Structure, Function, and Therapeutic Use of IgM Antibodies

Abstract
1. Introduction to Immunoglobulin M (IgM)
1.1. History and Discovery of IgM
1.2. Evolution of IgM Antibodies
1.3. Ontogeny of B Cells and IgM Antibodies
2. Biology of IgM
2.1. Innate Immunity
2.2. Early Adaptive Immunity
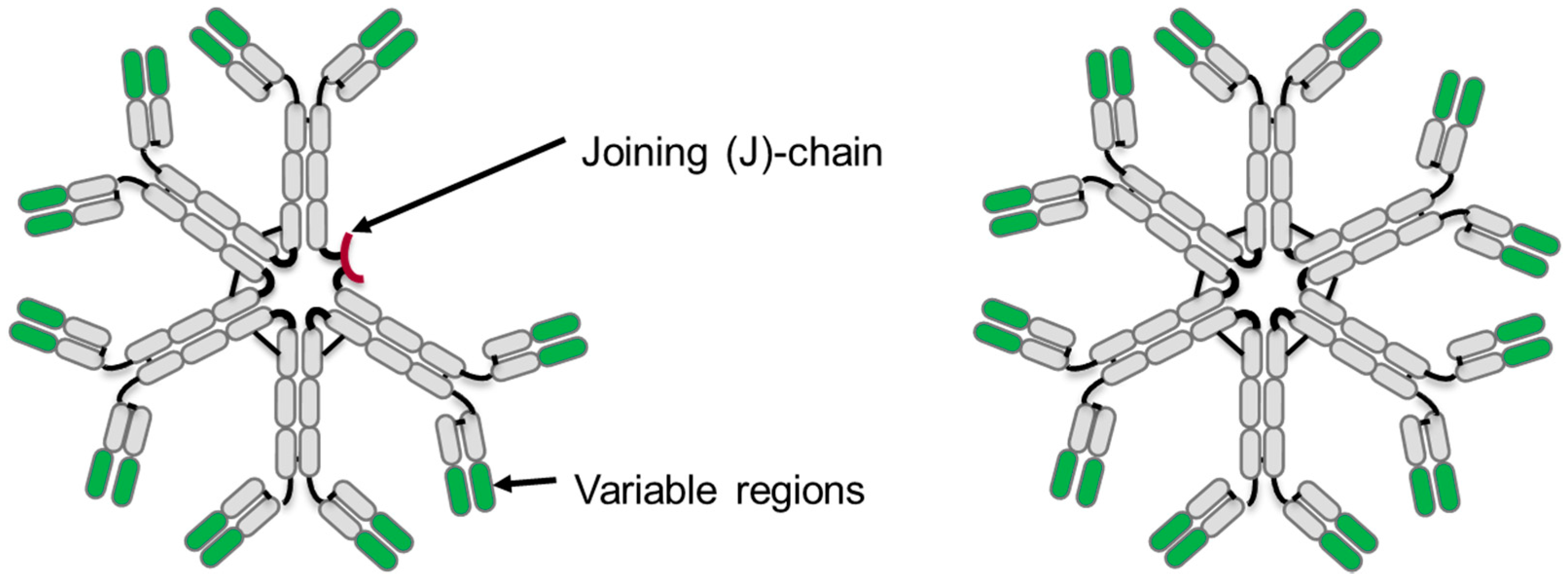
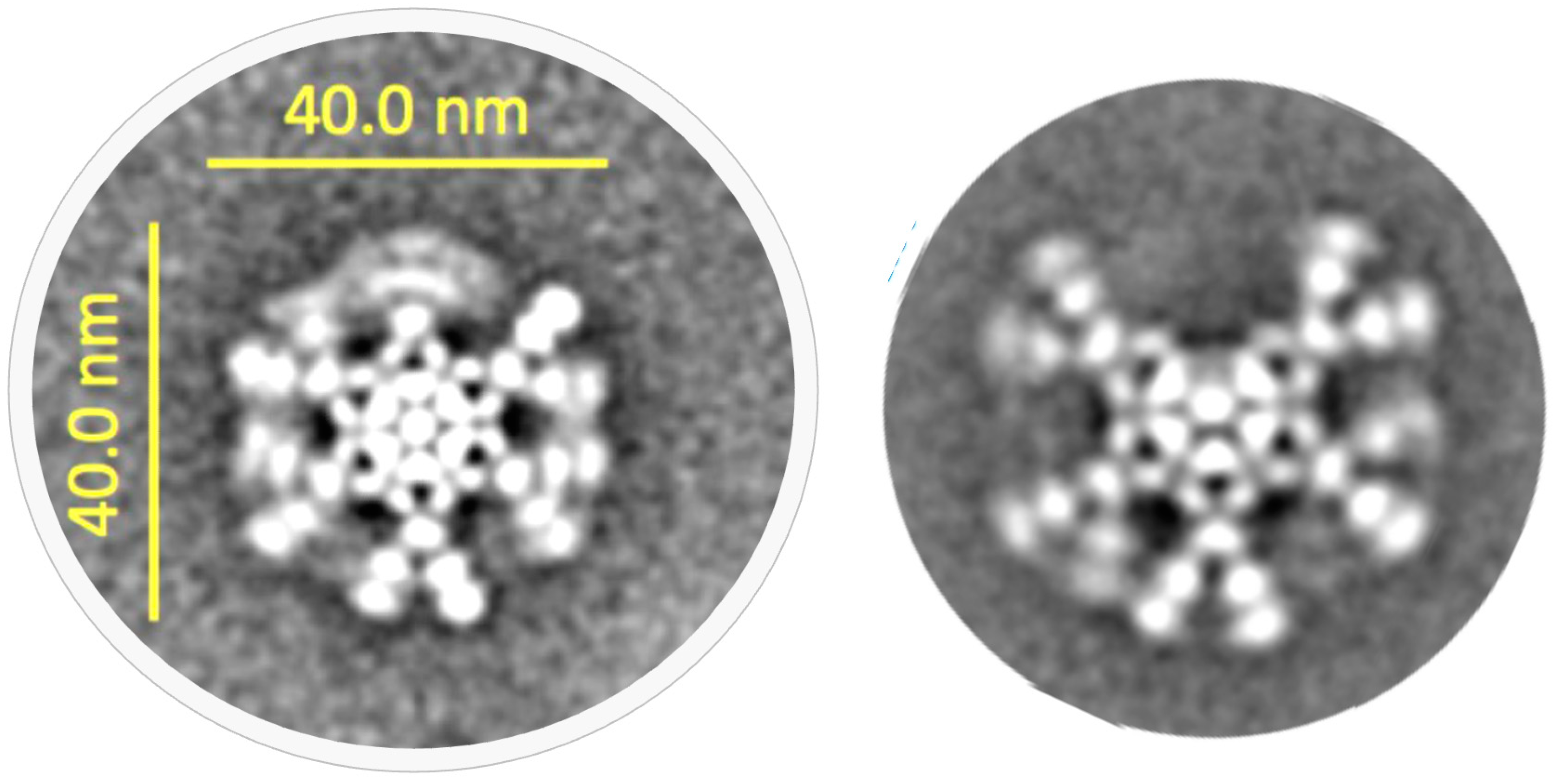
3. IgM Antibody Structure
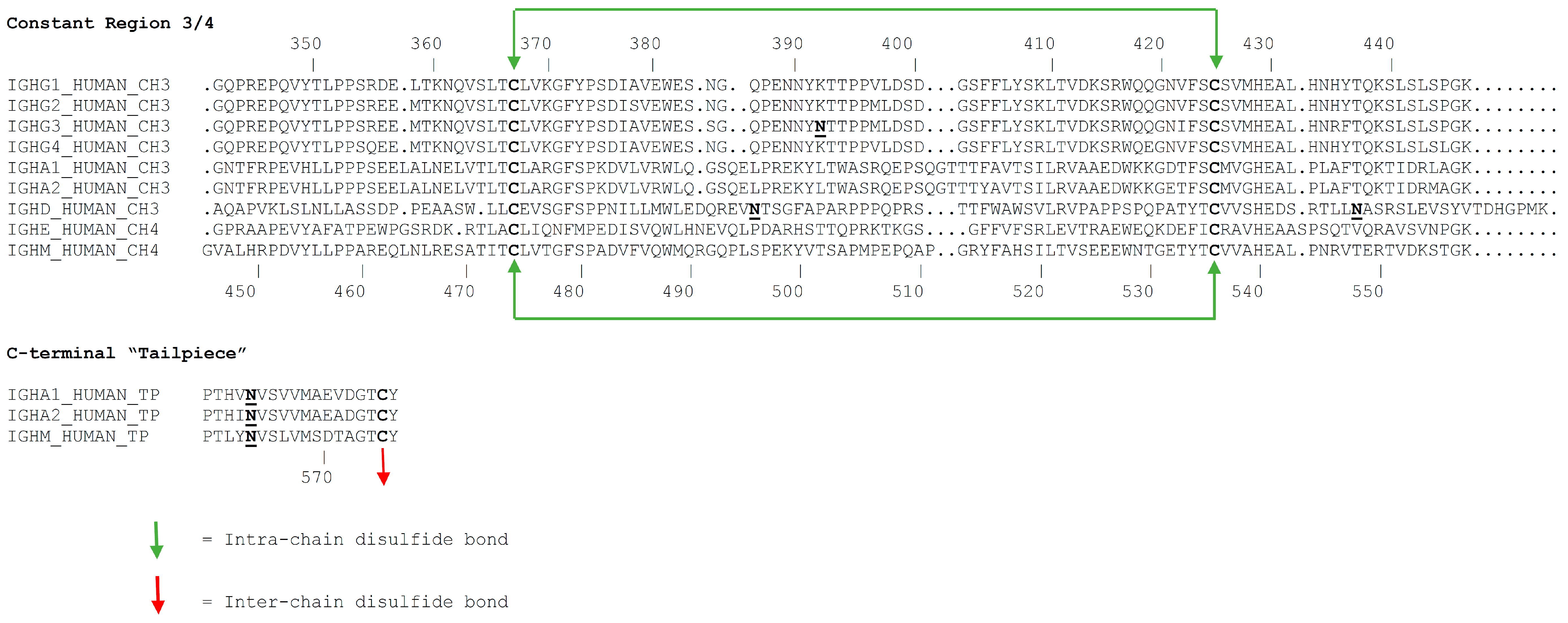
3.1. Primary Structure
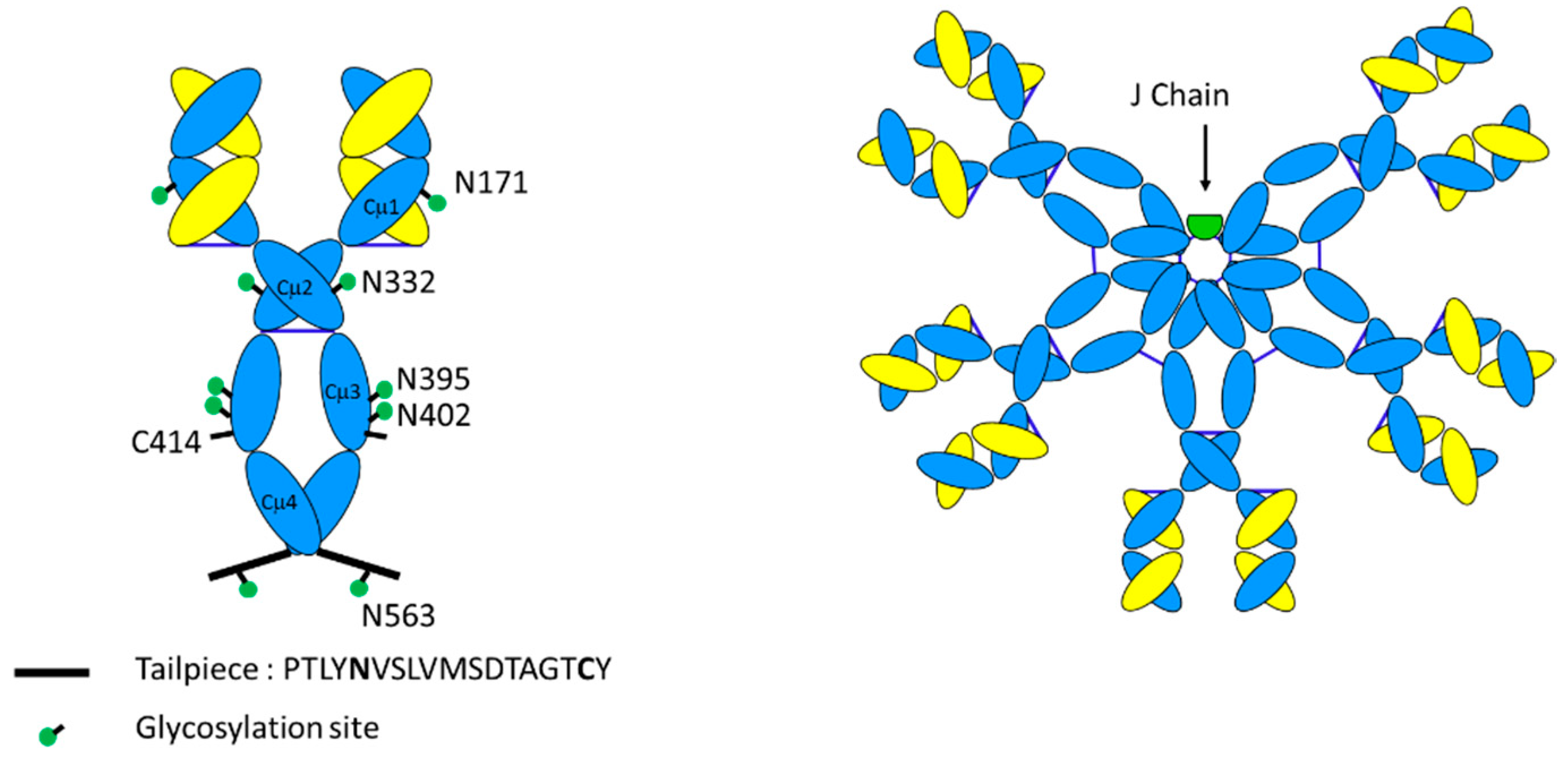
3.2. Glycosylation
3.3. Tertiary Structure
4. Function
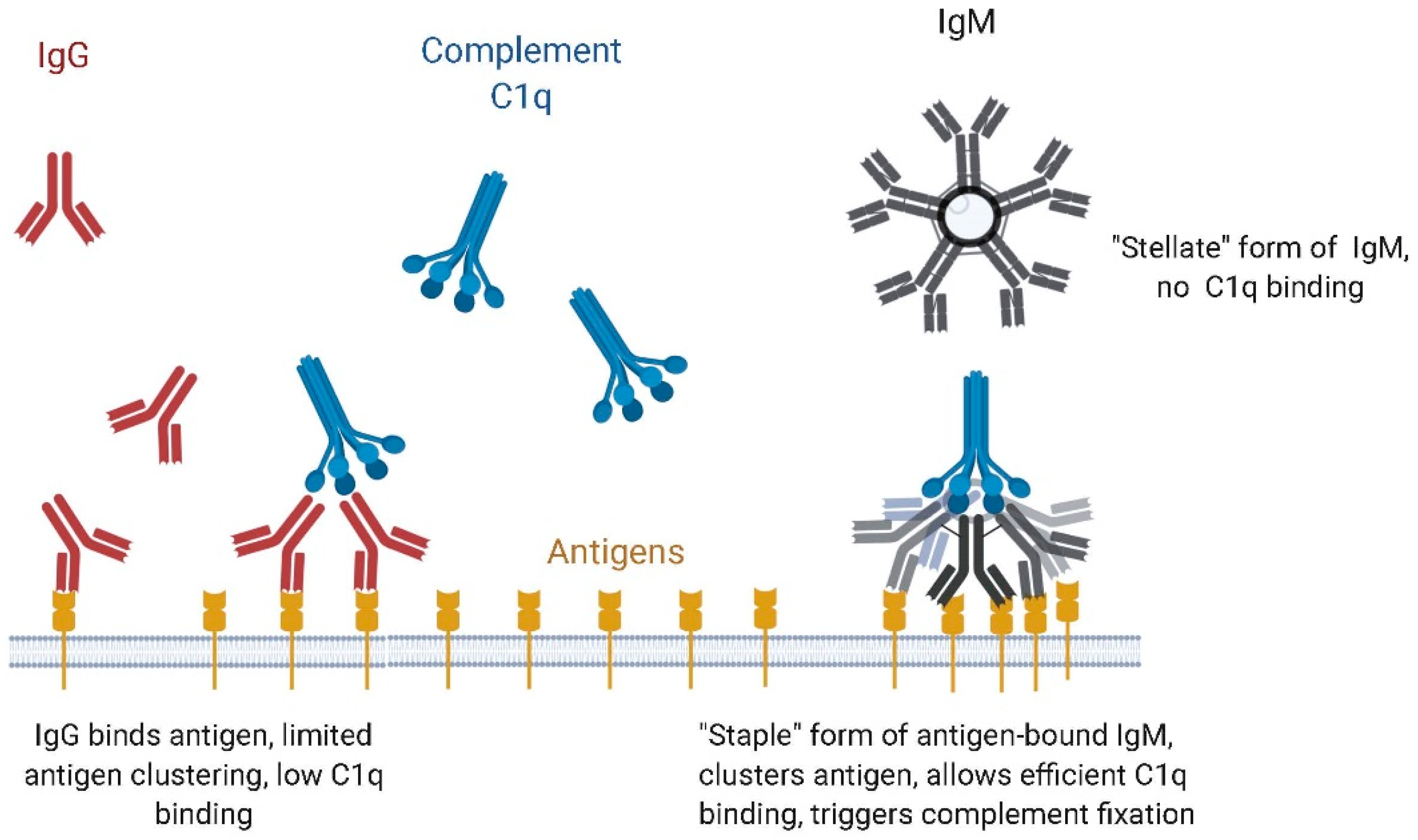
4.1. Binding to Microbial Antigens, Role of Avidity
4.2. IgM vs. IgG Function: Complement Dependent Cytotoxicity (CDC) vs. Antibody-Dependent Cell-Based Cytotoxicity (ADCC)
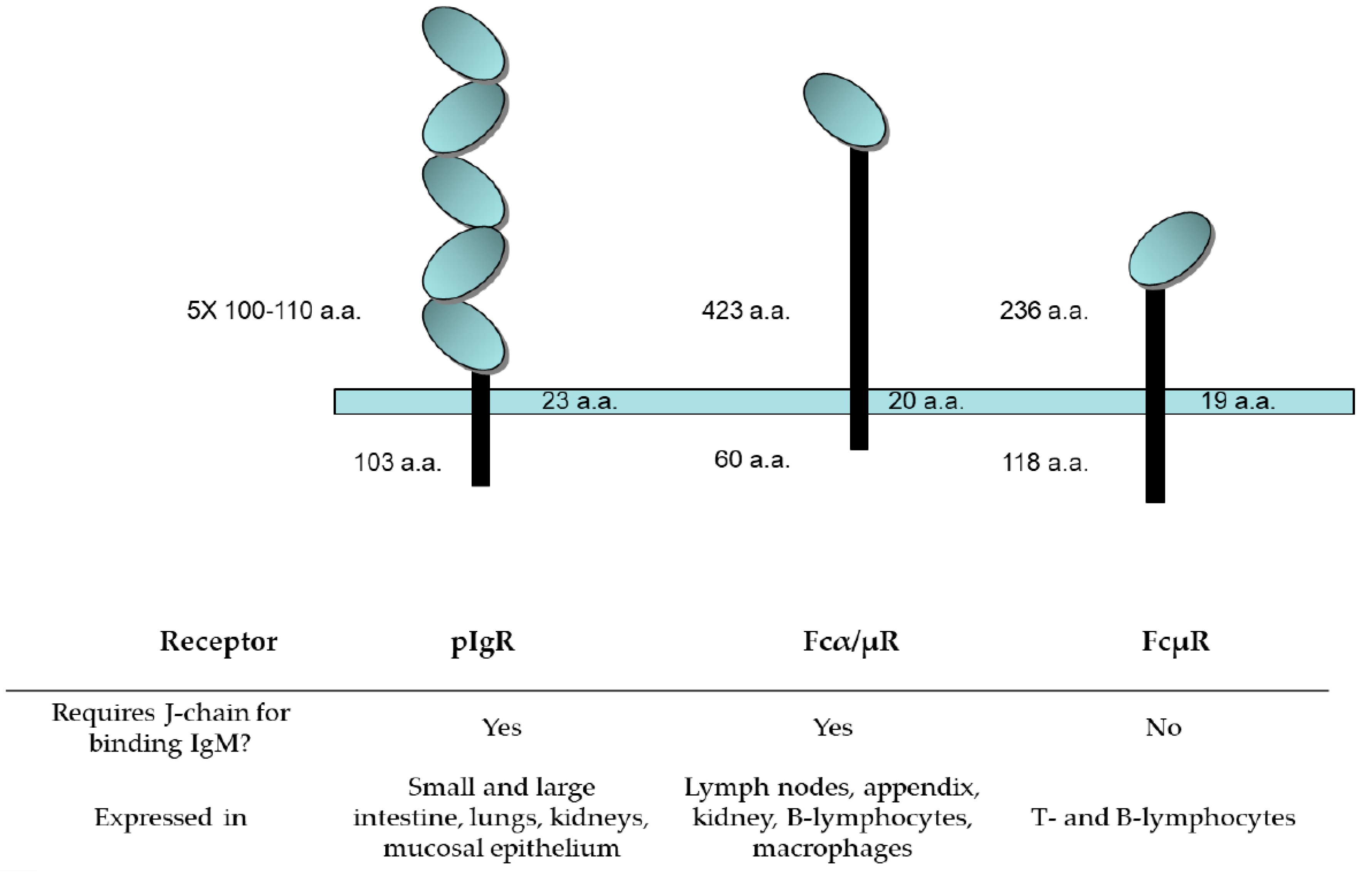
4.3. IgM Receptors: Structure and Tissue Distribution
4.3.1. Polymeric Ig Receptor (pIgR)
4.3.2. Fcα/µR
4.3.3. FcµR, the TOSO Receptor
5. Manufacturing Considerations
5.1. Expression of IgM
5.2. Purification of IgM
6. Therapeutic Uses of IgM Antibodies
6.1. IgM Clinical Trials
6.1.1. Lipopolysaccharide Antigens
6.1.2. Glycolipid and Proteolipid Antigens
6.1.3. Glycan Antigens
6.1.4. Protein Antigens
6.2. IgM Pharmacokinetics
6.3. IgM Safety and Immunogenicity
6.4. Other Oligomeric Antibody Forms
7. Future Applications of Therapeutic IgM
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nuttall, G. Experimente uber die bacterienfeindlichen Einflusse des theirischen Korpers. Z. Hyg. Infektionskr. 1888, 4, 353–395. [Google Scholar]
- Black, C.A. A brief history of the discovery of the immunoglobulins and the origin of the modern immunoglobulin nomenclature. Immunol. Cell Biol. 1997, 75, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Kabat, E.A. The Molecular Weight of Antibodies. J. Exp. Med. 1939, 69, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Waldenstrom, J. Incipient myelomatisis or “essential” hyperglobulinemia with fibrinogenopenia—A new syndrome? Acta Med. Scand. 1944, 67, 216–247. [Google Scholar]
- Wallenius, G.; Trautman, R.; Kunkel, H.G.; Franklin, E.C. Ultracentrifugal studies of major non-lipide electrophoretic components of normal human serum. J. Biol. Chem. 1957, 225, 253–267. [Google Scholar]
- Potter, M. The early history of plasma cell tumors in mice, 1954–1976. Adv. Cancer Res. 2007, 98, 17–51. [Google Scholar] [CrossRef]
- Ceppellini, R.; Dray, S.; Edelman, G.; Fahey, J.; Franek, F.; Franklin, E. Nomenclature for human immunoglobin. Bull World Health Org 1964, 30, 447–450. [Google Scholar]
- Flajnik, M.F.; Du Pasquier, L. Evolution of innate and adaptive immunity: Can we draw a line? Trends Immunol. 2004, 25, 640–644. [Google Scholar] [CrossRef]
- Litman, G.W.; Rast, J.P.; Fugmann, S.D. The origins of vertebrate adaptive immunity. Nat. Rev. Immunol. 2010, 10, 543–553. [Google Scholar] [CrossRef]
- Flajnik, M.F. Comparative analyses of immunoglobulin genes: Surprises and portents. Nat. Rev. Immunol. 2002, 2, 688–698. [Google Scholar] [CrossRef]
- Marchalonis, J.J.; Jensen, I.; Schluter, S.F. Structural, antigenic and evolutionary analyses of immunoglobulins and T cell receptors. J. Mol. Recognit. 2002, 15, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Dolder, F. Occurrence, isolation and interchain bridges of natural 7-S immunoglobulin M in human serum. Biochim. Biophys. Acta 1971, 236, 675–685. [Google Scholar] [PubMed]
- Eskeland, T.; Christensen, T.B. IgM molecules with and without J chain in serum and after purification, studied by ultracentrifugation, electrophoresis, and electron microscopy. Scand. J. Immunol. 1975, 4, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Hadji-Azimi, I.; Michea-Hamzehpour, M. Xenopus laevis 19S immunoglobulin. Ultrastructure and J chain isolation. Immunology 1976, 30, 587–591. [Google Scholar] [PubMed]
- Fillatreau, S.; Six, A.; Magadan, S.; Castro, R.; Sunyer, J.O.; Boudinot, P. The astonishing diversity of Ig classes and B cell repertoires in teleost fish. Front. Immunol. 2013, 4, 28. [Google Scholar] [CrossRef]
- Getahun, A.; Lundqvist, M.; Middleton, D.; Warr, G.; Pilstrom, L. Influence of the mu-chain C-terminal sequence on polymerization of immunoglobulin M. Immunology 1999, 97, 408–413. [Google Scholar] [CrossRef]
- Cattaneo, A.; Neuberger, M.S. Polymeric immunoglobulin M is secreted by transfectants of non-lymphoid cells in the absence of immunoglobulin J chain. EMBO J. 1987, 6, 2753–2758. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.C.; Roux, K.H.; Shulman, M.J. On the structure of polymeric IgM. Eur. J. Immunol. 1988, 18, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.C.; Shulman, M.J. IgM—Molecular requirements for its assembly and function. Immunol. Today 1989, 10, 118–122. [Google Scholar] [CrossRef]
- Collins, C.; Tsui, F.W.; Shulman, M.J. Differential activation of human and guinea pig complement by pentameric and hexameric IgM. Eur. J. Immunol. 2002, 32, 1802–1810. [Google Scholar] [CrossRef]
- Duramad, O.; Wang, B.; Zheng, F.; Keyt, L.; Repellin, C.; Beviglia, L.; Bhat, N.; Bieber, M.; Teng, N.; Keyt, B. Abstract 645: IGM-55.5, a novel monoclonal human recombinant IgM antibody with potent activity against B cell leukemia and lymphoma. J. Cancer Res. 2014, 74, 645. [Google Scholar] [CrossRef]
- Pettinello, R.; Dooley, H. The immunoglobulins of cold-blooded vertebrates. Biomolecules 2014, 4, 1045–1069. [Google Scholar] [CrossRef] [PubMed]
- Kaetzel, C.S. Coevolution of Mucosal Immunoglobulins and the Polymeric Immunoglobulin Receptor: Evidence That the Commensal Microbiota Provided the Driving Force. ISRN Immunol. 2014, 2014, 1–20. [Google Scholar] [CrossRef]
- Winkler, T.H.; Martensson, I.L. The Role of the Pre-B Cell Receptor in B Cell Development, Repertoire Selection, and Tolerance. Front. Immunol. 2018, 9, 2423. [Google Scholar] [CrossRef]
- Hobeika, E.; Maity, P.C.; Jumaa, H. Control of B Cell Responsiveness by Isotype and Structural Elements of the Antigen Receptor. Trends Immunol. 2016, 37, 310–320. [Google Scholar] [CrossRef]
- Deane, J.A.; Fruman, D.A. Phosphoinositide 3-kinase: Diverse roles in immune cell activation. Annu. Rev. Immunol. 2004, 22, 563–598. [Google Scholar] [CrossRef]
- Johnson, S.A.; Pleiman, C.M.; Pao, L.; Schneringer, J.; Hippen, K.; Cambier, J.C. Phosphorylated immunoreceptor signaling motifs (ITAMs) exhibit unique abilities to bind and activate Lyn and Syk tyrosine kinases. J. Immunol. 1995, 155, 4596–4603. [Google Scholar]
- Rolli, V.; Gallwitz, M.; Wossning, T.; Flemming, A.; Schamel, W.W.; Zurn, C.; Reth, M. Amplification of B cell antigen receptor signaling by a Syk/ITAM positive feedback loop. Mol. Cell 2002, 10, 1057–1069. [Google Scholar] [CrossRef]
- Werner, M.; Hobeika, E.; Jumaa, H. Role of PI3K in the generation and survival of B cells. Immunol. Rev. 2010, 237, 55–71. [Google Scholar] [CrossRef]
- Van Furth, R.; Schuit, H.R.; Hijmans, W. The immunological development of the human fetus. J. Exp. Med. 1965, 122, 1173–1188. [Google Scholar] [CrossRef]
- Alford, C.A., Jr.; Foft, J.W.; Blankenship, W.J.; Cassady, G.; Benton, J.W., Jr. Subclinical central nervous system disease of neonates: A prospective study of infants born with increased levels of IgM. J. Pediatr. 1969, 75, 1167–1178. [Google Scholar] [CrossRef]
- Stiehm, E.R.; Ammann, A.J.; Cherry, J.D. Elevated cord macroglobulins in the diagnosis of intrauterine infections. N. Engl. J. Med. 1966, 275, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Meffre, E.; Salmon, J.E. Autoantibody selection and production in early human life. J. Clin. Investig. 2007, 117, 598–601. [Google Scholar] [CrossRef] [PubMed]
- Weitkamp, J.H.; Lewis, D.B.; Levy, O. Immunology of the Fetus and Newborn. In Avery’s Diseases of the Newborn, 10th ed.; Gleason, C., Juul, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 36, pp. 453–481.e7. [Google Scholar]
- Gonzalez-Quintela, A.; Alende, R.; Gude, F.; Campos, J.; Rey, J.; Meijide, L.M.; Fernandez-Merino, C.; Vidal, C. Serum levels of immunoglobulins (IgG, IgA, IgM) in a general adult population and their relationship with alcohol consumption, smoking and common metabolic abnormalities. Clin. Exp. Immunol. 2008, 151, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.B.; Wilson, C.B. Developmental Immunology and Role of Host Defenses in Fetal and Neonatal Susceptibility to Infection. In Infectious Diseases of the Fetus and Newborn Infant, 6th ed.; Remington, J.S., Klein, J.O., Wilson, C.B., Baker, C.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 4, pp. 87–210. [Google Scholar]
- Boes, M. Role of natural and immune IgM antibodies in immune responses. Mol. Immunol. 2000, 37, 1141–1149. [Google Scholar] [CrossRef]
- Holodick, N.E.; Tumang, J.R.; Rothstein, T.L. Immunoglobulin secretion by B1 cells: Differential intensity and IRF4-dependence of spontaneous IgM secretion by peritoneal and splenic B1 cells. Eur. J. Immunol. 2010, 40, 3007–3016. [Google Scholar] [CrossRef]
- Jayasekera, J.P.; Moseman, E.A.; Carroll, M.C. Natural antibody and complement mediate neutralization of influenza virus in the absence of prior immunity. J. Virol. 2007, 81, 3487–3494. [Google Scholar] [CrossRef]
- Chen, Z.J.; Wheeler, C.J.; Shi, W.; Wu, A.J.; Yarboro, C.H.; Gallagher, M.; Notkins, A.L. Polyreactive antigen-binding B cells are the predominant cell type in the newborn B cell repertoire. Eur. J. Immunol. 1998, 28, 989–994. [Google Scholar] [CrossRef]
- Choi, Y.S.; Dieter, J.A.; Rothaeusler, K.; Luo, Z.; Baumgarth, N. B-1 cells in the bone marrow are a significant source of natural IgM. Eur. J. Immunol. 2012, 42, 120–129. [Google Scholar] [CrossRef]
- Forster, I.; Rajewsky, K. Expansion and functional activity of Ly-1+ B cells upon transfer of peritoneal cells into allotype-congenic, newborn mice. Eur. J. Immunol. 1987, 17, 521–528. [Google Scholar] [CrossRef]
- Lalor, P.A.; Herzenberg, L.A.; Adams, S.; Stall, A.M. Feedback regulation of murine Ly-1 B cell development. Eur. J. Immunol. 1989, 19, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Savage, H.P.; Baumgarth, N. Characteristics of natural antibody-secreting cells. Ann. N. Y. Acad. Sci. 2015, 1362, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Van Oudenaren, A.; Haaijman, J.J.; Benner, R. Frequencies of background cytoplasmic Ig-containing cells in various lymphoid organs of athymic and euthymic mice as a function of age and immune status. Immunology 1984, 51, 735–742. [Google Scholar] [PubMed]
- Reynolds, A.E.; Kuraoka, M.; Kelsoe, G. Natural IgM is produced by CD5- plasma cells that occupy a distinct survival niche in bone marrow. J. Immunol. 2015, 194, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.O.; Holodick, N.E.; Rothstein, T.L. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70. J. Exp. Med. 2011, 208, 67–80. [Google Scholar] [CrossRef]
- Gronwall, C.; Silverman, G.J. Natural IgM: Beneficial autoantibodies for the control of inflammatory and autoimmune disease. J. Clin. Immunol. 2014, 34 (Suppl. 1), S12–S21. [Google Scholar] [CrossRef]
- Casey, R.; Newcombe, J.; McFadden, J.; Bodman-Smith, K.B. The acute-phase reactant C-reactive protein binds to phosphorylcholine-expressing Neisseria meningitidis and increases uptake by human phagocytes. Infect. Immun. 2008, 76, 1298–1304. [Google Scholar] [CrossRef]
- Chou, M.Y.; Fogelstrand, L.; Hartvigsen, K.; Hansen, L.F.; Woelkers, D.; Shaw, P.X.; Choi, J.; Perkmann, T.; Backhed, F.; Miller, Y.I.; et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J. Clin. Investig. 2009, 119, 1335–1349. [Google Scholar] [CrossRef]
- Kearney, J.F.; Patel, P.; Stefanov, E.K.; King, R.G. Natural antibody repertoires: Development and functional role in inhibiting allergic airway disease. Annu. Rev. Immunol. 2015, 33, 475–504. [Google Scholar] [CrossRef]
- Shaw, P.X.; Horkko, S.; Chang, M.K.; Curtiss, L.K.; Palinski, W.; Silverman, G.J.; Witztum, J.L. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J. Clin. Investig. 2000, 105, 1731–1740. [Google Scholar] [CrossRef]
- Baumgarth, N. The double life of a B-1 cell: Self-reactivity selects for protective effector functions. Nat. Rev. Immunol. 2011, 11, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Gronwall, C.; Vas, J.; Silverman, G.J. Protective Roles of Natural IgM Antibodies. Front. Immunol. 2012, 3, 66. [Google Scholar] [CrossRef] [PubMed]
- Louis, A.G.; Gupta, S. Primary selective IgM deficiency: An ignored immunodeficiency. Clin. Rev. Allergy Immunol. 2014, 46, 104–111. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Elsner, R.A.; Baumgarth, N. Natural IgM prevents autoimmunity by enforcing B cell central tolerance induction. J. Immunol. 2015, 194, 1489–1502. [Google Scholar] [CrossRef] [PubMed]
- Capolunghi, F.; Rosado, M.M.; Sinibaldi, M.; Aranburu, A.; Carsetti, R. Why do we need IgM memory B cells? Immunol. Lett. 2013, 152, 114–120. [Google Scholar] [CrossRef]
- Bemark, M. Translating transitions—How to decipher peripheral human B cell development. J. Biomed. Res. 2015, 29, 264–284. [Google Scholar] [CrossRef]
- Huang, C.; Stewart, A.K.; Schwartz, R.S.; Stollar, B.D. Immunoglobulin heavy chain gene expression in peripheral blood B lymphocytes. J. Clin. Investig. 1992, 89, 1331–1343. [Google Scholar] [CrossRef]
- van Es, J.H.; Meyling, F.H.; Logtenberg, T. High frequency of somatically mutated IgM molecules in the human adult blood B cell repertoire. Eur. J. Immunol. 1992, 22, 2761–2764. [Google Scholar] [CrossRef]
- Klein, U.; Kuppers, R.; Rajewsky, K. Evidence for a large compartment of IgM-expressing memory B cells in humans. Blood 1997, 89, 1288–1298. [Google Scholar] [CrossRef]
- Krishnamurty, A.T.; Thouvenel, C.D.; Portugal, S.; Keitany, G.J.; Kim, K.S.; Holder, A.; Crompton, P.D.; Rawlings, D.J.; Pepper, M. Somatically Hypermutated Plasmodium-Specific IgM(+) Memory B Cells Are Rapid, Plastic, Early Responders upon Malaria Rechallenge. Immunity 2016, 45, 402–414. [Google Scholar] [CrossRef]
- Heyman, B. Antibodies as natural adjuvants. Curr. Top. Microbiol. Immunol. 2014, 382, 201–219. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhang, L.; Heyman, B. IgG-mediated immune suppression in mice is epitope specific except during high epitope density conditions. Sci. Rep. 2018, 8, 15292. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, J.J.; Heyman, B. IgG Suppresses Antibody Responses in Mice Lacking C1q, C3, Complement Receptors 1 and 2, or IgG Fc-Receptors. PLoS ONE 2015, 10, e0143841. [Google Scholar] [CrossRef] [PubMed]
- Sorman, A.; Westin, A.; Heyman, B. IgM is Unable to Enhance Antibody Responses in Mice Lacking C1q or C3. Scand. J. Immunol. 2017, 85, 381–382. [Google Scholar] [CrossRef] [PubMed]
- Sorman, A.; Zhang, L.; Ding, Z.; Heyman, B. How antibodies use complement to regulate antibody responses. Mol. Immunol. 2014, 61, 79–88. [Google Scholar] [CrossRef]
- Rutemark, C.; Bergman, A.; Getahun, A.; Hallgren, J.; Henningsson, F.; Heyman, B. Complement receptors 1 and 2 in murine antibody responses to IgM-complexed and uncomplexed sheep erythrocytes. PLoS ONE 2012, 7, e41968. [Google Scholar] [CrossRef]
- Johansen, F.E.; Braathen, R.; Brandtzaeg, P. Role of J chain in secretory immunoglobulin formation. Scand. J. Immunol. 2000, 52, 240–248. [Google Scholar] [CrossRef]
- Brandtzaeg, P. Secretory IgA: Designed for Anti-Microbial Defense. Front. Immunol. 2013, 4, 222. [Google Scholar] [CrossRef]
- Davis, A.C.; Roux, K.H.; Pursey, J.; Shulman, M.J. Intermolecular disulfide bonding in IgM: Effects of replacing cysteine residues in the mu heavy chain. EMBO J. 1989, 8, 2519–2526. [Google Scholar] [CrossRef]
- Fazel, S.; Wiersma, E.J.; Shulman, M.J. Interplay of J chain and disulfide bonding in assembly of polymeric IgM. Int. Immunol. 1997, 9, 1149–1158. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wiersma, E.J.; Shulman, M.J. Assembly of IgM. Role of disulfide bonding and noncovalent interactions. J. Immunol. 1995, 154, 5265–5272. [Google Scholar] [PubMed]
- Kabat, E.A.; Te Wu, T.; Perry, H.M.; Foeller, C.; Gottesman, K.S. Sequences of Proteins of Immunological Interest; U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health: Bethesda, MD, USA, 1991. [Google Scholar]
- Sorensen, V.; Sundvold, V.; Michaelsen, T.E.; Sandlie, I. Polymerization of IgA and IgM: Roles of Cys309/Cys414 and the secretory tailpiece. J. Immunol. 1999, 162, 3448–3455. [Google Scholar] [PubMed]
- Pasalic, D.; Weber, B.; Giannone, C.; Anelli, T.; Muller, R.; Fagioli, C.; Felkl, M.; John, C.; Mossuto, M.F.; Becker, C.F.W.; et al. A peptide extension dictates IgM assembly. Proc. Natl. Acad. Sci. USA 2017, 114, E8575–E8584. [Google Scholar] [CrossRef]
- Smith, R.I.; Coloma, M.J.; Morrison, S.L. Addition of a mu-tailpiece to IgG results in polymeric antibodies with enhanced effector functions including complement-mediated cytolysis by IgG4. J. Immunol. 1995, 154, 2226–2236. [Google Scholar] [PubMed]
- Frutiger, S.; Hughes, G.J.; Paquet, N.; Luthy, R.; Jaton, J.C. Disulfide bond assignment in human J chain and its covalent pairing with immunoglobulin M. Biochemistry 1992, 31, 12643–12647. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, V.; Rasmussen, I.B.; Sundvold, V.; Michaelsen, T.E.; Sandlie, I. Structural requirements for incorporation of J chain into human IgM and IgA. Int. Immunol. 2000, 12, 19–27. [Google Scholar] [CrossRef]
- Li, Y.; Wang, G.; Li, N.; Wang, Y.; Zhu, Q.; Chu, H.; Wu, W.; Tan, Y.; Yu, F.; Su, X.D.; et al. Structural insights into immunoglobulin M. Science 2020, 367, 1014–1017. [Google Scholar] [CrossRef]
- Kumar, N.; Arthur, C.P.; Ciferri, C.; Matsumoto, M.L. Structure of the secretory immunoglobulin a core. Science 2020, 367, 1008–1014. [Google Scholar] [CrossRef]
- Braathen, R.; Hohman, V.S.; Brandtzaeg, P.; Johansen, F.E. Secretory antibody formation: Conserved binding interactions between J chain and polymeric Ig receptor from humans and amphibians. J. Immunol. 2007, 178, 1589–1597. [Google Scholar] [CrossRef]
- Moh, E.S.; Lin, C.H.; Thaysen-Andersen, M.; Packer, N.H. Site-Specific N-Glycosylation of Recombinant Pentameric and Hexameric Human IgM. J. Am. Soc. Mass Spectrom. 2016, 27, 1143–1155. [Google Scholar] [CrossRef]
- Muraoka, S.; Shulman, M.J. Structural requirements for IgM assembly and cytolytic activity. Effects of mutations in the oligosaccharide acceptor site at Asn402. J. Immunol. 1989, 142, 695–701. [Google Scholar] [PubMed]
- Maiorella, B.L.; Winkelhake, J.; Young, J.; Moyer, B.; Bauer, R.; Hora, M.; Andya, J.; Thomson, J.; Patel, T.; Parekh, R. Effect of culture conditions on IgM antibody structure, pharmacokinetics and activity. Biotechnology (N. Y.) 1993, 11, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Colucci, M.; Stockmann, H.; Butera, A.; Masotti, A.; Baldassarre, A.; Giorda, E.; Petrini, S.; Rudd, P.M.; Sitia, R.; Emma, F.; et al. Sialylation of N-linked glycans influences the immunomodulatory effects of IgM on T cells. J. Immunol. 2015, 194, 151–157. [Google Scholar] [CrossRef]
- Feinstein, A.; Munn, E.A. Conformation of the free and antigen-bound IgM antibody molecules. Nature 1969, 224, 1307–1309. [Google Scholar] [CrossRef]
- Perkins, S.J.; Nealis, A.S.; Sutton, B.J.; Feinstein, A. Solution structure of human and mouse immunoglobulin M by synchrotron X-ray scattering and molecular graphics modelling. A possible mechanism for complement activation. J. Mol. Biol. 1991, 221, 1345–1366. [Google Scholar] [CrossRef]
- Czajkowsky, D.M.; Shao, Z. The human IgM pentamer is a mushroom-shaped molecule with a flexural bias. Proc. Natl. Acad. Sci. USA 2009, 106, 14960–14965. [Google Scholar] [CrossRef] [PubMed]
- Muller, R.; Grawert, M.A.; Kern, T.; Madl, T.; Peschek, J.; Sattler, M.; Groll, M.; Buchner, J. High-resolution structures of the IgM Fc domains reveal principles of its hexamer formation. Proc. Natl. Acad. Sci. USA 2013, 110, 10183–10188. [Google Scholar] [CrossRef]
- Hiramoto, E.; Tsutsumi, A.; Suzuki, R.; Matsuoka, S.; Arai, S.; Kikkawa, M.; Miyazaki, T. The IgM pentamer is an asymmetric pentagon with an open groove that binds the AIM protein. Sci. Adv. 2018, 4, eaau1199. [Google Scholar] [CrossRef] [PubMed]
- Zikan, J.; Novotny, J.; Trapane, T.L.; Koshland, M.E.; Urry, D.W.; Bennett, J.C.; Mestecky, J. Secondary structure of the immunoglobulin J chain. Proc. Natl. Acad. Sci. USA 1985, 82, 5905–5909. [Google Scholar] [CrossRef]
- Vollmers, H.P.; Brandlein, S. Natural IgM antibodies: The orphaned molecules in immune surveillance. Adv. Drug Deliv. Rev. 2006, 58, 755–765. [Google Scholar] [CrossRef]
- Wibroe, P.P.; Helvig, S.Y.; Moein Moghimi, S. The Role of Complement in Antibody Therapy for Infectious Diseases. Microbiol. Spectr. 2014, 2, 63–74. [Google Scholar] [CrossRef]
- Strohl, W.R.; Strohl, L.M. Therapeutic Antibody Engineering; Woodhead Publishing: Sawston, UK, 2012; pp. 197–223. [Google Scholar]
- Klimovich, V.B. IgM and its receptors: Structural and functional aspects. Biochemistry (Moscow) 2011, 76, 534–549. [Google Scholar] [CrossRef] [PubMed]
- Sharp, T.H.; Boyle, A.L.; Diebolder, C.A.; Kros, A.; Koster, A.J.; Gros, P. Insights into IgM-mediated complement activation based on in situ structures of IgM-C1-C4b. Proc. Natl. Acad. Sci. USA 2019, 116, 11900–11905. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, A.; Sakamoto, N.; Shimizu, Y.; Shibuya, K.; Osawa, M.; Hiroyama, T.; Eyre, H.J.; Sutherland, G.R.; Endo, Y.; Fujita, T.; et al. Fc alpha/mu receptor mediates endocytosis of IgM-coated microbes. Nat. Immunol. 2000, 1, 441–446. [Google Scholar] [CrossRef]
- Weinstein, J.R.; Quan, Y.; Hanson, J.F.; Colonna, L.; Iorga, M.; Honda, S.; Shibuya, K.; Shibuya, A.; Elkon, K.B.; Moller, T. IgM-Dependent Phagocytosis in Microglia Is Mediated by Complement Receptor 3, Not Fcalpha/mu Receptor. J. Immunol. 2015, 195, 5309–5317. [Google Scholar] [CrossRef]
- Mostov, K.E.; Blobel, G. A transmembrane precursor of secretory component. The receptor for transcellular transport of polymeric immunoglobulins. J. Biol. Chem. 1982, 257, 11816–11821. [Google Scholar]
- Krajci, P.; Solberg, R.; Sandberg, M.; Oyen, O.; Jahnsen, T.; Brandtzaeg, P. Molecular cloning of the human transmembrane secretory component (poly-Ig receptor) and its mRNA expression in human tissues. Biochem. Biophys. Res. Commun. 1989, 158, 783–789. [Google Scholar] [CrossRef]
- Mostov, K.E.; Friedlander, M.; Blobel, G. Structure and function of the receptor for polymeric immunoglobulins. Biochem. Soc. Symp. 1986, 51, 113–115. [Google Scholar]
- Mostov, K.E.; Altschuler, Y.; Chapin, S.J.; Enrich, C.; Low, S.H.; Luton, F.; Richman-Eisenstat, J.; Singer, K.L.; Tang, K.; Weimbs, T. Regulation of protein traffic in polarized epithelial cells: The polymeric immunoglobulin receptor model. Cold Spring Harb. Symp. Quant. Biol. 1995, 60, 775–781. [Google Scholar] [CrossRef][Green Version]
- Norderhaug, I.N.; Johansen, F.E.; Krajci, P.; Brandtzaeg, P. Domain deletions in the human polymeric Ig receptor disclose differences between its dimeric IgA and pentameric IgM interaction. Eur. J. Immunol. 1999, 29, 3401–3409. [Google Scholar] [CrossRef]
- Sakamoto, N.; Shibuya, K.; Shimizu, Y.; Yotsumoto, K.; Miyabayashi, T.; Sakano, S.; Tsuji, T.; Nakayama, E.; Nakauchi, H.; Shibuya, A. A novel Fc receptor for IgA and IgM is expressed on both hematopoietic and non-hematopoietic tissues. Eur. J. Immunol. 2001, 31, 1310–1316. [Google Scholar] [CrossRef]
- Kikuno, K.; Kang, D.W.; Tahara, K.; Torii, I.; Kubagawa, H.M.; Ho, K.J.; Baudino, L.; Nishizaki, N.; Shibuya, A.; Kubagawa, H. Unusual biochemical features and follicular dendritic cell expression of human Fcalpha/mu receptor. Eur. J. Immunol. 2007, 37, 3540–3550. [Google Scholar] [CrossRef]
- Ghumra, A.; Shi, J.; McIntosh, R.S.; Rasmussen, I.B.; Braathen, R.; Johansen, F.E.; Sandlie, I.; Mongini, P.K.; Areschoug, T.; Lindahl, G.; et al. Structural requirements for the interaction of human IgM and IgA with the human Fcalpha/mu receptor. Eur. J. Immunol. 2009, 39, 1147–1156. [Google Scholar] [CrossRef]
- Kubagawa, H.; Oka, S.; Kubagawa, Y.; Torii, I.; Takayama, E.; Kang, D.W.; Gartland, G.L.; Bertoli, L.F.; Mori, H.; Takatsu, H.; et al. Identity of the elusive IgM Fc receptor (FcmuR) in humans. J. Exp. Med. 2009, 206, 2779–2793. [Google Scholar] [CrossRef] [PubMed]
- Vire, B.; David, A.; Wiestner, A. TOSO, the Fcmicro receptor, is highly expressed on chronic lymphocytic leukemia B cells, internalizes upon IgM binding, shuttles to the lysosome, and is downregulated in response to TLR activation. J. Immunol. 2011, 187, 4040–4050. [Google Scholar] [CrossRef] [PubMed]
- Honjo, K.; Kubagawa, Y.; Kearney, J.F.; Kubagawa, H. Unique ligand-binding property of the human IgM Fc receptor. J. Immunol. 2015, 194, 1975–1982. [Google Scholar] [CrossRef] [PubMed]
- Honjo, K.; Kubagawa, Y.; Kubagawa, H. Is Toso/IgM Fc receptor (FcmuR) expressed by innate immune cells? Proc. Natl. Acad. Sci. USA 2013, 110, E2540–E2541. [Google Scholar] [CrossRef] [PubMed]
- Hensel, F.; Gagnon, P. An effective platform for purification of IgM monoclonal antibodies using Hydroxyapatite. In Proceedings of the 5th International Conference on Hydroxyapatite and Related Products, Rottach-Egern, Germany, 11–14 October 2009. [Google Scholar]
- Gagnon, P. Improved antibody aggregate removal by hydroxyapatite chromatography in the presence of polyethylene glycol. J. Immunol. Methods 2008, 336, 222–228. [Google Scholar] [CrossRef]
- Butler, M.; Meneses-Acosta, A. Recent advances in technology supporting biopharmaceutical production from mammalian cells. Appl. Microbiol. Biotechnol. 2012, 96, 885–894. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, Y.G.; Lee, G.M. CHO cells in biotechnology for production of recombinant proteins: Current state and further potential. Appl. Microbiol. Biotechnol. 2012, 93, 917–930. [Google Scholar] [CrossRef]
- Kunert, R.; Wolbank, S.; Stiegler, G.; Weik, R.; Katinger, H. Characterization of molecular features, antigen-binding, and in vitro properties of IgG and IgM variants of 4E10, an anti-HIV type 1 neutralizing monoclonal antibody. AIDS Res. Hum. Retrovir. 2004, 20, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Tchoudakova, A.; Hensel, F.; Murillo, A.; Eng, B.; Foley, M.; Smith, L.; Schoenen, F.; Hildebrand, A.; Kelter, A.R.; Ilag, L.L.; et al. High level expression of functional human IgMs in human PER.C6 cells. MAbs 2009, 1, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Loos, A.; Gruber, C.; Altmann, F.; Mehofer, U.; Hensel, F.; Grandits, M.; Oostenbrink, C.; Stadlmayr, G.; Furtmuller, P.G.; Steinkellner, H. Expression and glycoengineering of functionally active heteromultimeric IgM in plants. Proc. Natl. Acad. Sci. USA 2014, 111, 6263–6268. [Google Scholar] [CrossRef] [PubMed]
- Valasek, C.; Cole, F.; Hensel, F.; Ye, P.; Conner, M.A.; Ultee, M.E. Production and Purification of a PER.C6-Expressed IgM Antibody Therapeutic. BioProcess Int. 2011, 9, 10. [Google Scholar]
- Steindl, F.; Jungbauer, A.; Wenisch, E.; Himmler, G.; Katinger, H. Isoelectric precipitation and gel chromatography for purification of monoclonal IgM. Enzym. Microb. Technol. 1987, 9, 361–364. [Google Scholar] [CrossRef]
- Gagnon, P.; Hensel, F.; Richieri, R. Recent Advances in the Purification of IgM Monoclonal Antibodies. Available online: http://www.validated.com/revalbio/pdffiles/PUR07a.pdf (accessed on 11 October 2020).
- Gagnon, P.; Hensel, F.; Lee, S.; Zaidi, S. Chromatographic behavior of IgM:DNA complexes. J. Chromatogr. A 2011, 1218, 2405–2412. [Google Scholar] [CrossRef]
- Hanala, S. The new ParaDIgm: IgM from bench to clinic: November 15-16, 2011, Frankfurt, Germany. MAbs 2012, 4, 555–561. [Google Scholar] [CrossRef][Green Version]
- Gong, S.; Tomusange, K.; Kulkarni, V.; Adeniji, O.S.; Lakhashe, S.K.; Hariraju, D.; Strickland, A.; Plake, E.; Frost, P.A.; Ratcliffe, S.J.; et al. Anti-HIV IgM protects against mucosal SHIV transmission. Aids 2018, 32, F5–F13. [Google Scholar] [CrossRef]
- Hale, G.; Bright, S.; Chumbley, G.; Hoang, T.; Metcalf, D.; Munro, A.J.; Waldmann, H. Removal of T cells from bone marrow for transplantation: A monoclonal antilymphocyte antibody that fixes human complement. Blood 1983, 62, 873–882. [Google Scholar] [CrossRef]
- Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/ (accessed on 11 October 2020).
- Vollmers, H.P.; Brandlein, S. Nature’s best weapons to fight cancer. Revival of human monoclonal IgM antibodies. Hum. Antibodies 2002, 11, 131–142. [Google Scholar] [CrossRef]
- Heimburg-Molinaro, J.; Rittenhouse-Olson, K. Development and characterization of antibodies to carbohydrate antigens. Methods Mol. Biol. 2009, 534, 341–357. [Google Scholar] [CrossRef] [PubMed]
- Harkonen, S.; Scannon, P.; Mischak, R.P.; Spitler, L.E.; Foxall, C.; Kennedy, D.; Greenberg, R. Phase I study of a murine monoclonal anti-lipid A antibody in bacteremic and nonbacteremic patients. Antimicrob. Agents Chemother. 1988, 32, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.J., Jr.; Zimmerman, J.; Khazaeli, M.B.; Albertson, T.E.; Dellinger, R.P.; Panacek, E.A.; Foulke, G.E.; Dating, C.; Smith, C.R.; LoBuglio, A.F. Initial evaluation of human monoclonal anti-lipid A antibody (HA-1A) in patients with sepsis syndrome. Crit. Care Med. 1990, 18, 1311–1315. [Google Scholar] [CrossRef] [PubMed]
- Daifuku, R.; Haenftling, K.; Young, J.; Groves, E.S.; Turrell, C.; Meyers, F.J. Phase I study of antilipopolysaccharide human monoclonal antibody MAB-T88. Antimicrob. Agents Chemother. 1992, 36, 2349–2351. [Google Scholar] [CrossRef]
- Saravolatz, L.D.; Markowitz, N.; Collins, M.S.; Bogdanoff, D.; Pennington, J.E. Safety, pharmacokinetics, and functional activity of human anti-Pseudomonas aeruginosa monoclonal antibodies in septic and nonseptic patients. J. Infect. Dis. 1991, 164, 803–806. [Google Scholar] [CrossRef]
- Lu, Q.; Rouby, J.J.; Laterre, P.F.; Eggimann, P.; Dugard, A.; Giamarellos-Bourboulis, E.J.; Mercier, E.; Garbino, J.; Luyt, C.E.; Chastre, J.; et al. Pharmacokinetics and safety of panobacumab: Specific adjunctive immunotherapy in critical patients with nosocomial Pseudomonas aeruginosa O11 pneumonia. J. Antimicrob. Chemother. 2011, 66, 1110–1116. [Google Scholar] [CrossRef]
- Irie, R.F.; Ollila, D.W.; O’Day, S.; Morton, D.L. Phase I pilot clinical trial of human IgM monoclonal antibody to ganglioside GM3 in patients with metastatic melanoma. Cancer Immunol. Immunother. 2004, 53, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Safety Study of Human IgM (MORAb-028) to Treat Metastatic Melanoma. Available online: https://clinicaltrials.gov/ct2/show/NCT01123304 (accessed on 11 October 2020).
- Eisen, A.; Greenberg, B.M.; Bowen, J.D.; Arnold, D.L.; Caggiano, A.O. A double-blind, placebo-controlled, single ascending-dose study of remyelinating antibody rHIgM22 in people with multiple sclerosis. Mult. Scler. J. Exp. Transl. Clin. 2017, 3, 2055217317743097. [Google Scholar] [CrossRef] [PubMed]
- Line, B.R.; Breyer, R.J.; McElvany, K.D.; Earle, D.C.; Khazaeli, M.B. Evaluation of human anti-mouse antibody response in normal volunteers following repeated injections of fanolesomab (NeutroSpec), a murine anti-CD15 IgM monoclonal antibody for imaging infection. Nucl. Med. Commun. 2004, 25, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Hensel, F.; Timmermann, W.; von Rahden, B.H.; Rosenwald, A.; Brandlein, S.; Illert, B. Ten-year follow-up of a prospective trial for the targeted therapy of gastric cancer with the human monoclonal antibody PAT-SC1. Oncol. Rep. 2014, 31, 1059–1066. [Google Scholar] [CrossRef][Green Version]
- Liedtke, M.; Twist, C.J.; Medeiros, B.C.; Gotlib, J.R.; Berube, C.; Bieber, M.M.; Bhat, N.M.; Teng, N.N.; Coutre, S.E. Phase I trial of a novel human monoclonal antibody mAb216 in patients with relapsed or refractory B-cell acute lymphoblastic leukemia. Haematologica 2012, 97, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Duell, J.; Castro, I.C.; Dubljevic, V.; Chatterjee, M.; Knop, S.; Hensel, F.; Rosenwald, A.; Einsele, H.; Topp, M.S.; et al. GRP78-directed immunotherapy in relapsed or refractory multiple myeloma—Results from a phase 1 trial with the monoclonal immunoglobulin M antibody PAT-SM6. Haematologica 2015, 100, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Friend, P.J.; Hale, G.; Waldmann, H.; Gore, S.; Thiru, S.; Joysey, V.; Evans, D.B.; Calne, R.Y. Campath-1M--prophylactic use after kidney transplantation. A randomized controlled clinical trial. Transplantation 1989, 48, 248–253. [Google Scholar] [CrossRef]
- Haisma, H.J.; Pinedo, H.M.; Kessel, M.A.; van Muijen, M.; Roos, J.C.; Plaizier, M.A.; Martens, H.J.; DeJager, R.; Boven, E. Human IgM monoclonal antibody 16.88: Pharmacokinetics and immunogenicity in colorectal cancer patients. J. Natl. Cancer Inst. 1991, 83, 1813–1819. [Google Scholar] [CrossRef] [PubMed]
- Deeg, H.J.; Blazar, B.R.; Bolwell, B.J.; Long, G.D.; Schuening, F.; Cunningham, J.; Rifkin, R.M.; Abhyankar, S.; Briggs, A.D.; Burt, R.; et al. Treatment of steroid-refractory acute graft-versus-host disease with anti-CD147 monoclonal antibody ABX-CBL. Blood 2001, 98, 2052–2058. [Google Scholar] [CrossRef]
- Getts, D.R.; Kramer, W.G.; Wiseman, A.C.; Flechner, S.M. The pharmacokinetics and pharmacodynamics of TOL101, a murine IgM anti-human alphabeta T cell receptor antibody, in renal transplant patients. Clin. Pharm. 2014, 53, 649–657. [Google Scholar] [CrossRef]
- Matsubara, T.; Okuda, K.; Chiba, J.; Takayama, A.; Inoue, H.; Sakurai, T.; Wakabayashi, H.; Kaneko, A.; Sugimoto, K.; Yamazaki, H.; et al. A phase I/II clinical trial of intra-articular administration of ARG098, an anti-FAS IGM monoclonal antibody, in knee joint synovitis of japanese patients with rheumatoid arthritis. J. Ann. Rheum. Dis. 2013, 71, 384. [Google Scholar] [CrossRef]
- Teng, N.N.; Kaplan, H.S.; Hebert, J.M.; Moore, C.; Douglas, H.; Wunderlich, A.; Braude, A.I. Protection against gram-negative bacteremia and endotoxemia with human monoclonal IgM antibodies. Proc. Natl. Acad. Sci. USA 1985, 82, 1790–1794. [Google Scholar] [CrossRef]
- Marks, L. The birth pangs of monoclonal antibody therapeutics: The failure and legacy of Centoxin. MAbs 2012, 4, 403–412. [Google Scholar] [CrossRef]
- Daifuku, R.; Panacek, E.A.; Haenftling, K.; Swenson, W.K.; Prescott, A.W.; Johnson, J.L. Pilot study of anti-lipopolysaccharide human monoclonal antibody MAB-T88 in patients with gram-negative sepsis. Hum. Antibodies Hybrid. 1993, 4, 36–39. [Google Scholar] [CrossRef]
- Lazar, H.; Horn, M.P.; Zuercher, A.W.; Imboden, M.A.; Durrer, P.; Seiberling, M.; Pokorny, R.; Hammer, C.; Lang, A.B. Pharmacokinetics and safety profile of the human anti-Pseudomonas aeruginosa serotype O11 immunoglobulin M monoclonal antibody KBPA-101 in healthy volunteers. Antimicrob. Agents Chemother. 2009, 53, 3442–3446. [Google Scholar] [CrossRef] [PubMed]
- AR-105: Broadly Active Human Monoclonal Antibody (mAb) Against Pseudomonas aeruginosa. Available online: https://aridispharma.com/ar-105/ (accessed on 11 October 2020).
- IgM-enriched Immunoglobulin Attenuates Systemic Endotoxin Activity in Early Severe Sepsis. Available online: https://clinicaltrials.gov/ct2/show/NCT02444871 (accessed on 11 October 2020).
- Effects on Microcirculation of IgGAM in Severe Septic/Septic Shock Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT02655133 (accessed on 11 October 2020).
- Reichert, J.M.; Dewitz, M.C. Anti-infective monoclonal antibodies: Perils and promise of development. Nat. Rev. Drug Discov. 2006, 5, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Hoon, D.S.; Wang, Y.; Sze, L.; Kanda, H.; Watanabe, T.; Morrison, S.L.; Morton, D.L.; Irie, R.F. Molecular cloning of a human monoclonal antibody reactive to ganglioside GM3 antigen on human cancers. Cancer Res. 1993, 53, 5244–5250. [Google Scholar] [PubMed]
- Azuma, Y.; Ishikawa, Y.; Kawai, S.; Tsunenari, T.; Tsunoda, H.; Igawa, T.; Iida, S.; Nanami, M.; Suzuki, M.; Irie, R.F.; et al. Recombinant human hexamer-dominant IgM monoclonal antibody to ganglioside GM3 for treatment of melanoma. Clin. Cancer Res. 2007, 13, 2745–2750. [Google Scholar] [CrossRef] [PubMed]
- A Safety and MORAb-028 Dose Determination Study in Subjects with Metastatic Melanoma. Available online: https://clinicaltrials.gov/ct2/show/NCT01212276 (accessed on 11 October 2020).
- MORAb 028. Available online: https://adisinsight.springer.com/drugs/800025741 (accessed on 11 October 2020).
- Mitsunaga, Y.; Ciric, B.; Van Keulen, V.; Warrington, A.E.; Paz Soldan, M.; Bieber, A.J.; Rodriguez, M.; Pease, L.R. Direct evidence that a human antibody derived from patient serum can promote myelin repair in a mouse model of chronic-progressive demyelinating disease. FASEB J. 2002, 16, 1325–1327. [Google Scholar] [CrossRef] [PubMed]
- An Intravenous Infusion Study of rHIgM22 in Patients with Multiple Sclerosis (M22). Available online: https://clinicaltrials.gov/ct2/show/NCT01803867 (accessed on 11 October 2020).
- An Intravenous Infusion Study of rHIgM22 in Patients with Multiple Sclerosis Immediately Following a Relapse. Available online: https://clinicaltrials.gov/ct2/show/NCT02398461 (accessed on 11 October 2020).
- Sterner, E.; Flanagan, N.; Gildersleeve, J.C. Perspectives on Anti-Glycan Antibodies Gleaned from Development of a Community Resource Database. ACS Chem. Biol. 2016, 11, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Bhat, N.M.; Bieber, M.M.; Chapman, C.J.; Stevenson, F.K.; Teng, N.N. Human anti-lipid A monoclonal antibodies bind to human B cells and the i antigen on cord red blood cells. J. Immunol. 1993, 151, 5011–5021. [Google Scholar]
- Bhat, N.M.; Bieber, M.M.; Spellerberg, M.B.; Stevenson, F.K.; Teng, N.N. Recognition of auto- and exoantigens by V4-34 gene encoded antibodies. Scand. J. Immunol. 2000, 51, 134–140. [Google Scholar] [CrossRef]
- Pascual, V.; Victor, K.; Spellerberg, M.; Hamblin, T.J.; Stevenson, F.K.; Capra, J.D. VH restriction among human cold agglutinins. The VH4-21 gene segment is required to encode anti-I and anti-i specificities. J. Immunol. 1992, 149, 2337–2344. [Google Scholar]
- Bhat, N.M.; Bieber, M.M.; Stevenson, F.K.; Teng, N.N. Rapid cytotoxicity of human B lymphocytes induced by VH4-34 (VH4.21) gene-encoded monoclonal antibodies. Clin. Exp. Immunol. 1996, 105, 183–190. [Google Scholar] [CrossRef]
- Bhat, N.M.; Bieber, M.M.; Teng, N.N. Cytotoxicity of murine B lymphocytes induced by human VH4-34 (VH4.21) gene-encoded monoclonal antibodies. Clin. Immunol. Immunopathol. 1997, 84, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xu, Y.; Gu, J.; Schlossman, S.F. A cell surface receptor defined by a mAb mediates a unique type of cell death similar to oncosis. Proc. Natl. Acad. Sci. USA 1998, 95, 6290–6295. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.L.; Fong, W.J.; Lee, E.H.; Yap, M.; Choo, A. mAb 84, a cytotoxic antibody that kills undifferentiated human embryonic stem cells via oncosis. Stem Cells 2009, 27, 1792–1801. [Google Scholar] [CrossRef]
- Vollmers HP1, O.C.R.; Müller, J.; Kirchner, T.; Müller-Hermelink, H.K. SC-1, a functional human monoclonal antibody against autologous stomach carcinoma cells. Cancer Res. 1989, 49, 6. [Google Scholar]
- Pohle, T.; Brandlein, S.; Ruoff, N.; Muller-Hermelink, H.K.; Vollmers, H.P. Lipoptosis: Tumor-specific cell death by antibody-induced intracellular lipid accumulation. Cancer Res. 2004, 64, 3900–3906. [Google Scholar] [CrossRef] [PubMed]
- PAT SC1. Available online: http://www.patrys.com/pat-sc1/ (accessed on 11 October 2020).
- NeutroSpec. Available online: https://medlibrary.org/lib/rx/meds/neutrospec/ (accessed on 11 October 2020).
- Waldmann, H.; Hale, G. CAMPATH: From concept to clinic. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 1707–1711. [Google Scholar] [CrossRef] [PubMed]
- Hale, G.; Cobbold, S.P.; Waldmann, H.; Easter, G.; Matejtschuk, P.; Coombs, R.R. Isolation of low-frequency class-switch variants from rat hybrid myelomas. J. Immunol. Methods 1987, 103, 59–67. [Google Scholar] [CrossRef]
- Riechmann, L.; Clark, M.; Waldmann, H.; Winter, G. Reshaping human antibodies for therapy. Nature 1988, 332, 323–327. [Google Scholar] [CrossRef]
- Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/103948s5160_5165lbl.pdf (accessed on 11 October 2020).
- Macmillan, M.L.; Couriel, D.; Weisdorf, D.J.; Schwab, G.; Havrilla, N.; Fleming, T.R.; Huang, S.; Roskos, L.; Slavin, S.; Shadduck, R.K.; et al. A phase 2/3 multicenter randomized clinical trial of ABX-CBL versus ATG as secondary therapy for steroid-resistant acute graft-versus-host disease. Blood 2007, 109, 2657–2662. [Google Scholar] [CrossRef]
- Flechner, S.M.; Mulgoankar, S.; Melton, L.B.; Waid, T.H.; Agarwal, A.; Miller, S.D.; Fokta, F.; Getts, M.T.; Frederick, T.J.; Herrman, J.J.; et al. First-in-human study of the safety and efficacy of TOL101 induction to prevent kidney transplant rejection. Am. J. Transplant. 2014, 14, 1346–1355. [Google Scholar] [CrossRef][Green Version]
- DE 098. Available online: https://adisinsight.springer.com/drugs/800016731 (accessed on 11 October 2020).
- Haspel, M.V.; McCabe, R.P.; Pomato, N.; Janesch, N.J.; Knowlton, J.V.; Peters, L.C.; Hoover, H.C., Jr.; Hanna, M.G., Jr. Generation of tumor cell-reactive human monoclonal antibodies using peripheral blood lymphocytes from actively immunized colorectal carcinoma patients. Cancer Res. 1985, 45, 3951–3961. [Google Scholar] [PubMed]
- Haisma, H.J.; Kessel, M.A.; Silva, C.; van Muijen, M.; Roos, J.C.; Bril, H.; Martens, H.J.; McCabe, R.; Boven, E. Human IgM monoclonal antibody 16.88: Pharmacokinetics and distribution in mouse and man. Br. J. Cancer Suppl. 1990, 10, 40–43. [Google Scholar] [PubMed]
- Rosenblum, M.G.; Levin, B.; Roh, M.; Hohn, D.; McCabe, R.; Thompson, L.; Cheung, L.; Murray, J.L. Clinical pharmacology and tissue disposition studies of 131I-labeled anticolorectal carcinoma human monoclonal antibody LiCO 16.88. Cancer Immunol. Immunother. 1994, 39, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Barth, W.F.; Wochner, R.D.; Waldmann, T.A.; Fahey, J.L. METABOLISM OF HUMAN GAMMA MACROGLOBULINS. J. Clin. Investig. 1964, 43, 1036–1048. [Google Scholar] [CrossRef] [PubMed]
- Ryman, J.T.; Meibohm, B. Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.J.; Kearns, G.L.; Kaplan, S.L.; Jacobs, R.F.; Killian, A.; Bradley, J.S.; Moss, M.M.; Van Dyke, R.; Rodriguez, W.; Straube, R.C. Single-dose pharmacokinetics and safety of HA-1A, a human IgM anti-lipid-A monoclonal antibody, in pediatric patients with sepsis syndrome. J. Pediatrics 1993, 122, 974–981. [Google Scholar] [CrossRef]
- Meng, Y.G.; Wong, T.; Saravolatz, L.D.; Pennington, J.E. Pharmacokinetics of an IgM human monoclonal antibody against Pseudomonas aeruginosa in nonseptic patients. J. Infect. Dis. 1993, 167, 784–785. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and Research. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/103928Orig1s000Lbl.pdf (accessed on 11 October 2020).
- Mimura, Y.; Katoh, T.; Saldova, R.; O’Flaherty, R.; Izumi, T.; Mimura-Kimura, Y.; Utsunomiya, T.; Mizukami, Y.; Yamamoto, K.; Matsumoto, T.; et al. Glycosylation engineering of therapeutic IgG antibodies: Challenges for the safety, functionality and efficacy. Protein Cell 2018, 9, 47–62. [Google Scholar] [CrossRef]
- Maiorella, B.L.; Ferris, R.; Thomson, J.; White, C.; Brannon, M.; Hora, M.; Henriksson, T.; Triglia, R.; Kunitani, M.; Kresin, L.; et al. Evaluation of product equivalence during process optimization for manufacture of a human IgM monoclonal antibody. Biologicals 1993, 21, 197–205. [Google Scholar] [CrossRef]
- Angus, D.C.; Birmingham, M.C.; Balk, R.A.; Scannon, P.J.; Collins, D.; Kruse, J.A.; Graham, D.R.; Dedhia, H.V.; Homann, S.; MacIntyre, N. E5 murine monoclonal antiendotoxin antibody in gram-negative sepsis: A randomized controlled trial. E5 Study Investigators. JAMA 2000, 283, 1723–1730. [Google Scholar] [CrossRef]
- Greenman, R.L.; Schein, R.M.; Martin, M.A.; Wenzel, R.P.; MacIntyre, N.R.; Emmanuel, G.; Chmel, H.; Kohler, R.B.; McCarthy, M.; Plouffe, J.; et al. A controlled clinical trial of E5 murine monoclonal IgM antibody to endotoxin in the treatment of gram-negative sepsis. The XOMA Sepsis Study Group. JAMA 1991, 266, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Brun-Buisson, C. The HA-1A saga: The scientific and ethical dilemma of innovative and costly therapies. Intensive Care Med. 1994, 20, 314–316. [Google Scholar] [CrossRef] [PubMed]
- Derkx, B.; Wittes, J.; McCloskey, R. Randomized, placebo-controlled trial of HA-1A, a human monoclonal antibody to endotoxin, in children with meningococcal septic shock. European Pediatric Meningococcal Septic Shock Trial Study Group. Clin. Infect. Dis. 1999, 28, 770–777. [Google Scholar] [CrossRef] [PubMed]
- De Jong, R.N.; Beurskens, F.J.; Verploegen, S.; Strumane, K.; van Kampen, M.D.; Voorhorst, M.; Horstman, W.; Engelberts, P.J.; Oostindie, S.C.; Wang, G.; et al. A Novel Platform for the Potentiation of Therapeutic Antibodies Based on Antigen-Dependent Formation of IgG Hexamers at the Cell Surface. PLoS Biol. 2016, 14, e1002344. [Google Scholar] [CrossRef]
- GEN1029 (HexaBody®-DR5/DR5) Safety Trial in Patients with Malignant Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT03576131 (accessed on 11 October 2020).
- Piao, X.; Ozawa, T.; Hamana, H.; Shitaoka, K.; Jin, A.; Kishi, H.; Muraguchi, A. TRAIL-receptor 1 IgM antibodies strongly induce apoptosis in human cancer cells in vitro and in vivo. Oncoimmunology 2016, 5, e1131380. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Kothambalwala, T.; Hinton, P.; Ng, D.; Saini, A.; Baliga, R.; Keyt, B. Abstract 1702: Multimeric anti-DR5 IgM antibody displays potent cytotoxicity in vitro and promotes tumor regression in vivo. In Proceedings of the American Association for Cancer Research Annual Meeting, Washington, DC, USA, 1–5 April 2017; Volume 77, p. 1702. [Google Scholar]
- Wang, B.; Kothambawala, T.; Wang, L.; Saini, A.; Baliga, R.; Sinclair, A.; Keyt, B. Abstract 3050: Multimeric IgM antibodies targeting DR5 are potent and rapid inducers of tumor cell apoptosis and cell death in vitro and in vivo. In Proceedings of the American Association for Cancer Research Annual Meeting 2019, Atlanta, GA, USA, 29 March–3 April 2019; Volume 79, p. 3050. [Google Scholar]
- Shen, C.; Zhang, M.; Chen, Y.; Zhang, L.; Wang, G.; Chen, J.; Chen, S.; Li, Z.; Wei, F.; Chen, J.; et al. An IgM antibody targeting the receptor binding site of influenza B blocks viral infection with great breadth and potency. Theranostics 2019, 9, 210–231. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Baliga, R.; Li, K.; Manlusoc, M.; Hinton, P.; Ng, D.; Tran, M.; Shan, B.; Lu, H.; Saini, A.; Rahman, S.; et al. IGM-2323: High Avidity IgM-Based CD20xCD3 Bispecific Antibody (IGM-2323) for Enhanced T-Cell Dependent Killing with Minimal Cytokine Release. In Proceedings of the American Society of Hematology Meeting, Orlando, FL, USA, 7–10 December 2019; p. 1574. [Google Scholar]
- A Safety and Pharmacokinetic Study of IGM-2323 in Subjects with Relapsed/Refractory Non-Hodgkin Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT04082936 (accessed on 11 October 2020).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IgM Form | Molecular Weight | Peptides in IgM Complex | Inter-Chain Disulfide Bonds | N-Linked Sites of Glycosylation |
|---|---|---|---|---|
| Pentamer (with J-chain) | 950 kD | 21 | 27 | 51 |
| Hexamer (without J-chain) | 1150 kD | 24 | 30 | 60 |
| Antibody (Name) | Company | IgM Source | Antigen | Indication | Most Advanced Clinical Development |
|---|---|---|---|---|---|
| Campath-1M | Academic (MRC-RDCT) | Rat | CD52 | Graft vs. host disease | Phase 2 |
| E5 (Xomen-E5) | XOMA | Mouse | J5 lipid A | Sepsis | Phase 3 |
| HA-1A (Centoxin) | Centocor | Human | J5 lipid A | Sepsis | Phase 3 |
| Fanolesomab-Tc99 (NeutroSpec) | Palatin | Mouse | CD15 | Appendicitis | Phase 3 |
| IgM cocktail (5) | Cutter/Miles | Human | LPS | Sepsis | Phase 1 |
| Mab 16.88 | Academic (Free University Hospital) | Human | Colon cancer antigen | Colorectal cancer | Phase 1 |
| MAB-T88 | Chiron | Human | LPS | Neutropenia | Phase 1 |
| PAT-SC1 | Patrys | Human | CD55 isoform | Gastric cancer | Phase 1 |
| ABX-CBL | Abgenix | Mouse | CD147 | Graft vs. host disease | Phase 2/3 |
| L612 | Chugai | Human | Ganglioside GM3 | Melanoma | Phase 1 |
| MORAb-028 | Morphotek/Eisai | Human | Ganglioside GD2 | Melanoma | Phase 1 |
| AR-101 | Aridis | Human | LPS | Nosocomial P. a. pneumonia | Phase 2a |
| mAb216 | Academic (Stanford) | Human | CDIM | B-lineage ALL | Phase 1 |
| PAT-SM6 | Patrys | Human | GRP78 | Multiple myeloma | Phase 1/2a |
| ARG098 | Argenes | Mouse/Human (chimeric) | FAS | Rheumatoid arthritis | Phase 1/2 |
| rHIgM22 | Acorda | Human | CNS myelin | Multiple sclerosis/neuronal degeneration | Phase 1 |
| TOL101 | Tolera | Mouse | αβ TCR | Renal transplant | Phase 2 |
| Target Antigen Class | Antibody | Antigen | IgM Source | Production Cell | Indication | Clinical Trial | Dose | Immunogenicity in Humans | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Lipopolysaccharide | E5 | J5 lipid A | Mouse B cells | Hybridoma with mouse myeloma | Sepsis | Phase 1 | 0.1, 0.5, 2, 7.5, 15 mg/kg | 3 of 9 subjects | Harkonen 1988 [129] |
| HA-1A | J5 lipid A | Human B cells | Heteromyeloma with lymphoma spleen cells | Sepsis | Phase 1 | 25, 100, 250 mg | 0 of 34 subjects | Fisher 1990 [130] | |
| MAB-T88 | LPS | Human B cells | Hybridoma with mouse myeloma | Neutropenia | Phase 1 | 1, 4, 8 mg/kg single dose | 0 of 9 subjects | Daifuku 1992 [131] | |
| Mab cocktail (5 IgM) | LPS | Human B cells | Normal adults P. aeruginosa bacteremia | Phase 1 | 0.75 to 3.0 mg/kg | 12 subjects 8 subjects | Saravolatz 1991 [132] | ||
| AR-101 (KBPA-101) | LPS | Human B cells | Hybridoma with mouse myeloma | Nosocomial P. aeruginosa pneumonia | Phase 2a | 1.2 mg/kg × 3 | 0 of 18 subjects | Lu 2011 [133] | |
| Glycolipid/ Proteolipid | L612 | Ganglioside GM3 | Human B cells | EBV-transformed patient B cells | Melanoma | Phase 1 | 960, 1440, 1920 mg 48 h infusion | 0 of 9 subjects | Irie 2004 [134] |
| MORAb-028 | Ganglioside GD2 | Human B cells | Hybridoma with human/mouse myeloma | Melanoma | Phase 1 | 1 or 2 mg/cm2/day × 5 days, repeated 2× | 18 subjects | NCT-01123304 [135] | |
| rHIgM22 | CNS myelin proteolipid | Human B cells | Hybridoma with mouse myeloma | Multiple sclerosis/neuronal degeneration diseases | Phase 1 | 0.025 to 2 mg/kgsingle dose | 55 subjects | Eisen 2017 [136] | |
| Glycan | Fanolesomab-Tc99 | CD15 (carbohydrate) | Mouse B cells | Hybridoma with mouse myeloma | Healthy volunteers | Phase 1 | 125 µg × 2 (21 days apart) | 5 of 30 subjects | Line 2004 [137] |
| PAT-SC1 | CD55 (glycan isoform) | Human B cells | Recombinant production Per.C6 cells | Gastric cancer | Phase 1 | 20 mg single dose | 51 subjects | Hensel 2014 [138] | |
| mAb216 | CDIM (carbohydrate) | Human B cells | Heteromyeloma with lymphoma spleen cells | B-lineage ALL | Phase 1 | 1.25 mg/kg to 5 mg/kg 3 + 2 dose escalation | 0 of 13 subjects | Liedtke 2012 [139] | |
| PAT-SM6 | GRP78 (O-linked glycan) | Human B cells | Recombinant production Per.C6 cells | Multiple myeloma | Phase 1 | 0.3, 1 3 or 6 mg/kg 4 doses over 2 weeks | 0 of 12 subjects | Rasche 2015 [140] | |
| Protein | Campath-1M | CD52 | Rat B cells | Hybridoma with rat myeloma | Graft vs. host disease | Phase 2 | 25 mg bid × 10 | (not tested) | Friend 1989 [141] |
| Mab 16.88 | Colon cancer antigen | Human B cells | Hybridoma with mouse myeloma | Colorectal cancer | Phase 1 | 8 mg, then 200, 500 or 1000 mg | 0 of 20 subjects | Haisma 1991 [142] | |
| ABX-CBL | CD147 | Mouse B cells | Hybridoma with mouse myeloma | Graft vs. host disease | Phase 1 | 0.2 to 0.3 mg/kg 9 doses | 0 of 51 subjects | Deeg 2001 [143] | |
| TOL101 | αβ TCR | Mouse B cells | Hybridoma with mouse myeloma | Renal transplant | Phase 2 | 0.3, 1.4, 7, 14, 28, 42 mg 5 daily doses | 1 of 36 subjects | Getts 2014 [144] | |
| ARG098 | FAS | Mouse: Human B cells (chimeric) | Hybridoma with mouse myeloma | Rheumatoid arthritis | Phase 1/2 | up to 10 μg/knee (intraarticular) | 43 subjects | Matsubara 2013 [145] |
| Antibody | Antigen | Indication | Model | Terminal Half-Life | Reference |
|---|---|---|---|---|---|
| Serum IgM (hu) I131-labeled | - | Humans | Two-compartment | 5.1 days (122 h) | Barth 1964 [183] |
| E5 (mu) | LPS (Lipid A) | Sepsis | One-compartment | 19.3 h | Harkonen 1988 [129] |
| HA-1A (hu) | LPS (Lipid A) | Sepsis | One-compartment | 15.9 h | Fisher 1990 [130] |
| Sepsis | One-compartment | 14.5 h | Romano 1993 [185] | ||
| MAB-T88 | Lipopolysaccharide | neutropenia | Two-compartment | 41.5 h | Daifuku 1992 [131] |
| AR-101 | Lipopolysaccharide | Nosocomial pneumonia | Two-compartment | 102 h (after 3rd dose) | Lu 2011 [133] |
| 5G2 | LPS (O-side chain) | Sepsis | One-compartment | 56 h | Meng 1993 [186] |
| rHIgM22 | CNS myelin proteolipid | Multiple sclerosis | (not stated) | 99 h (2 mg/kg) | Eisen 2017 [136] |
| ABX-CBL | CD147 | GvHD | Two-compartment | 15–19 h | Deeg 2001 [143] |
| TOL101 | ab TCR | Renal transplant | One-compartment | 23.8 h | Getts 2014 [144] |
| PAT-SM6 | GRP-78 | Multiple myeloma | (not stated) | 5.9 to 8.4 h | Rasche 2015 [140] |
| Fanolesomab-Tc99 | CD15 | Healthy volunteers | Two-compartment | 8 h | Package insert [187] |
| Mab 16.88 | Colon cancer antigen | Cancer | (not stated) | 20 h | Haisma 1990 [181] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keyt, B.A.; Baliga, R.; Sinclair, A.M.; Carroll, S.F.; Peterson, M.S. Structure, Function, and Therapeutic Use of IgM Antibodies. Antibodies 2020, 9, 53. https://doi.org/10.3390/antib9040053
Keyt BA, Baliga R, Sinclair AM, Carroll SF, Peterson MS. Structure, Function, and Therapeutic Use of IgM Antibodies. Antibodies. 2020; 9(4):53. https://doi.org/10.3390/antib9040053
Chicago/Turabian StyleKeyt, Bruce A., Ramesh Baliga, Angus M. Sinclair, Stephen F. Carroll, and Marvin S. Peterson. 2020. "Structure, Function, and Therapeutic Use of IgM Antibodies" Antibodies 9, no. 4: 53. https://doi.org/10.3390/antib9040053
APA StyleKeyt, B. A., Baliga, R., Sinclair, A. M., Carroll, S. F., & Peterson, M. S. (2020). Structure, Function, and Therapeutic Use of IgM Antibodies. Antibodies, 9(4), 53. https://doi.org/10.3390/antib9040053