Considerations for Optimizing Dosing of Immunoglobulins Based on Pharmacokinetic Evidence


Abstract
1. Introduction
2. Dosing of Immunoglobulins in Patients with PID
3. Pharmacokinetics (PK) of Immunoglobuins
4. Factors Influencing PK and Dosing of Immunoglobulins
4.1. Obesity
4.2. Precision-Dosing of IgG
4.3. Autoimmunity
4.4. Pregnancy
5. Discussion
- A mean dose of 100 mg/kg/month (n = 14; all subjects received 100 mg/kg/month dose)
- A mean dose 157 mg/kg/month (n = 14; dose ranging from 100–300 mg/kg/month)
- A mean dose 336 mg/kg/month (n = 10; dose ranging from 175–690 mg/kg/month)
- A mean dose 346 mg/kg/month (n = 7; dose ranging from 161–670 mg/kg/month)
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Disclaimer
References
- Bruton, O.C. Agammaglobulinemia. Pediatrics 1952, 9, 722–728. [Google Scholar] [PubMed]
- King, D.J. Applications and Engineering of Monoclonal Antibodies; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Fehr, J.; Hofmann, V.; Rappeler, U. Transient Reversal of Thrombocytopenia in Idiopathic Thrombocytopenic Purpura by High-Dose Intravenous Gamma Globulin. N. Engl. J. Med. 1982, 306, 1254–1258. [Google Scholar] [CrossRef]
- Imbach, P.; d’Apuzzo, V.; Hirt, A.; Rossi, E.; Vest, M.; Barandun, S.; Baumgartner, C.; Morell, A.; Schöni, M.; Wagner, H.P. High-Dose Intravenous Gammaglobulin for Idiopathic Thrombocytopenic Purpura in Childhood. Lancet 1981, 1, 1228–1231. [Google Scholar] [CrossRef]
- Hartung, H.P.; Mouthon, L.; Ahmed, R.; Jordan, S.; Laupland, K.B.; Jolles, S. Clinical Applications of Intravenous Immunoglobulins (Ivig)--Beyond Immunodeficiencies and Neurology. Clin. Exp. Immunol. 2009, 158 (Suppl. 1), 23–33. [Google Scholar] [CrossRef] [PubMed]
- Ouwehand, W.H.; Smith, G.; Ranasinghe, E. Management of Severe Alloimmune Thrombocytopenia in the Newborn. Arch. Dis. Child Fetal Neonatal Ed. 2000, 82, F173–F175. [Google Scholar] [CrossRef] [PubMed]
- Stanley, V.T.; Grimwood, K. Classical Kawasaki Disease in a Neonate. Arch. Dis. Child Fetal Neonatal Ed. 2002, 86, F135–F136. [Google Scholar] [CrossRef]
- Koleba, T.; Ensom, M.H. Pharmacokinetics of Intravenous Immunoglobulin: A Systematic Review. Pharmacother. J. Human Pharmacol. Drug Ther. 2006, 26, 813–827. [Google Scholar] [CrossRef]
- Kerr, J.; Quinti, I.; Eibl, M.; Chapel, H.; Späth, P.J.; Sewell, W.A.; Salama, A.; Van Schaik, I.N.; Kuijpers, T.W.; Peter, H.H. Is Dosing of Therapeutic Immunoglobulins Optimal? A Review of a Three-Decade Long Debate in Europe. Front. Immunol. 2014, 5, 629. [Google Scholar] [CrossRef]
- Bruton, O.C. A Decade with Agammaglobulinemia. J. Pediatr. 1962, 60, 672–676. [Google Scholar] [CrossRef]
- Jolles, S.; Orange, J.S.; Gardulf, A.; Stein, M.R.; Shapiro, R.; Borte, M.; Berger, M. Current Treatment Options with Immunoglobulin G for the Individualization of Care in Patients with Primary Immunodeficiency Disease. Clin. Exp. Immunol. 2015, 179, 146–160. [Google Scholar] [CrossRef]
- FDA. Guidance for Industry: Safety, Efficacy, and Pharmacokinetic Studies to Support Marketing of Immune Globulin Intravenous (Human) as Replacement Therapy for Primary Humoral Immunodeficiency. Available online: https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Blood/ucm078526.pdf (accessed on 10 October 2019).
- Committee for Medicinal Products for Human Use (Chmp): Guideline on the Clinical Investigation of Human Normal Immunoglobulin for Intravenous Administration (Ivig). Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-human-normal-immunoglobulin-intravenous-administration-ivig-rev-3_en.pdf (accessed on 25 November 2019).
- Fda Package Insert: Panzyga, (Immune Globulin Intravenous, Human-Ifas) 10% Liquid Preparation. Available online: https://www.fda.gov/downloads/BiologicsBloodVaccines/BloodBloodProducts/ApprovedProducts/LicensedProductsBLAs/FractionatedPlasmaProducts/UCM615706.pdf (accessed on 25 November 2019).
- Fda Package Insert: Cuvitru, Immune Globulin Subcutaneous(Human), 20% Solution. Available online: https://www.fda.gov/downloads/BiologicsBloodVaccines/BloodBloodProducts/ApprovedProducts/LicensedProductsBLAs/FractionatedPlasmaProducts/UCM520587.pdf (accessed on 25 November 2019).
- Fda Package Insert: Gammaplex, I Mmune Globulin Intravenous [Human], 5% Liquid, for Intravenous Use. Available online: https://www.fda.gov/downloads/biologicsbloodvaccines/ucm182963.pdf (accessed on 25 November 2019).
- Fda Package Insert: Gammagard, Liquid, Immune Globulin Infusion (Human), 10% Solution, for Intravenous and Subcutaneous Administration. Available online: https://www.fda.gov/downloads/BiologicsBloodVaccines/BloodBloodProducts/ApprovedProducts/LicensedProductsBLAs/FractionatedPlasmaProducts/ucm070010.pdf (accessed on 25 November 2019).
- Noya, F.J.; Rench, M.A.; Garcia-Prats, J.A.; Jones, T.M.; Baker, C.J. Disposition of an Immunoglobulin Intravenous Preparation in Very Low Birth Weight Neonates. J. Pediatrics 1988, 112, 278–283. [Google Scholar] [CrossRef]
- Weisman, L.E.; Fischer, G.W.; Hemming, V.G.; Peck, C.C. Pharmacokinetics of Intravenous Immunoglobulin (Sandoglobulin) in Neonates. Pediatr. Infect. Dis. 1986, 5, S185–S188. [Google Scholar] [CrossRef] [PubMed]
- Noya, F.J.; Rench, M.A.; Courtney, J.T.; Feldman, S.T.U.A.R.T.; Baker, C.J. Pharmacokinetics of Intravenous Immunoglobulin in Very Low Birth Weight Neonates. Pediatr. Infect. Dis. J. 1989, 8, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Weisman, L.E.; Fischer, G.W.; Marinelli, P.; Hemming, V.G.; Pierce, J.R.; Golden, S.M.; Peck, C.C. Pharmacokinetics of Intravenous Immunoglobulin in Neonates. Vox. Sang. 1989, 57, 243–248. [Google Scholar] [CrossRef]
- Orange, J.S.; Belohradsky, B.H.; Berger, M.; Borte, M.; Hagan, J.; Jolles, S.; Wasserman, R.L.; Baggish, J.S.; Saunders, R.; Grimbacher, B. Evaluation of Correlation between Dose and Clinical Outcomes in Subcutaneous Immunoglobulin Replacement Therapy. Clin. Exp. Immunol. 2012, 169, 172–181. [Google Scholar] [CrossRef]
- Orange, J.S.; Grossman, W.J.; Navickis, R.J.; Wilkes, M.M. Impact of Trough Igg on Pneumonia Incidence in Primary Immunodeficiency: A Meta-Analysis of Clinical Studies. Clin. Immunol. 2010, 137, 21–30. [Google Scholar] [CrossRef]
- Hodkinson, J.P. Considerations for Dosing Immunoglobulin in Obese Patients. Clin. Exp. Immunol. 2017, 188, 353–362. [Google Scholar] [CrossRef]
- Shapiro, R. Subcutaneous Immunoglobulin (16 or 20%) Therapy in Obese Patients with Primary Immunodeficiency: A Retrospective Analysis of Administration by Infusion Pump or Subcutaneous Rapid Push. Clin. Exp. Immunol. 2013, 173, 365–371. [Google Scholar] [CrossRef]
- Stump, S.E.; Schepers, A.J.; Jones, A.R.; Alexander, M.D.; Auten, J.J. Comparison of Weight-Based Dosing Strategies for Intravenous Immunoglobulin in Patients with Hematologic Malignancies. Pharmacotherapy 2017, 37, 1530–1536. [Google Scholar] [CrossRef] [PubMed]
- Kuitwaard, K.; Van Doorn, P.A.; Vermeulen, M.; Van den Berg, L.H.; Brusse, E.; Van der Kooi, A.J.; Van der Pol, W.L.; van Schaik, I.N.; Notermans, N.; Tio-Gillen, A.P.; et al. Serum Igg Levels in Iv Immunoglobulin Treated Chronic Inflammatory Demyelinating Polyneuropathy. J. Neurol. Neurosurg. Psychiatry 2013, 84, 859–861. [Google Scholar] [CrossRef][Green Version]
- Mosca, M.; Strigini, F.; Carmignani, A.; D’ascanio, A.; Genazzani, A.R.; Bombardieri, S. Pregnant Patient with Dermatomyositis Successfully Treated with Intravenous Immunoglobulin Therapy. Arthritis Rheum. 2005, 53, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Perricone, R.; De Carolis, C.; Kröegler, B.; Greco, E.; Giacomelli, R.; Cipriani, P.; Fontana, L.; Perricone, C. Intravenous Immunoglobulin Therapy in Pregnant Patients Affected with Systemic Lupus Erythematosus and Recurrent Spontaneous Abortion. Rheumatology (Oxford) 2008, 47, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Triolo, G.; Ferrante, A.; Ciccia, F.; Accardo-Palumbo, A.; Perino, A.; Castelli, A.; Giarratano, A.; Licata, G. Randomized Study of Subcutaneous Low Molecular Weight Heparin Plus Aspirin Versus Intravenous Immunoglobulin in the Treatment of Recurrent Fetal Loss Associated with Antiphospholipid Antibodies. Arthritis Rheum. 2003, 48, 728–731. [Google Scholar] [CrossRef]
- Coulam, C.B.; Krysa, L.W.; Bustillo, M. Intravenous Immunoglobulin for in-Vitro Fertilization Failure. Hum. Reprod. 1994, 9, 2265–2269. [Google Scholar] [CrossRef] [PubMed]
- Intravenous Immunoglobulin (Ivig) and Recurrent Spontaneous Pregnancy Loss. Fertility and Sterility; The Practice Committee of the American Society for Reproductive Medicine: Amsterdam, The Netherlands, 2006; pp. 226–227. [Google Scholar]
- Ensom, M.H.; Stephenson, M.D. A Two-Center Study on the Pharmacokinetics of Intravenous Immunoglobulin before and During Pregnancy in Healthy Women with Poor Obstetrical Histories. Hum. Reprod. 2011, 26, 2283–2288. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.; Conklin, J.; Hunter, S.K.; Empey, R.; Tyler, E.M.; Christensen, A.; Talcott, K.M.; Ballas, Z.K.; Santillan, D.A.; Santillan, M.K. Defining Normal Igg Changes Throughout Pregnancy. Proc. Obstet. Gynecol. 2013, 3, 1–2. [Google Scholar] [CrossRef]
- Schaffer, F.M.; Newton, J.A. Intravenous Gamma Globulin Administration to Common Variable Immunodeficient Women During Pregnancy: Case Report and Review of the Literature. J. Perinatol. 1994, 14, 114–117. [Google Scholar]
- Ledford, D.K. Igg Dosing During Pregnancy; American Academy of Allergy Asthma & Immunology: Milwaukee, WI, USA, 2019. [Google Scholar]
- Lingman-Framme, J.; Fasth, A. Subcutaneous Immunoglobulin for Primary and Secondary Immunodeficiencies: An Evidence-Based Review. Drugs 2013, 73, 1307–1319. [Google Scholar] [CrossRef]
- Brambell, F.W. The Transmission of Immunity from Mother to Young and the Catabolism of Immunoglobulins. Lancet 1966, 2, 1087–1093. [Google Scholar] [CrossRef]
- Brambell, F.W.; Hemmings, W.A.; Morris, I.G. A Theoretical Model of Gamma-Globulin Catabolism. Nature 1964, 203, 1352–1354. [Google Scholar] [CrossRef]
- Simister, N.E.; Mostov, K.E. An Fc Receptor Structurally Related to Mhc Class I Antigens. Nature 1989, 337, 184–187. [Google Scholar] [CrossRef]
- Junghans, R.P. Finally! The Brambell Receptor (Fcrb). Mediator of Transmission of Immunity and Protection from Catabolism for Igg. Immunol. Res. 1997, 16, 29–57. [Google Scholar] [CrossRef] [PubMed]
- Roopenian, D.C.; Akilesh, S. Fcrn: The Neonatal Fc Receptor Comes of Age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Schiff, R.I.; Rudd, C. Alterations in the Half-Life and Clearance of Igg During Therapy with Intravenous Gamma-Globulin in 16 Patients with Severe Primary Humoral Immunodeficiency. J. Clin. Immunol. 1986, 6, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A.; Strober, W. Metabolism of Immunoglobulins. Prog. Allergy 1969, 13, 1–110. [Google Scholar] [PubMed]
- Hansen, R.J.; Balthasar, J.P. Effects of Intravenous Immunoglobulin on Platelet Count and Antiplatelet Antibody Disposition in a Rat Model of Immune Thrombocytopenia. Blood 2002, 100, 2087–2093. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.J.; Balthasar, J.P. Intravenous Immunoglobulin Mediates an Increase in Anti-Platelet Antibody Clearance Via the Fcrn Receptor. Thromb. Haemost. 2002, 88, 898–899. [Google Scholar] [CrossRef] [PubMed]



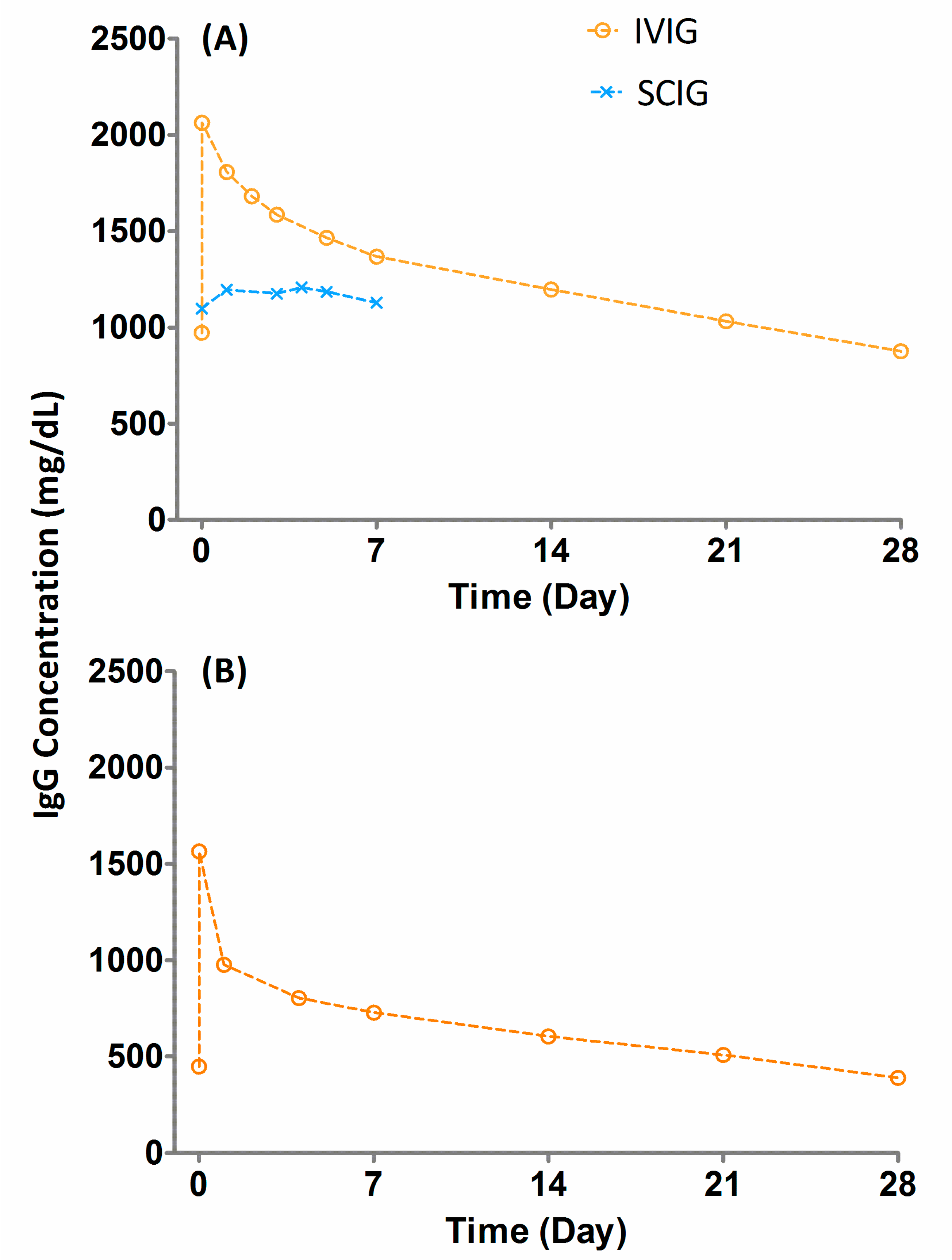{kind=link}
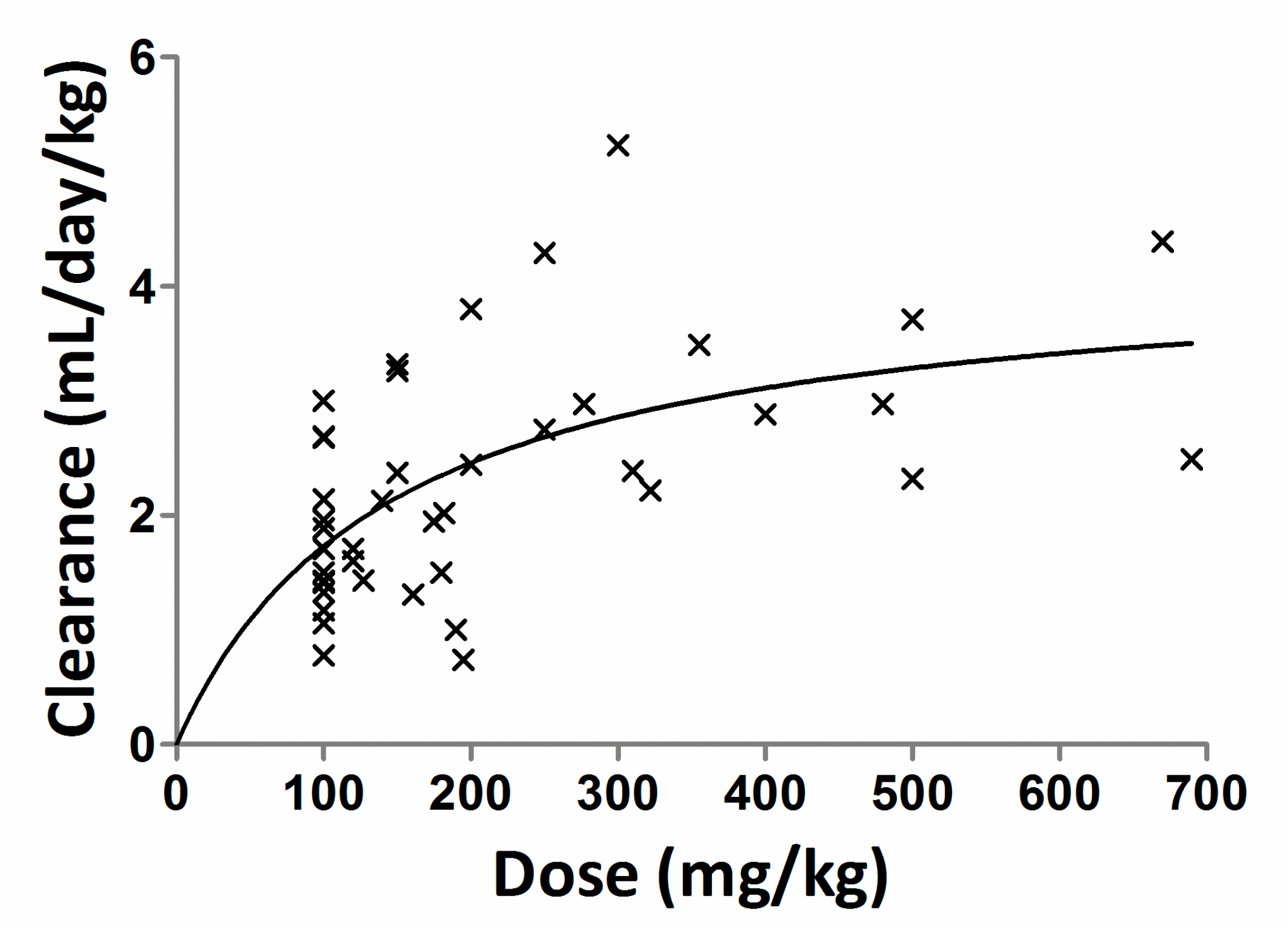{kind=link}
| Products | Half-Life (Days) | Clearance (mL/day/kg) | Half-Life (Days) | Clearance (mL/Day/kg) | Dosing Interval |
|---|---|---|---|---|---|
| Baseline-Uncorrected | Baseline-Corrected | ||||
| Asceniv | 29 ± 5 | 1.68 ± 0.40 | 6 ± 2 | 9 ± 4 | 3 weeks |
| Asceniv | 40 ± 12 | 1.47 ± 0.50 | 10 ± 8 | 8 ± 5 | 4 weeks |
| Panzyga | 32 ± 12 | 1.44 ± 0.24 | 5 ± 2 | 7.2 ± 1.7 | 3 weeks |
| Panzyga | 45 ± 21 | 1.44 ± 0.48 | 8 ± 5 | 7.4 ± 3.1 | 4 weeks |
| Gammaplex 5% | NA | NA | 6 ± 6 | 7.3 ± 6.9 | 3 weeks |
| Gammaplex 5% | NA | NA | 6 ± 6 | 5.3 ± 5.5 | 4 weeks |
| Privigen | 28 ± 6 | 1.30 ± 0.10 | NA | NA | 3 weeks |
| Privigen | 45 ± 19 | 1.30 ± 0.30 | NA | NA | 4 weeks |
| Bivigam | 20 ± 4 | 1.97 ± 0.22 | NA | NA | 3 weeks |
| Bivigam | 33 ± 11 | 1.41 ± 0.46 | NA | NA | 4 weeks |
| Dose (mg/kg) | Weight (kg) | Age | Half-Life (days) | CL (mL/day/kg) |
|---|---|---|---|---|
| Sandoglobulin (preterm) [19] | ||||
| 500 | 2.6 ± 0.6 | 35.8 ± 2.9 weeks | 11.3 ± 0.6 | 4.2 ± 1.0 |
| Venoglobulin (preterm) [20] | ||||
| 1000 | 1.12 ± 0.21 | 29.3 ± 1.8 days | 19.6 * | 5.2 ± 1.5 |
| 750 | 1.06 ± 0.24 | 28.9 ± 2.0 days | 28.7 | 5.6 ± 0.5 |
| 500 | 1.16 ±0.14 | 29.2 ± 2.3 days | 22.1 | 3.7 ± 0.8 |
| Gammagard (preterm) [18] | ||||
| 750 | 1.09 ± 0.23 | 28.6 ± 2.9 weeks | 22 ± 5 | 3.4 ± 0.9 |
| 500 | 1.12 ± 0.24 | 29.2 ± 1.6 weeks | 22 ± 6 | 2.6 ± 0.8 |
| Gamimune [21] | ||||
| 250 (term) | 2.87 ± 0.74 | 37.4 ± 2.6 weeks | 15 ** | 2.8 ** |
| 500 (term) | 3.04 ± 0.57 | 37.6 ± 2.4 weeks | 21 | 4.1 |
| 1000 (preterm) | 2.52 ± 0.90 | 34.8 ± 3.0 weeks | 35 | 3.6 |
| Products | SC | IV | ||
|---|---|---|---|---|
| Cmax (mg/dL) | Cmin (mg/dL) | Cmax (mg/dL) | Cmin (mg/dL) | |
| Cuvitru | 1809 | 1477 | 2521 | 1019 |
| Hizentra | 1616 | 1448 | 2564 | 1127 |
| CUTAQUIG | 1400 ± 440 | 1200 ± 350 | 1970 ± 560 | 1050 ± 260 |
| Gammagard | 1393 ± 289 | 1202 ± 282 | 2240 ± 536 | 1050 ± 260 |
| Gamunex-C | NA | 1140 | NA | 958 |
| Dose (mg/kg/Month) | Trough (mg/dL) | Half-Life (Days) | CL (mL/Day/kg) |
|---|---|---|---|
| 100 | 191 ± 112 | 43 ± 23 | 1.83 ± 0.64 |
| 157 | 225 ±168 | 39 ± 14 | 2.28 ± 1.27 |
| 336 | 390 ± 158 | 33 ± 9 | 2.40 ± 0.89 |
| 346 | 427 ± 138 | 33 ± 16 | 2.43 ± 1.27 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmood, I.; Tegenge, M.A.; Golding, B. Considerations for Optimizing Dosing of Immunoglobulins Based on Pharmacokinetic Evidence. Antibodies 2020, 9, 24. https://doi.org/10.3390/antib9020024
Mahmood I, Tegenge MA, Golding B. Considerations for Optimizing Dosing of Immunoglobulins Based on Pharmacokinetic Evidence. Antibodies. 2020; 9(2):24. https://doi.org/10.3390/antib9020024
Chicago/Turabian StyleMahmood, Iftekhar, Million A. Tegenge, and Basil Golding. 2020. "Considerations for Optimizing Dosing of Immunoglobulins Based on Pharmacokinetic Evidence" Antibodies 9, no. 2: 24. https://doi.org/10.3390/antib9020024
APA StyleMahmood, I., Tegenge, M. A., & Golding, B. (2020). Considerations for Optimizing Dosing of Immunoglobulins Based on Pharmacokinetic Evidence. Antibodies, 9(2), 24. https://doi.org/10.3390/antib9020024