Bemporad, F.; Pellegrino, S.; Chiti, F.; Bolognesi, M.; Ricagno, S. Edge strand engineering prevents native-like aggregation in Sulfolobus solfataricus acylphosphatase. FEBS J. 2014, 281, 4072–4084. [Google Scholar] [CrossRef]
- Cheung, Y.-Y.; Lam, S.Y.; Chu, W.-K.; Allen, M.D.; Bycroft, M.; Wong, K.-B. Crystal Structure of a Hyperthermophilic Archaeal Acylphosphatase fromPyrococcus horikoshiiStructural Insights into Enzymatic Catalysis, Thermostability, and Dimerization†, ‡. Biochemistry 2005, 44, 4601–4611. [Google Scholar] [CrossRef] [PubMed]
- Bemporad, F.; De Simone, A.; Chiti, F.; Dobson, C.M. Characterizing Intermolecular Interactions That Initiate Native-Like Protein Aggregation. Biophys. J. 2012, 102, 2595–2604. [Google Scholar] [CrossRef] [PubMed]
- Chiti, T.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Konieczny, L.; Brylinski, M.; Roterman, I. Gauss-function-Based model of hydrophobicity density in proteins. Silico Boil. 2006, 6, 15–22. [Google Scholar]
- Kalinowska, B.; Banach, M.; Konieczny, L.; Roterman, A.I. Application of Divergence Entropy to Characterize the Structure of the Hydrophobic Core in DNA Interacting Proteins. Entropy 2015, 17, 1477–1507. [Google Scholar] [CrossRef]
- Zuccotti, S.; Rosano, C.; Ramazzotti, M.; Degl’Innocenti, D.; Stefani, M.; Manao, G.; Bolognesi, M. Three-dimensional structural characterization of a novel Drosophila melanogaster acylphosphatase. Acta Crystallogr. Sect. D Boil. Crystallogr. 2004, 60, 1177–1179. [Google Scholar] [CrossRef]
- Fusco, G.; De Simone, A.; Hsu, S.-T.D.; Bemporad, F.; Vendruscolo, M.; Chiti, F.; Dobson, C.M. 1H, 13C and 15N resonance assignments of human muscle acylphosphatase. Biomol. NMR Assign. 2011, 6, 27–29. [Google Scholar] [CrossRef]
- Pagano, K.; Ramazzotti, M.; Viglino, P.; Esposito, G.; Degl’Innocenti, D.; Taddei, N.; Corazza, A. NMR solution structure of the acylphosphatase from Escherichia coli. J. Biomol. NMR 2006, 36, 199–204. [Google Scholar] [CrossRef]
- Pastore, A.; Saudek, V.; Ramponi, G.; Williams, R.J. Three-dimensional structure of acylphosphatase. J. Mol. Boil. 1992, 224, 427–440. [Google Scholar] [CrossRef]
- Thunnissen, M.M.; Taddei, N.; Liguri, G.; Ramponi, G.; Nordlund, P. Crystal structure of common type acylphosphatase from bovine testis. Structure 1997, 5, 69–79. [Google Scholar] [CrossRef]
- Zuccotti, S.; Rosano, C.; Bemporad, F.; Stefani, M.; Bolognesi, M. Preliminary characterization of two different crystal forms of acylphosphatase from the hyperthermophile archaeon Sulfolobus solfataricus. Acta Crystallogr. Sect. F Struct. Boil. Cryst. Commun. 2004, 61, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Soldi, G.; Plakoutsi, G.; Taddei, N.; Chiti, F. Stabilization of a Native Protein Mediated by Ligand Binding Inhibits Amyloid Formation Independently of the Aggregation Pathway. J. Med. Chem. 2006, 49, 6057–6064. [Google Scholar] [CrossRef] [PubMed]
- Bemporad, F.; Ramazzotti, M. From the Evolution of Protein Sequences Able to Resist Self-Assembly to the Prediction of Aggregation Propensity. In International Review of Cell and Molecular Biology; Elsevier BV: Amsterdam, The Netherlands, 2017; pp. 1–47. [Google Scholar]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Homepage of CATH Database. Available online: http://www.cathdb.info/ (accessed on 4 May 2011).
- Dawson, N.L.; Lewis, T.E.; Das, S.; Lees, J.G.; Lee, D.; Ashford, P.; Orengo, C.A.; Sillitoe, I. CATH: An expanded resource to predict protein function through structure and sequence. Nucleic Acids Res. 2016, 45, D289–D295. [Google Scholar] [CrossRef]
- Kullback, S.; Leibler, R.A. On Information and Sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Banach, M.; Fabian, P.; Stapor, K.; Konieczny, L.; Roterman, I. Structure of the Hydrophobic Core Determines the 3D Protein Structure—Verification by Single Mutation Proteins. Biomolecules 2020, 10, 767. [Google Scholar] [CrossRef]
- Banach, M.; Konieczny, L.; Roterman, A.I. The Amyloid as a Ribbon-Like Micelle in Contrast to Spherical Micelles Represented by Globular Proteins. Molecules 2019, 24, 4395. [Google Scholar] [CrossRef]
- Fabian, P.; Banach, M.; Stapor, K.; Konieczny, L.; Roterman, I. Structure of amyloid versus structure of globular proteins. Int. J. Mol. Sci. 2020, in press.
- Prymula, K.; Jadczyk, T.; Roterman, A.I. Catalytic residues in hydrolases: Analysis of methods designed for ligand-binding site prediction. J. Comput. Mol. Des. 2010, 25, 117–133. [Google Scholar] [CrossRef]
- Mohanta, D.; Jana, M. Can 2,2,2-trifluoroethanol be an efficient protein denaturant than methanol and ethanol under thermal stress? Phys. Chem. Chem. Phys. 2018, 20, 9886–9896. [Google Scholar] [CrossRef]
- Khan, M.V.; Rabbani, G.; Ishtikhar, M.; Khan, S.; Saini, G.; Khan, R. Non-fluorinated cosolvents: A potent amorphous aggregate inducer of metalloproteinase-conalbumin (ovotransferrin). Int. J. Boil. Macromol. 2015, 78, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Molla, A.R.; Mandal, D.K. Trifluoroethanol-induced conformational change of tetrameric and monomeric soybean agglutinin: Role of structural organization and implication for protein folding and stability. Biochim. 2013, 95, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Jäntschi, L. Structure–property relationships for solubility of monosaccharides. Appl. Water Sci. 2019, 9, 38. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Abbreviations Used (PDB ID) | Sequence Identity | Length (aa) | Secondary Structure | Ref. |
|---|---|---|---|---|---|
| Acylphosphatase Drosphila Melanogaster | AclDM (1URR) | LOW | 98 | 3.30.70.100 Alpha Beta 2-Layer Sandwich | [16] |
| Acylphosphatase Sulfolobus solfataricus | AclSS (2BJD) | 101 | 3.30.70.100 Alpha Beta 2-Layer Sandwich | [8] | |
| AclSS(V84D) (4OJG) | V84D | 101 | 3.30.70.100 | [10] | |
| AclSS(V84P) (4OJ3) | V84P | 101 | 3.30.70.100 | [10] | |
| AclSS(V86E) (4OJH) | Y86E | 101 | 3.30.70.100 | [10] | |
| AclSS ΔN11 (4OXI) | ΔN11 | 101 | 3.30.70.100 | [10] |
| AclDM [16] (1URR) | Fragment | CC | RD | RD | CC | Fragment | AclSS [8] (2BJD) |
|---|---|---|---|---|---|---|---|
| AclDM | 2-98 | 0.617 | 0.418 | 0.422 | 0.613 | 12-101 | AclSS [8]Beta |
| Helix | |||||||
| Beta | 6-16 | 0.346 | 0.590 | 0.545 | 0.612 | 12-23 | Beta |
| Helix | 21-34 | 0.498 | 0.434 | 0.458 | 0.474 | 28-41 | Beta |
| Beta | 36-42 | 0.810 | 0.337 | 0.474 | 0.487 | 43-49 | Helix |
| Beta | 47-53 | 0.465 | 0.431 | 0.473 | 0.59 | 53-61 | |
| Helix | 54-67 | 0.473 | 0.464 | 0.317 | 0.715 | 62-75 | Beta |
| Beta | 74-81 | 0.542 | 0.561 | 0.492 | Beta | ||
| Beta | 84-86 | 0.194 | 0.496 | 0.292 | 0.793 | 80-89 | |
| Beta | 94-96 | 0.960 | 0.154 | 0.200 | 0.955 | 97-101 | β-sheet |
| β-sheet | 0.607 | 0.444 | 0.389 | 0.696 | P–P | ||
| Ligand bind. | 10−12,79−81 | 0.956 | 0.101 | 0.469 | 0.497 | 15,17,19,49,50 | Enzymatic |
| 85-87,92,94 | 30R | ||||||
| Enzymatic | 18-28 | 48N | |||||
| 23R | 36-46 | 0.198 | 0.640 | 0.570 | 0.259 | 25-35 | |
| 41N | 0.853 | 0.222 | 0.508 | 0.504 | 43-53 |
| Protein ID | 2BJD | 4OIX | 4OJG | 4OJ3 | 4OJH | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Name | AclSS | ΔN11 | ΔN11 | ΔN11 | ΔN11 | |||||
| Mutation | V84D | V84P | Y86E | |||||||
| RD | CC | RD | CC | RD | CC | RD | CC | RD | CC | |
| Chain | 0.422 | 0.613 | 0.432 | 0.595 | 0.438 | 0.575 | 0.447 | 0.577 | 0.467 | 0.567 |
| 23-12 | 0.545 | 0.612 | 0.531 | 0.636 | 0.640 | 0.520 | 0.558 | 0.613 | 0.605 | 0.604 |
| 28-41 H | 0.458 | 0.474 | 0.537 | 0.317 | 0.504 | 0.388 | 0.523 | 0.365 | 0.530 | 0.358 |
| 43-49 | 0.474 | 0.487 | 0.522 | 0.355 | 0.507 | 0.394 | 0.515 | 0.373 | 0.522 | 0.371 |
| 53-61 | 0.473 | 0.590 | 0.471 | 0.578 | 0.505 | 0.541 | 0.463 | 0.595 | 0.512 | 0.563 |
| 62-75 H | 0.317 | 0.715 | 0.333 | 0.697 | 0.322 | 0.724 | 0.313 | 0.726 | 0.301 | 0.73 |
| 80-89 | 0.292 | 0.793 | 0.252 | 0.821 | 0.372 | 0.636 | 0.468 | 0.454 | 0.460 | 0.547 |
| 97-101 | 0.200 | 0.955 | 0.165 | 0.950 | 0.198 | 0.950 | 0.186 | 0.963 | 0.155 | 0.960 |
| Beat-sheet | 0.389 | 0.696 | 0.393 | 0.685 | 0.423 | 0.640 | 0.408 | 0.660 | 0.431 | 0.658 |
| Mutant | 0.390 | 0.601 | 0.482 | 0.300 | 0.532 | 0.131 | 0.486 | 0.434 | ||
| Protein | Complete | P–P | NoP–P | Edge Fragment | ||
|---|---|---|---|---|---|---|
| Chain A | Chain B | Chains A+B | ||||
| AclSS (2BJD) | 0.684 | 0.488 (11/17) | 0.651 | 0.754 | 0.848 | 0.810 |
| 4OJG (V84D) | 0.676 | 0.281 (13/22) | 0.629 | 0.411 | 0.416 | 0.414 |
| 4OJ3 (V84P) | 0.650 | 0.532 (10/22) | 0.608 | 0.722 | 0.635 | 0.687 |
| 4OJH (Y86E) | 0.670 | 0.502 (2/14) | 0.630 | 0.498 | 0.763 | 0.837 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banach, M.; Fabian, P.; Stapor, K.; Konieczny, L.; Ptak-Kaczor, M.; Roterman, I. The Status of Edge Strands in Ferredoxin-Like Fold. Symmetry 2020, 12, 1032. https://doi.org/10.3390/sym12061032
Banach M, Fabian P, Stapor K, Konieczny L, Ptak-Kaczor M, Roterman I. The Status of Edge Strands in Ferredoxin-Like Fold. Symmetry. 2020; 12(6):1032. https://doi.org/10.3390/sym12061032
Chicago/Turabian StyleBanach, Mateusz, Piotr Fabian, Katarzyna Stapor, Leszek Konieczny, Magdalena Ptak-Kaczor, and Irena Roterman. 2020. "The Status of Edge Strands in Ferredoxin-Like Fold" Symmetry 12, no. 6: 1032. https://doi.org/10.3390/sym12061032
APA StyleBanach, M., Fabian, P., Stapor, K., Konieczny, L., Ptak-Kaczor, M., & Roterman, I. (2020). The Status of Edge Strands in Ferredoxin-Like Fold. Symmetry, 12(6), 1032. https://doi.org/10.3390/sym12061032