1. Introduction
Antibodies and antibody-like molecules offer the potential to develop highly efficacious drugs against a wide range of disorders from cancers to autoimmune diseases to rheumatic and cardiovascular disease. Although the beginnings of this potential have been glimpsed, truly harnessing the superior targeting properties of these molecules requires a platform from which to effectively identify the best candidates for drug development.
The idea of an immunotherapeutic treatment strategy for cancer emerged in the 1920s focused around the treatment of Hodgkin’s Lymphoma with lymph node extract. However, it is only more recently that immunotherapies have become an established treatment modality, leading to the development of several novel therapeutics for hematological cancers and solid tumors [
1]. Over the past 20 years, antibody-based therapies have seen particular success with nearly 20 antibodies gaining US Food and Drug Administration (FDA) approval for use in oncologic care since 1997 [
1]. The more recent emergence of chimeric, humanized, and human monoclonal antibodies, has led to a rapid increase in antibody-based therapeutics, which, with 75 Billion USD global sales in 2013, are now the dominant class of molecules within the global biopharmaceutical market [
2].
However, antibodies by themselves can, depending on their mechanism of action, display low therapeutic efficacy, meaning alternative approaches are required to increase the potency of antibody-based therapeutics. To address such limitations, antibody-drug conjugates (ADCs) have emerged as a promising therapeutic approach, which combine the selectivity of a targeted treatment with the cytotoxic potency of chemotherapy agents.
The first ADC gemtuzumab ocogamicin (Mylotarg
®) gained clinical approval in 2000 [
3], paving the way for three further ADCs, brentuximab vedotin (Adectris
®), ado-trastuzumab emtansine (Kadcyla
®), and Inotuzumab ozogamicin (Besponsa
®), which were licensed for the treatment of Hodgkin’s and anaplastic large-cell lymphomas, HER-2 positive breast cancer, and relapsed or refractory B-cell precursor acute lymphoblastic leukemia, respectively [
4,
5,
6]. The need to develop efficacious, novel antibody-based therapies means that over 50 different ADCs are currently in preclinical or clinical development [
7,
8].
In such a competitive marketplace, there is an increasing focus on the potential developability of early-stage molecules to prevent costly late-stage failures. This responsibility falls on analytical techniques, which are used to study structural and functional properties including affinity, kinetics, potency, aggregation, solubility, stability, immunogenicity, and pharmacokinetics, as well as cell-based assays to study toxicity and off-target effects.
One such technology, rapidly adopted to study antibody-antigen interactions following its introduction in 1990, is surface plasmon resonance (SPR) [
9]. First applied to the study of antibody-antigen interactions and epitope mapping [
10], SPR has several advantages over traditional immunoassays such as enzyme-linked immunosorbent assay (ELISA) or radioimmune assay (RIA). It is a label-free technique that monitors the formation and dissociation of biomolecular complexes in real-time, allowing binding kinetics and affinities to be measured. It is also sensitive, requires small sample volumes, works well with crude samples, and has the automation and throughput capability required to support high throughput screening and characterization studies [
11,
12,
13].
Current uses for SPR technology include early-stage screening of hybridoma/phage libraries to monitor expression and triage antibodies based on binding affinity, profiling binding specificity, and providing a detailed understanding of binding kinetics and affinity to characterize antibody-antigen interactions. During therapeutic antibody development, SPR is part of a suite of analytical methods used to study stability, drug-target binding interactions, and binding to Fc receptors, complement and the neonatal receptor (FcRn) to assess the critical quality attributes that determine the efficacy and clinical safety of the final product. As a core technology in analytical and Quality Control (QC) labs, SPR is also used to monitor batch-to-batch variation, support Chemistry, Manufacturing, and Controls (CMC), and as a potency assay to support clinical batch and final product release.
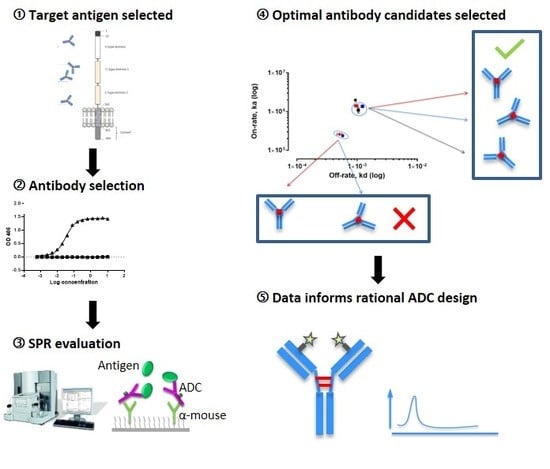
Given the prevalent use of SPR in the selection and development of standard antibody biotherapeutics, together with its increasing use in many aspects of ADC development, including target selection, antibody kinetics characterization, epitope mapping, and optimization [
14,
15], we explored how the technology could be used within our lab for the selection and characterization of next-generation ADCs. The importance of the antibody component of an ADC to therapeutic efficacy means that careful consideration must be given to the selection of antibodies for this purpose. Previous work within our laboratory demonstrated variability in the efficacy of antibodies characterized using standard immunoassay techniques such as ELISA, which led us to investigate ADC characteristics using SPR (unpublished).
Aiming to streamline the design and development of ADCs, we study multiple aspects of effective ADC design, each assessed by SPR. Biacore™ technology is employed to characterize four ADCs that target the Receptor for Advanced Glycation End Products (RAGE), a multi-ligand signaling system that drives innate immune inflammatory responses via nuclear factor-kappa beta (NF-kB) mediated gene activation and is associated with gynecological disease [
16]. We demonstrate that a wide range of antibody characteristics can be evaluated including specificity, kinetics/affinity, the effect of linker binding, the impact of drug to antibody ratio (DAR), and the effect of endosomal pH on antibody-antigen binding. In doing so, this study demonstrates the application of SPR Biacore™ technology as a platform, where detailed information can be obtained, supporting the requirements for rapid and high-throughput screening that will enable enhanced ADC development.
2. Materials and Methods
2.1. Antibody Production
Monoclonal antibody production was performed by Bio-Rad Antibodies (formerly AbD Serotec, Bio-Rad Laboratories, Oxford, UK). All procedures were performed in accordance with the Animals (Scientific Procedures) Act 1986, and the guidance issued in ‘Responsibility in the case of Animals in Bioscience research: expectations of the major research council and charitable funding bodies.’ Monoclonal antibodies against RAGE were produced using standard protocols for monoclonal antibody production [
17,
18]. Briefly, BALB/c mice, obtained from Charles River, Oxford, UK, were immunized with keyhole limpet hemocyanin (KLH)-conjugated RAGE, or KLH-conjugated peptides corresponding to amino acids (aa) 198–217 or 327–344 of the RAGE protein. Clones were selected based on a positive ELISA screen using bovine serum albumin (BSA)-conjugated peptides. Post-fusion, individual clones were then selected by limiting dilution and clonal expansion to identify genetically stable, antibody-producing cells for subsequent antibody production. One clone with an affinity for the full-length rRAGE protein (RBGO1), two clones with an affinity for aa198–217 (RBGO2 and RBGO3), and one with an affinity for aa327–344 (RBGO4) were selected for antibody production. Antibodies were purified from the tissue culture medium using protein G affinity purification.
2.2. Antibody-Drug Conjugation
Murine antibodies against RAGE were reconstituted in 10 mM Tris/HCl (Sigma, Dorset, UK) and 2 mM EDTA (Sigma) pH 8.0. Antibodies were reduced with 3.5 equivalents TCEP:Ab (10 mM in water, Sigma) for 2 h at 37 °C. Without purification the reduced antibody was split in two equal-volume aliquots and each aliquot alkylated with 6.5 equivalents of drug linker: Ab (10 mM MC-ValCit-PAB-MMAE or MC-MMAF, see
Figure S1, in DMA with additional DMA added to achieve 5%
v/
v final DMA, ADC Biotechnology, St Asaf, UK) for 2 h at 22 °C. Following alkylation, N-acetyl cysteine (Sigma) was used to quench any unreacted toxin linker. The conjugates were purified using a HiTrap™ G25 column (GE Healthcare, Uppsala, Sweden) equilibrated in 5 mM histidine/HCl, 50 mM trehalose (Sigma), 0.01%
w/
v polysorbate 20 (Sigma), pH 6.0. Conjugates were analysed by size exclusion chromatography (SEC) for monomeric content and concentration using a calibration curve of naked antibody. Running conditions: Agilent 1100 High Pressure Liquid Chromatography (HPLC), Tosoh TSKgel® G3000SWXL 7.8 mm × 30 cm, 5 μm column (Tosoh Bioscience, Reading, UK), 0.5 mL/min in, 0.2 M Potassium Phosphate, 0.25 M Potassium Chloride, 10% isopropyl alcohol (IPA), pH 6.95. Drug loading of the conjugates was confirmed using a combination of hydrophobic interaction chromatography (HIC) and reverse phase chromatography. HIC was carried out using a TOSOH Butyl-NPR 4.6 mm × 3.5 cm, 2.5 μm column (Tosoh Bioscience) run at 0.8 mL/min with a 12 min linear gradient between A—1.5 M (NH4)2SO4, 25 mM NaPi, pH 6.95±0.05 and B—75% 25 mM NaPi, pH 6.95 ± 0.05, 25% IPA. Reverse phase analysis was performed on a Polymer Lab’s polymeric reversed phase (PLRP) 2.1 mm × 5 cm, 5 μm column (Tosoh Bioscience) run at 1 mL/min at 80 °C with a 25 min linear gradient between 0.05% trifluoracetic acid (TFA)/H
2O and 0.04% TFA/CH
3CN. Samples were first reduced by incubation with 1, 4-Dithiothritol (DTT, Sigma) at pH 8.0 at 37 °C for 15 min.
2.3. Surface Plasmon Resonance
SPR reagents used were Series S Sensor Chip CM5, HBS-EP+ buffer (10 mM Hepes, 150 mM NaCl, 3 mM ethylenediaminetetraacetic acid, and 0.05% Surfactant P20, pH 7.4), Amine Coupling Kit, Mouse Antibody Capture Kit, including 10 mM glycine-HCl pH 1.7 regeneration solution (all from GE Healthcare).
SPR analysis was performed using a Biacore™ T200 system (GE Healthcare) and HBS-EP+ buffer was used as sample and running buffer. The analysis temperature and sample compartment were set to 25 °C. Immobilization of α-mouse antibody was performed using the Amine Coupling Kit in accordance with the manufacturer’s instructions. The anti-mouse antibody was immobilized in all flow cells, but flow cells 1 and 3 were used as reference cells for antibodies captured in flow cells 2 and 4, respectively. Antibody capture levels were typically in the range 500–1000 RU for the kinetic experiments. Protein or peptide was injected for 60 s in order of increasing concentration over reference and active flow cells, applying a single cycle kinetics procedure using five concentrations. Following each binding cycle, the surface was regenerated with a 180 s injection of regeneration solution from the capture kit, removing the bound antibody. Blank cycles (antibody + buffer injections + regeneration) were performed between each antibody. Data were double referenced by first subtracting responses from the reference flow cell and then subtracting the blank cycles. Data were fitted to a one-to-one binding model using Biacore™ T200 Evaluation Software 2.0.
2.4. Enzyme Linked Immunosorbent Assay (ELISA)
The 96-well micro plates were coated with peptide-BSA conjugates (10 µg/mL) in 20 mM carbonate-bicarbonate buffer (Sigma) at room temperature (RT) for 2 h. After coating, plates were washed (×3) with washing solution, which comprised phosphate buffered saline (PBS; Sigma) containing 0.1% Tween-20 (Sigma). Plates were then blocked with PBS containing 0.1% Tween-20 and 0.2% (w/v) Gelatin (Sigma) for 30 min at RT. After blocking, plates were washed (×3) with washing solution and doubling-dilutions of primary antibody (10 to 0.0006 µg/mL) prepared in PBS containing 0.1% Tween-20. Primary antibody dilutions were added to the appropriate wells of the plate in 100 µL volumes and plates incubated at RT for 2 h. At the end of the period, plates were washed (×4) with washing solution and horseradish peroxidase (HRP) conjugated α-mouse IgG1 added at a dilution of 1:2000. Plates were incubated at RT for 30 min before washing (×4) and the addition of HRP substrate in accordance with the manufacturer’s instructions (3,3′,5,5′-Tetramethylbenzidine, TMB, liquid substrate system, BD Biosciences, Oxford, UK). After sufficient color development, the TMB substrate reaction was stopped by the addition of 2 M sulphuric acid (BD Biosciences) and plates read at 405 nm using a FLUOstar Omega (BMG Labtech, Aylesbury, UK) spectrophotometer. Data were fitted to a 4-parameter logistic model using MARS data analysis software v3.01R2 (BMG Labtech).
2.5. Protein Analysis
Protein or peptides (100 µg/mL) were immobilized onto activated polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Watford, UK) by spotting the desired volume onto the membrane and allowing air-drying at RT. Non-specific sites were blocked with 5% bovine serum albumin (BSA) in tris-buffered saline containing 0.05% Tween-20 (TBS-T) at RT for 1 h. After blocking, membranes were incubated with primary antibody (1 µg/mL) in BSA/TBS-T at RT for 2 h. Membranes were then washed (3 × 5 min) in TBS-T before incubation with HRP-conjugated α-mouse IgG at RT for 1 h. Membranes were then washed with TBS-T (1 × 15 min, 2 × 5 min), then once with TBS (5 min) before visualization using luminol reagent in accordance with the manufacturer’s instructions (Bio-Rad). Images were acquired using a ChemiDoc™ MP imaging system (Bio-Rad) and analyzed with Image Lab™ (Version 3.0) software (Bio-Rad).
2.6. Cell Culture and α-RAGE Antibody Cell Surface Binding and Internalization
The HEC1A (endometrial cancer) cell line was obtained from the European Collection of Authenticated Cell Cultures (ECACC, Public Health England, UK). Cells were grown to 80% confluence before passage in complete medium, which comprised a 1:1 mixture of Dulbecco’s Modified Eagle Medium and Ham’s F-12 nutrient medium (DMEM/F12, Thermo Fisher, Gloucester, UK) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Thermo Fisher), 100 units/mL penicillin, and 100 µg/mL streptomycin (Thermo Fisher). Cells were maintained in a humidified, 5% CO2 in air atmosphere incubator at 37 °C, and the culture medium was changed every 48 h.
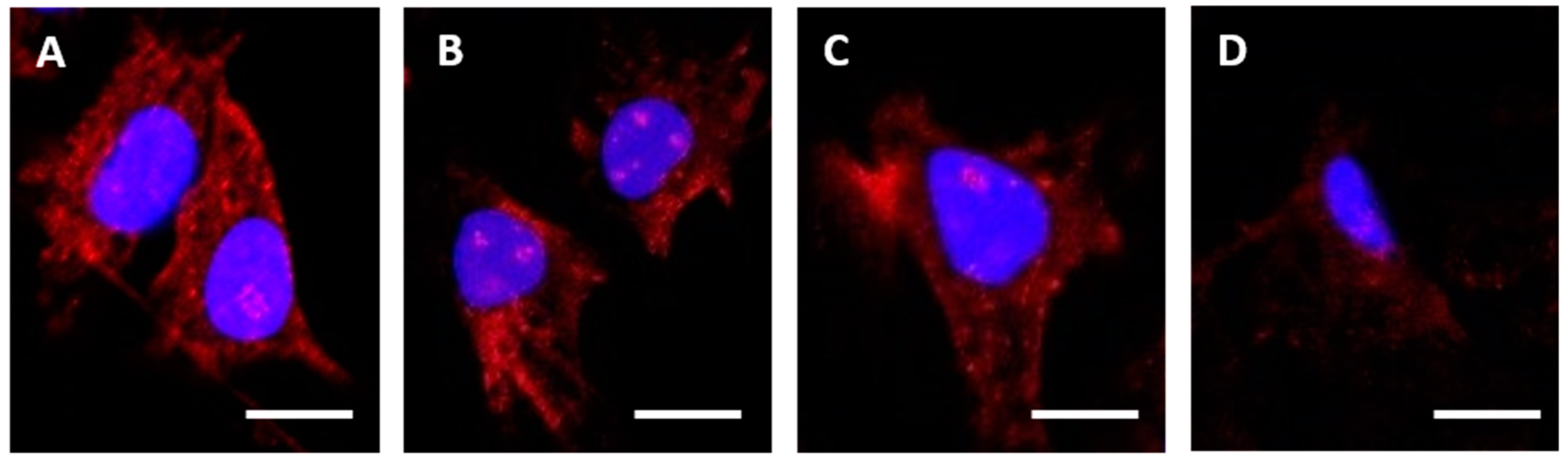
HEC1A cells were seeded (1 × 105 cells/mL) in 8-well chamber slides (BD Biosciences, Oxford, UK) in 200 µL of stripped medium (complete medium using charcoal stripped FBS) and cultured for 24 h in a humidified, 5% CO2 in air atmosphere incubator at 37 °C. After culture, cells were washed in pre-warmed (37 °C) Dulbecco’s phosphate buffered saline (DPBS) and slides were placed on ice. Cells were treated with control medium or medium containing one of the α-RAGE antibodies at 10 µg/mL, and the 8-well chamber slides were incubated on ice for 30 min. Slides were then transferred to the incubator at 37 °C for 240 min, before washing in DPBS and then fixing in 4% paraformaldehyde at 4 °C for 20 min. Where appropriate, cells were permeabilized following fixation, by incubation in 0.01% Triton X-100 in DPBS at 4 °C for 10 min. Cells were then washed and stained with goat anti-mouse IgG-Alexafluor488 diluted 1:1000 in DPBS before nucleus staining with 4′,6-Diamidine-2′-phenylindole dihydrochloride (DAPI).
Images were acquired on a Zeiss LSM 710 confocal microscope (Carl Zeiss Microscopy, Jena, Germany), and analyzed using the Zen 2012 (blue edition) image analysis software v10 (Carl Zeiss).
2.7. RAGE-ADC in vitro Efficacy Screening
HEC1A endometrial cancer cells were seeded (5 × 102 cells/mL) in 96-well tissue culture plates (TPP) in 100 µL of stripped medium and cultured for 24 h in a humidified, 5% CO2 in air atmosphere incubator at 37 °C. After culturing was carried out, cells were treated with control medium or medium containing ADCs (0.01–100 µg/mL) for 96 h. Positive controls were cells treated with 0.01% Triton X-100 in stripped medium for the last 4 h of the experiment. Cell growth was monitored over a 96-h period using the RealTime-Glo™ MT Cell Viability Assay (Promega, Southampton, UK) in accordance with the manufacturer’s instructions. Fluorescence was measured using a FLUOstar Omega microplate reader (BMG Labtech, Aylesbury, UK).
2.8. Statistical Analyses
Statistical analyses were performed using IBM SPSS Statistics 22 (IBM Corp. Armonk, NY, USA). Initially, the data were tested for homogeneity. Data were analysed by a Student’s t-test and are represented mean (SD). A p-value < 0.05 was considered statistically significant.
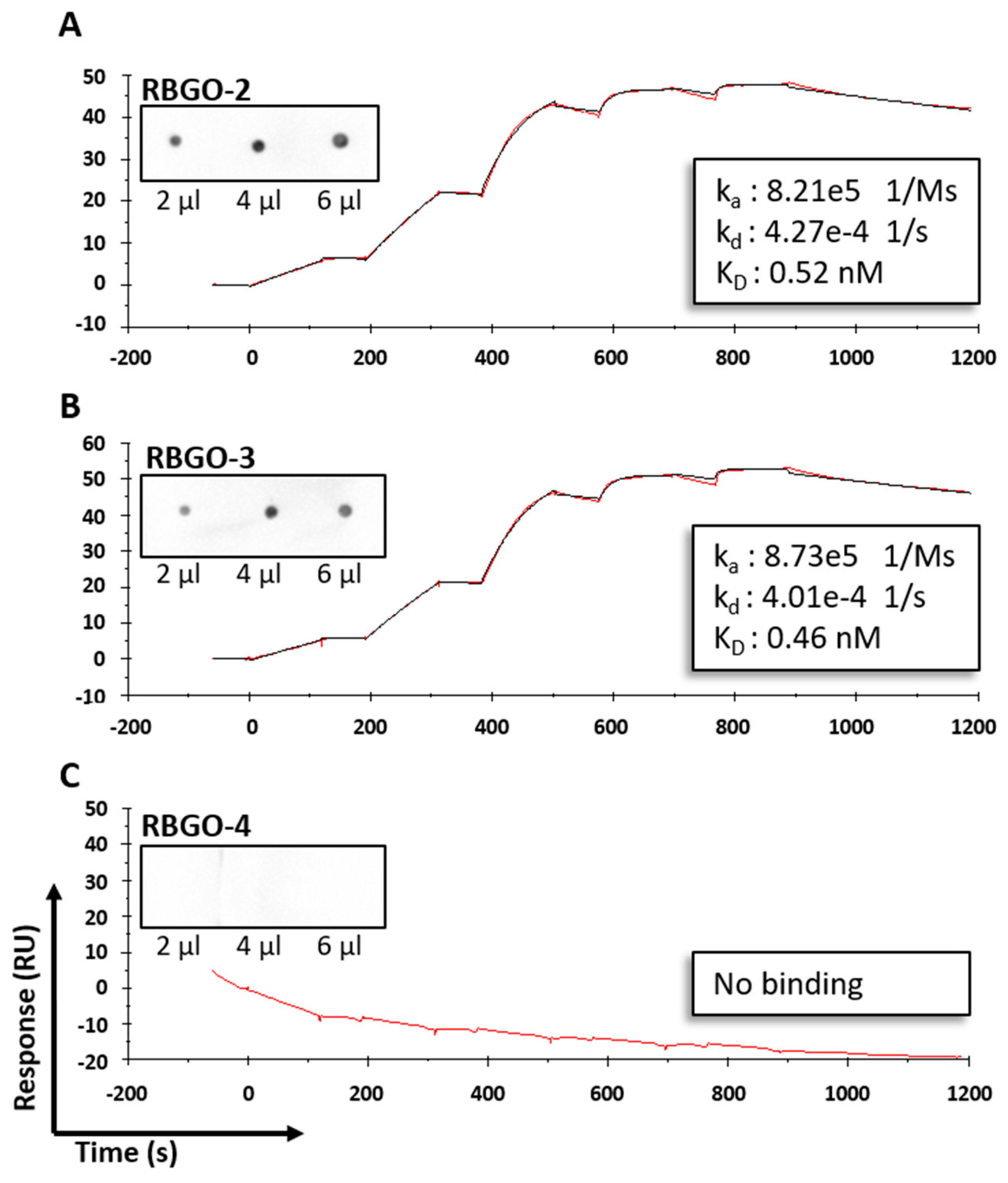
4. Discussion
Antibody–drug conjugates are a proven example of the type of novel, precision medicines required to combat the increasing incidence of diseases such as the gynecological cancers [
24,
25]. Aiming to streamline the design and development of ADCs within our laboratory, we evaluated antibodies being developed as ADCs, using SPR technology. This technology enabled us to study ADC characteristics such as specificity, antigen kinetics/affinity, any effects that the pH or our choice of linker type and DAR may have on antigen binding kinetics and demonstrate the applicability of this technology to ADC design and development. SPR analysis within our lab had indicated that antibodies raised against our therapeutic target, RAGE, bound poorly to the full-length protein, data that we verified using dot blot analysis and confocal microscopy. Clone selection based on ELISA against the immunization peptide conjugated to BSA is typical, so we compared ELISA data to kinetics data from SPR analysis using the same peptides. Whilst two of the antibodies in question, RBGO2 and RBGO3, bound with high-affinity to the immunization peptide, the RBGO4 antibody did not, suggesting that the use of ELISA alone might lack the specificity required for the selection of effective, high-affinity binding antibodies for therapeutic development. Indeed, dot blot analysis verified the SPR data, confirming that the RBGO4 antibody did not bind to the aa327-344 peptide. It is unclear why a positive ELISA screen was obtained for the RBGO4 antibody, although interestingly, repeat ELISAs using a non-BSA conjugated aa327-344 or BSA alone were negative, see
Figure S4, suggesting that the RBGO4 epitope may span the BSA-peptide junction.
A variable effect of conjugation on antibody characteristics is known, but this can range from a large effect to none depending on the antibody and conjugation chemistry employed. Our cell-based data demonstrated variability in the cell killing efficacy of our ADCs. An effect that was variable for each antibody and so could not be attributed to the drug or linker being used. Non-specific conjugation often alters the electrostatic properties and hydrophobicity of an antibody with implications for ADC stability and pharmacokinetics [
26]. Thiol conjugation, in particular, has a dramatic, DAR-related effect on antibody thermostability compared to alternative techniques such as amine or carbohydrate conjugation; however, the effect is not consistent [
21]. Our data showed antibody to antibody variability regarding the effect of conjugation. Although antibody-antigen affinity was comparable for different types of linker and DARs, conjugation caused a four-fold reduction of the affinity (primarily due to a 10-fold reduction in the association rate) for the RBGO3 antibody, whilst the RBGO1 antibody was unaffected despite both antibodies having similar DARs. Utilizing inter-chain, disulfide bridge cysteines is an effective, inexpensive strategy for ADC production. However, variability in the effect of conjugation on antibody binding kinetics means it is important to quantify the impact of conjugation. An important consideration when developing ADCs is also that affinity is not the whole story. ADCs with similar affinity may, in fact, have very different kinetics and, depending on the receptor being targeted, it may be acceptable to have a reduced on-rate if the off-rate is slow enough. The strength of using SPR as a development tool is that affinity can be dissolved into kinetics to obtain the desired kinetic profile for the ADC being developed.
The efficacy of an ADC is dependent on internalization and the release of the cytotoxic payload. Consequently, recent developments in antibody therapeutics have included the design of antibodies that, in addition to binding with high-affinity at the extracellular pH 7.4, also dissociate at a higher rate under endosomal pH 6.0 conditions [
27]. Although this concept is yet to be demonstrated for ADCs, such an approach is plausible and could be beneficial for ADCs targeting antibodies that are rapidly recycled back to the cell surface such as HER2 and RAGE [
27,
28,
29]. To explore this aspect of our ADCs, we used an SPR-based method to qualitatively assess the effect of pH on antibody-antigen dissociation. Whilst association was performed at extracellular pH 7.4, it was possible to monitor and compare dissociation at pH 7.4 or endosomal pH 6.0. Interestingly, our lead ADC candidate, based upon the RBGO1 antibody, exhibits high-binding affinity at pH 7.4 and increased dissociation rate at pH 6.0 compared to the dissociation rate at pH 7.4.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}