Nanobody Engineering: Toward Next Generation Immunotherapies and Immunoimaging of Cancer



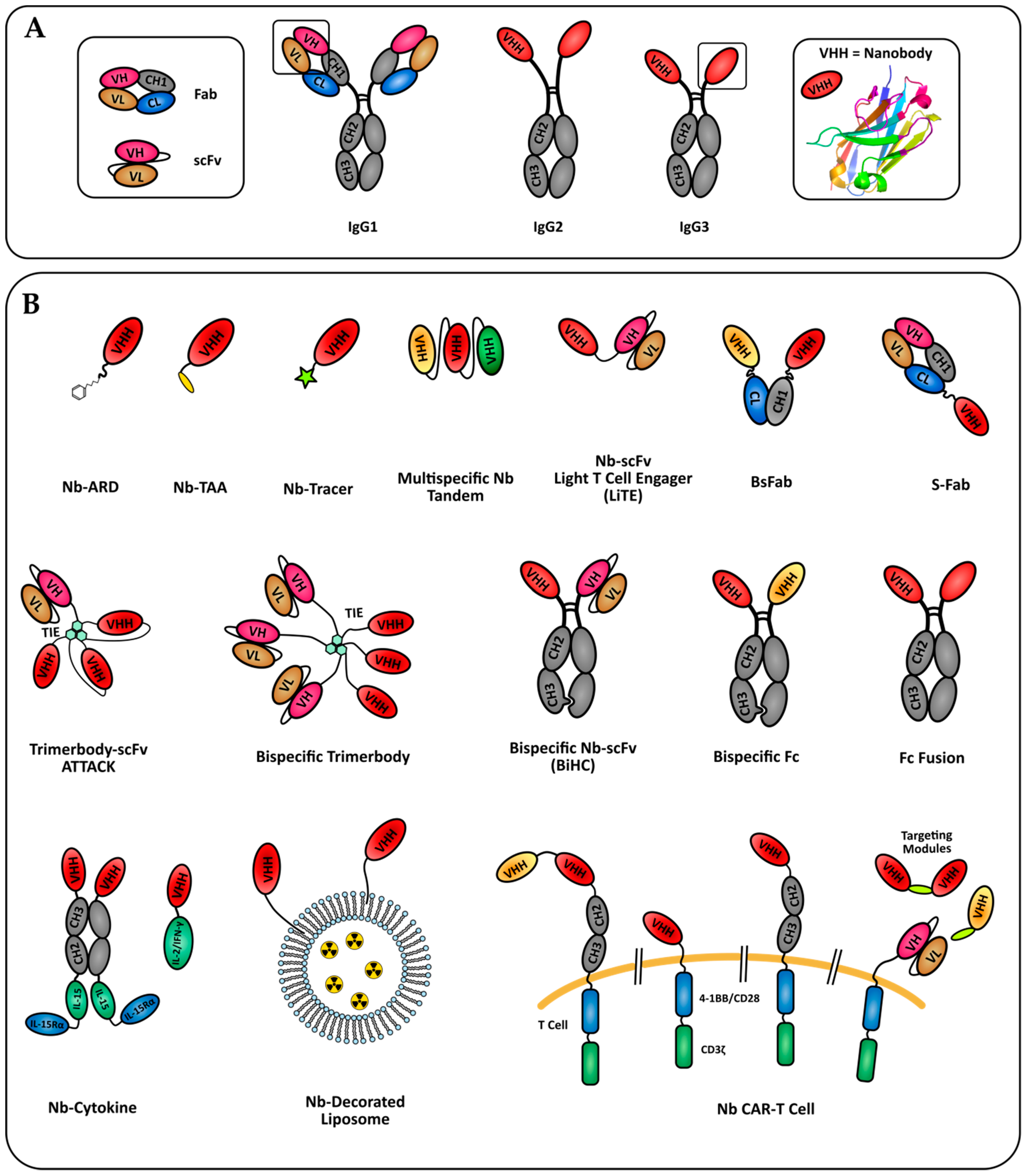{kind=link}
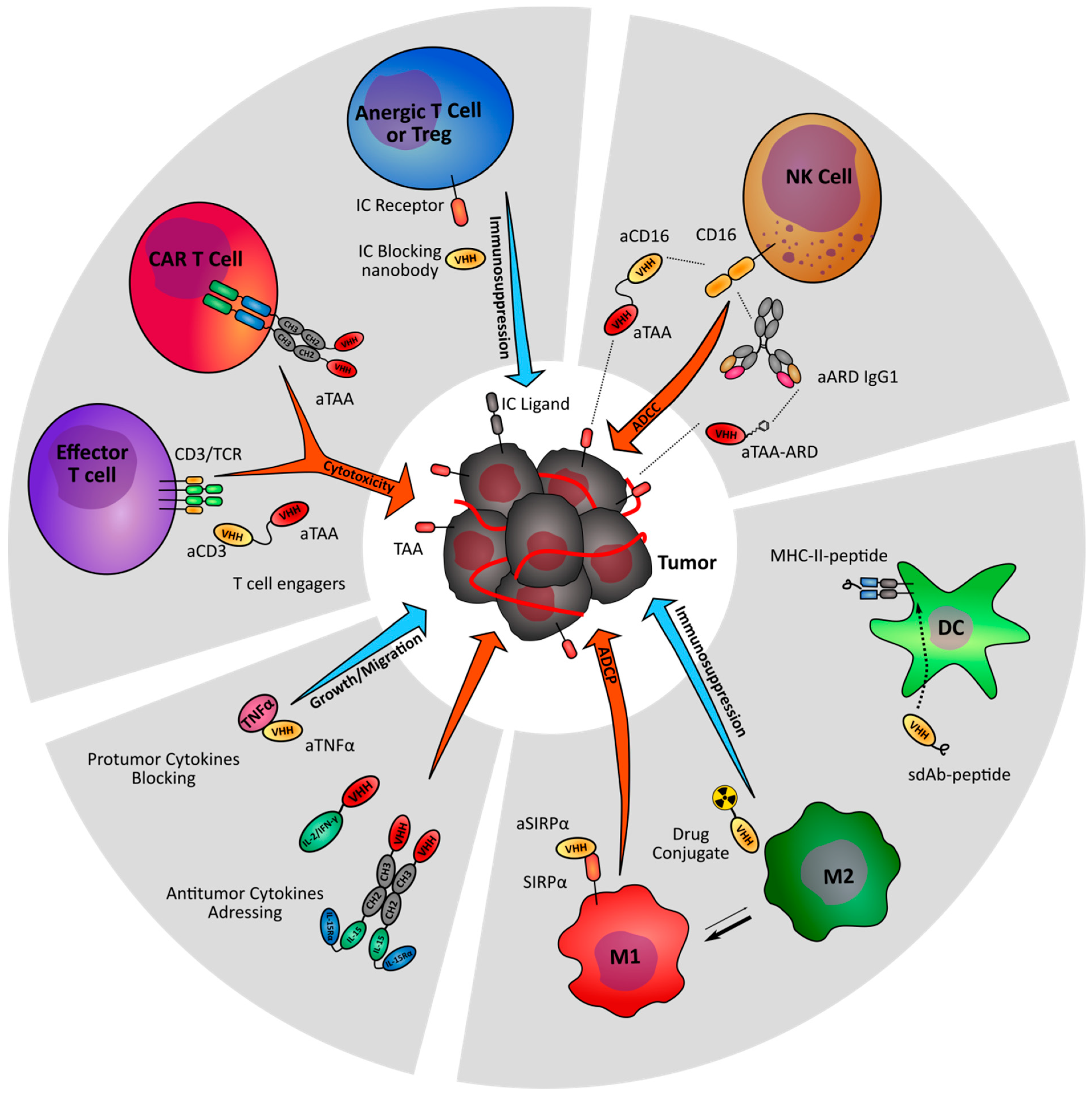{kind=link}
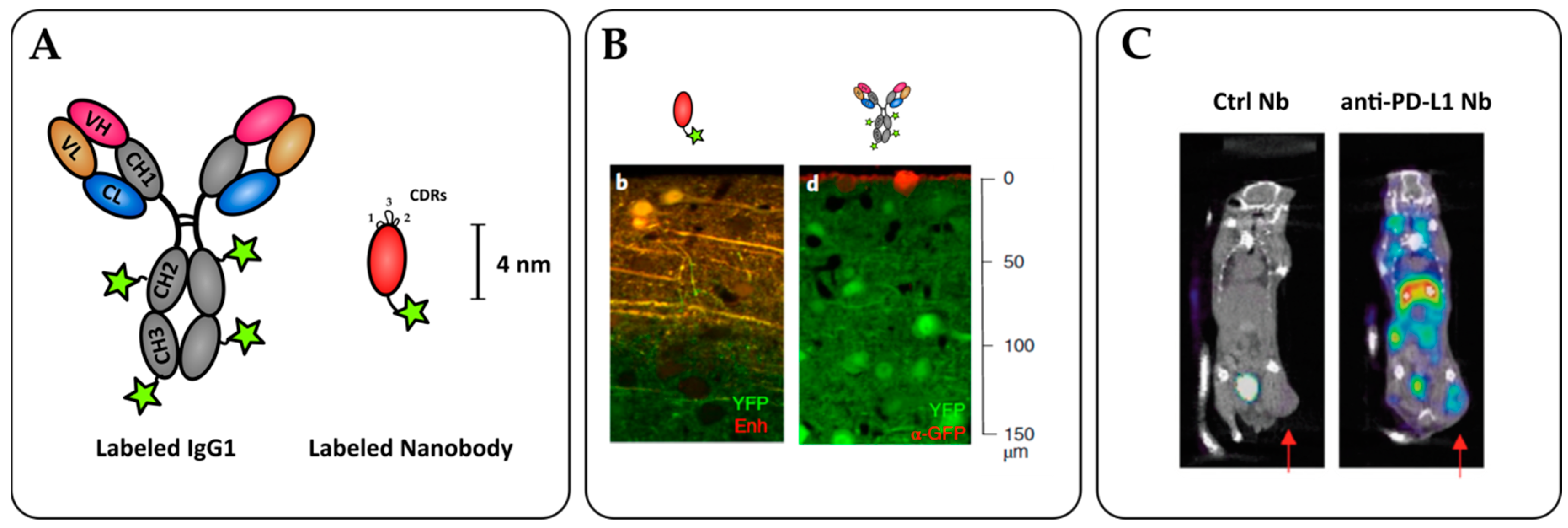{kind=link}
Abstract
1. Introduction
2. Targeting T Cell Activation and Cytotoxicity
2.1. CD3 Bispecific Nanobodies: BiTE-Like Formats
2.2. γδT Cell Activation
2.3. Engineering Nanobody-Derived TCR in CAR-T Cell Therapy
2.4. Immune Checkpoint Blockade
3. Enhancing NK Cell-Mediated Antitumor Activity
3.1. Anti-CD16 Bispecific Antibodies
3.2. Nanobody Coupling to An Antibody Recruiting Domain
4. Modulation of Antigen Presenting Cells
4.1. Innate Immune Checkpoint Blockade
4.2. Nanobody-Based Immunization Strategies
4.3. Drug Delivery
4.4. APC Reprogramming
5. Targeting the Tumor Environment Cytokines and Chemokines
5.1. Pro-Tumor Cytokines Targeting: TNF/G-CSF
5.2. Modulating the CXCR4/CXCR7/CXCL12 Chemokines Axis in Solid Cancers
5.3. Nanobody as Carriers for Cytokine Delivery
6. Nanobodies as Potent Imaging Tools
6.1. Importance of Molecular Imaging for Cancer Diagnostics
6.2. Cancer Cell Detection
6.3. Monitoring of Immune Infiltration
6.3.1. Tumor Infiltrating Lymphocyte Monitoring
6.3.2. APC Monitoring
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stambrook, P.J.; Maher, J.; Farzaneh, F. Cancer Immunotherapy: Whence and Whither. Mol. Cancer Res. 2017, 15, 635–650. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.W.; Gill, D.M.; Pal, S.K.; Agarwal, N. The future of immune checkpoint cancer therapy after PD-1 and CTLA-4. Immunotherapy 2017, 9, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed]
- Del Bano, J.; Chames, P.; Baty, D.; Kerfelec, B. Taking up Cancer Immunotherapy Challenges: Bispecific Antibodies, the Path Forward? Antibodies 2016, 5, 1. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. mAbs 2017, 9, 182–212. [Google Scholar] [CrossRef]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Vincke, C.; Loris, R.; Saerens, D.; Martinez-Rodriguez, S.; Muyldermans, S.; Conrath, K. General Strategy to Humanize a Camelid Single-domain Antibody and Identification of a Universal Humanized Nanobody Scaffold. J. Biol. Chem. 2009, 284, 3273–3284. [Google Scholar] [CrossRef]
- Duggan, S. Caplacizumab: First Global Approval. Drugs 2018, 78, 1639–1642. [Google Scholar] [CrossRef]
- Genst, E.D.; Silence, K.; Decanniere, K.; Conrath, K.; Loris, R.; Kinne, J.; Muyldermans, S.; Wyns, L. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. USA 2006, 103, 4586–4591. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, L.S.; Colwell, L.J. Analysis of nanobody paratopes reveals greater diversity than classical antibodies. Protein Eng. Des. Sel. 2018, 31, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, A.N.R.; Griggs, J.; Davis, D.M. The immune synapse clears and excludes molecules above a size threshold. Nat. Commun. 2014, 5, 5479. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, M.; Ververken, C.; Beirnaert, E.; Hoefman, S.; Kolkman, J.; Vierboom, M.; Breedveld, E.; ‘t Hart, B.; Poelmans, S.; Bontinck, L.; et al. The preclinical pharmacology of the high affinity anti-IL-6R Nanobody® ALX-0061 supports its clinical development in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 135. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef]
- Martin, V.; Cappuzzo, F.; Mazzucchelli, L.; Frattini, M. HER2 in solid tumors: More than 10 years under the microscope; where are we now? Future Oncol. 2014, 10, 1469–1486. [Google Scholar] [CrossRef]
- Lin, L.; Li, L.; Zhou, C.; Li, J.; Liu, J.; Shu, R.; Dong, B.; Li, Q.; Wang, Z. A HER2 bispecific antibody can be efficiently expressed in Escherichia coli with potent cytotoxicity. Oncol. Lett. 2018, 16, 1259–1266. [Google Scholar]
- Ridgway, J.B.B.; Presta, L.G.; Carter, P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. Des. Sel. 1996, 9, 617–621. [Google Scholar] [CrossRef]
- Xing, J.; Lin, L.; Li, J.; Liu, J.; Zhou, C.; Pan, H.; Shu, R.; Dong, B.; Cao, D.; Li, Q.; et al. BiHC, a T-Cell–Engaging Bispecific Recombinant Antibody, Has Potent Cytotoxic Activity Against Her2 Tumor Cells. Transl. Oncol. 2017, 10, 780–785. [Google Scholar] [CrossRef]
- Hammarström, S. The carcinoembryonic antigen (CEA) family: Structures, suggested functions and expression in normal and malignant tissues. Semin. Cancer Biol. 1999, 9, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; He, P.; Zhou, C.; Jing, L.; Dong, B.; Chen, S.; Zhang, N.; Liu, Y.; Miao, J.; Wang, Z.; et al. A novel bispecific antibody, S-Fab, induces potent cancer cell killing. J. Immunother. 2015, 38, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Liu, J.; Deng, W.; Xing, J.; Li, Q.; Wang, Z. Site-specific PEGylation of an anti-CEA/CD3 bispecific antibody improves its antitumor efficacy. Int. J. Nanomed. 2018, 13, 3189–3201. [Google Scholar] [CrossRef] [PubMed]
- Mølgaard, K.; Harwood, S.L.; Compte, M.; Merino, N.; Bonet, J.; Alvarez-Cienfuegos, A.; Mikkelsen, K.; Nuñez-Prado, N.; Alvarez-Mendez, A.; Sanz, L.; et al. Bispecific light T-cell engagers for gene-based immunotherapy of epidermal growth factor receptor (EGFR)-positive malignancies. Cancer Immunol. Immunother. 2018, 67, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Harwood, S.L.; Alvarez-Cienfuegos, A.; Nuñez-Prado, N.; Compte, M.; Hernández-Pérez, S.; Merino, N.; Bonet, J.; Navarro, R.; Bergen en Henegouwen, P.M.; Lykkemark, S.; et al. ATTACK, a novel bispecific T cell-recruiting antibody with trivalent EGFR binding and monovalent CD3 binding for cancer immunotherapy. Oncoimmunology 2018, 7, e1377874. [Google Scholar] [CrossRef] [PubMed]
- Compte, M.; Harwood, S.L.; Muñoz, I.G.; Navarro, R.; Zonca, M.; Perez-Chacon, G.; Erce-Llamazares, A.; Merino, N.; Tapia-Galisteo, A.; Cuesta, A.M.; et al. A tumor-targeted trimeric 4-1BB-agonistic antibody induces potent anti-tumor immunity without systemic toxicity. Nat. Commun. 2018, 9, 4809. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, F.; Pende, D.; Burgio, V.L.; Castelli, C.; Spada, M.; Venditti, M.; Luciani, F.; Lugini, L.; Federici, C.; Ramoni, C.; et al. Effect of human natural killer and gammadelta T cells on the growth of human autologous melanoma xenografts in SCID mice. Cancer Res. 2004, 64, 378–385. [Google Scholar] [CrossRef]
- Duault, C.; Betous, D.; Bezombes, C.; Roga, S.; Cayrol, C.; Girard, J.-P.; Fournié, J.-J.; Poupot, M. IL-33-expanded human Vγ9Vδ2 T cells have anti-lymphoma effect in a mouse tumor model. Eur. J. Immunol. 2017, 47, 2137–2141. [Google Scholar] [CrossRef]
- Beck, B.H.; Kim, H.-G.; Kim, H.; Samuel, S.; Liu, Z.; Shrestha, R.; Haines, H.; Zinn, K.; Lopez, R.D. Adoptively-transferred ex vivo expanded γδ-T cells mediate in vivo antitumor activity in preclinical mouse models of breast cancer. Breast Cancer Res. Treat. 2010, 122, 135–144. [Google Scholar] [CrossRef]
- Pauza, C.D.; Liou, M.-L.; Lahusen, T.; Xiao, L.; Lapidus, R.G.; Cairo, C.; Li, H. Gamma Delta T Cell Therapy for Cancer: It Is Good to be Local. Front. Immunol. 2018, 9, 1305. [Google Scholar] [CrossRef]
- de Bruin, R.C.G.; Lougheed, S.M.; van der Kruk, L.; Stam, A.G.; Hooijberg, E.; Roovers, R.C.; van Bergen en Henegouwen, P.M.P.; Verheul, H.M.W.; de Gruijl, T.D.; van der Vliet, H.J. Highly specific and potently activating Vγ9Vδ2-T cell specific nanobodies for diagnostic and therapeutic applications. Clin. Immunol. 2016, 169, 128–138. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, R.C.G.; Veluchamy, J.P.; Lougheed, S.M.; Schneiders, F.L.; Lopez-Lastra, S.; Lameris, R.; Stam, A.G.; Sebestyen, Z.; Kuball, J.; Molthoff, C.F.M.; et al. A bispecific nanobody approach to leverage the potent and widely applicable tumor cytolytic capacity of Vγ9Vδ2-T cells. OncoImmunology 2018, 7, e1375641. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Iri-Sofla, F.J.; Rahbarizadeh, F.; Ahmadvand, D.; Rasaee, M.J. Nanobody-based chimeric receptor gene integration in Jurkat cells mediated by PhiC31 integrase. Exp. Cell Res. 2011, 317, 2630–2641. [Google Scholar] [CrossRef] [PubMed]
- Khaleghi, S.; Rahbarizadeh, F.; Ahmadvand, D.; Rasaee, M.J.; Pognonec, P. A caspase 8-based suicide switch induces apoptosis in nanobody-directed chimeric receptor expressing T cells. Int. J. Hematol. 2012, 95, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Cartellieri, M.; Feldmann, A.; Koristka, S.; Arndt, C.; Loff, S.; Ehninger, A.; von Bonin, M.; Bejestani, E.P.; Ehninger, G.; Bachmann, M.P. Switching CAR T cells on and off: A novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016, 6, e458. [Google Scholar] [CrossRef] [PubMed]
- Albert, S.; Arndt, C.; Feldmann, A.; Bergmann, R.; Bachmann, D.; Koristka, S.; Ludwig, F.; Ziller-Walter, P.; Kegler, A.; Gärtner, S.; et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. OncoImmunology 2017, 6, e1287246. [Google Scholar] [CrossRef]
- Albert, S.; Arndt, C.; Koristka, S.; Berndt, N.; Bergmann, R.; Feldmann, A.; Schmitz, M.; Pietzsch, J.; Steinbach, J.; Bachmann, M. From mono- to bivalent: Improving theranostic properties of target modules for redirection of UniCAR T cells against EGFR-expressing tumor cells in vitro and in vivo. Oncotarget 2018, 9, 25597–25616. [Google Scholar] [CrossRef]
- De Munter, S.; Ingels, J.; Goetgeluk, G.; Bonte, S.; Pille, M.; Weening, K.; Kerre, T.; Abken, H.; Vandekerckhove, B. Nanobody Based Dual Specific CARs. Int. J. Mol. Sci. 2018, 19, 403. [Google Scholar] [CrossRef]
- Jamnani, F.R.; Rahbarizadeh, F.; Shokrgozar, M.A.; Mahboudi, F.; Ahmadvand, D.; Sharifzadeh, Z.; Parhamifar, L.; Moghimi, S.M. T cells expressing VHH-directed oligoclonal chimeric HER2 antigen receptors: Towards tumor-directed oligoclonal T cell therapy. Biochim. Biophys. Acta 2014, 1840, 378–386. [Google Scholar] [CrossRef]
- Li, N.; Fu, H.; Hewitt, S.M.; Dimitrov, D.S.; Ho, M. Therapeutically targeting glypican-2 via single-domain antibody-based chimeric antigen receptors and immunotoxins in neuroblastoma. Proc. Natl. Acad. Sci. USA 2017, 114, E6623–E6631. [Google Scholar] [CrossRef]
- An, N.; Hou, Y.N.; Zhang, Q.X.; Li, T.; Zhang, Q.L.; Fang, C.; Chen, H.; Lee, H.C.; Zhao, Y.J.; Du, X. Anti-Multiple Myeloma Activity of Nanobody-Based Anti-CD38 Chimeric Antigen Receptor T Cells. Mol. Pharm. 2018, 15, 4577–4588. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Ingram, J.R.; Blomberg, O.S.; Rashidian, M.; Ali, L.; Garforth, S.; Fedorov, E.; Fedorov, A.A.; Bonanno, J.B.; Gall, C.L.; Crowley, S.; et al. Anti–CTLA-4 therapy requires an Fc domain for efficacy. Proc. Natl. Acad. Sci. USA 2018, 115, 3912–3917. [Google Scholar] [CrossRef]
- Wan, R.; Liu, A.; Hou, X.; Lai, Z.; Li, J.; Yang, N.; Tan, J.; Mo, F.; Hu, Z.; Yang, X.; et al. Screening and antitumor effect of an anti-CTLA-4 nanobody. Oncol. Rep. 2018, 39, 511–518. [Google Scholar] [CrossRef]
- Zhang, F.; Wei, H.; Wang, X.; Bai, Y.; Wang, P.; Wu, J.; Jiang, X.; Wang, Y.; Cai, H.; Xu, T.; et al. Structural basis of a novel PD-L1 nanobody for immune checkpoint blockade. Cell Discov. 2017, 3, 17004. [Google Scholar] [CrossRef]
- Dahan, R.; Sega, E.; Engelhardt, J.; Selby, M.; Korman, A.J.; Ravetch, J.V. FcγRs Modulate the Anti-tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis. Cancer Cell 2015, 28, 285–295. [Google Scholar] [CrossRef]
- Homayouni, V.; Ganjalikhani-hakemi, M.; Rezaei, A.; Khanahmad, H.; Behdani, M.; Lomedasht, F.K. Preparation and characterization of a novel nanobody against T-cell immunoglobulin and mucin-3 (TIM-3). Iran. J. Basic Med. Sci. 2016, 19, 1201–1208. [Google Scholar]
- Imai, K.; Matsuyama, S.; Miyake, S.; Suga, K.; Nakachi, K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: An 11-year follow-up study of a general population. Lancet 2000, 356, 1795–1799. [Google Scholar] [CrossRef]
- Rusakiewicz, S.; Semeraro, M.; Sarabi, M.; Desbois, M.; Locher, C.; Mendez, R.; Vimond, N.; Concha, A.; Garrido, F.; Isambert, N.; et al. Immune Infiltrates Are Prognostic Factors in Localized Gastrointestinal Stromal Tumors. Cancer Res. 2013, 73, 3499–3510. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Sousa, C.R. e NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef]
- Krebs, P.; Barnes, M.J.; Lampe, K.; Whitley, K.; Bahjat, K.S.; Beutler, B.; Janssen, E.; Hoebe, K. NK cell–mediated killing of target cells triggers robust antigen-specific T cell–mediated and humoral responses. Blood 2009, 113, 6593–6602. [Google Scholar] [CrossRef]
- Kelly, J.M.; Darcy, P.K.; Markby, J.L.; Godfrey, D.I.; Takeda, K.; Yagita, H.; Smyth, M.J. Induction of tumor-specific T cell memory by NK cell–mediated tumor rejection. Nat. Immunol. 2002, 3, 83–90. [Google Scholar] [CrossRef]
- Cartron, G.; Dacheux, L.; Salles, G.; Solal-Celigny, P.; Bardos, P.; Colombat, P.; Watier, H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene. Blood 2002, 99, 754–758. [Google Scholar] [CrossRef]
- Arnould, L.; Gelly, M.; Penault-Llorca, F.; Benoit, L.; Bonnetain, F.; Migeon, C.; Cabaret, V.; Fermeaux, V.; Bertheau, P.; Garnier, J.; et al. Trastuzumab-based treatment of HER2-positive breast cancer: An antibody-dependent cellular cytotoxicity mechanism? Br. J. Cancer 2006, 94, 259–267. [Google Scholar] [CrossRef]
- Maréchal, R.; De Schutter, J.; Nagy, N.; Demetter, P.; Lemmers, A.; Devière, J.; Salmon, I.; Tejpar, S.; Van Laethem, J.-L. Putative contribution of CD56 positive cells in cetuximab treatment efficacy in first-line metastatic colorectal cancer patients. BMC Cancer 2010, 10, 340. [Google Scholar] [CrossRef]
- Veeramani, S.; Wang, S.-Y.; Dahle, C.; Blackwell, S.; Jacobus, L.; Knutson, T.; Button, A.; Link, B.K.; Weiner, G.J. Rituximab infusion induces NK activation in lymphoma patients with the high-affinity CD16 polymorphism. Blood 2011, 118, 3347–3349. [Google Scholar] [CrossRef]
- Sondermann, P.; Szymkowski, D.E. Harnessing Fc receptor biology in the design of therapeutic antibodies. Curr. Opin. Immunol. 2016, 40, 78–87. [Google Scholar] [CrossRef]
- Trotta, R.; Kanakaraj, P.; Perussia, B. Fc gamma R-dependent mitogen-activated protein kinase activation in leukocytes: A common signal transduction event necessary for expression of TNF-alpha and early activation genes. J. Exp. Med. 1996, 184, 1027–1035. [Google Scholar] [CrossRef]
- Lee, H.-R.; Son, C.-H.; Koh, E.-K.; Bae, J.-H.; Kang, C.-D.; Yang, K.; Park, Y.-S. Expansion of cytotoxic natural killer cells using irradiated autologous peripheral blood mononuclear cells and anti-CD16 antibody. Sci. Rep. 2017, 7, 11075. [Google Scholar] [CrossRef]
- Behar, G.; Sibéril, S.; Groulet, A.; Chames, P.; Pugnière, M.; Boix, C.; Sautès-Fridman, C.; Teillaud, J.-L.; Baty, D. Isolation and characterization of anti-FcγRIII (CD16) llama single-domain antibodies that activate natural killer cells. Protein Eng. Des. Sel. 2008, 21, 1–10. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, C.; Li, J.; Liu, J.; Lin, L.; Li, L.; Cao, D.; Li, Q.; Wang, Z. Single domain based bispecific antibody, Muc1-Bi-1, and its humanized form, Muc1-Bi-2, induce potent cancer cell killing in muc1 positive tumor cells. PLoS ONE 2018, 13, e0191024. [Google Scholar] [CrossRef]
- Dong, B.; Zhou, C.; He, P.; Li, J.; Chen, S.; Miao, J.; Li, Q.; Wang, Z. A novel bispecific antibody, BiSS, with potent anti-cancer activities. Cancer Biol. Ther. 2016, 17, 364–370. [Google Scholar] [CrossRef]
- Rozan, C.; Cornillon, A.; Pétiard, C.; Chartier, M.; Behar, G.; Boix, C.; Kerfelec, B.; Robert, B.; Pèlegrin, A.; Chames, P.; et al. Single-Domain Antibody–Based and Linker-Free Bispecific Antibodies Targeting FcγRIII Induce Potent Antitumor Activity without Recruiting Regulatory T Cells. Mol. Cancer Ther. 2013, 12, 1481–1491. [Google Scholar] [CrossRef]
- Turini, M.; Chames, P.; Bruhns, P.; Baty, D.; Kerfelec, B. A FcγRIII-engaging bispecific antibody expands the range of HER2-expressing breast tumors eligible to antibody therapy. Oncotarget 2014, 5, 5304–5319. [Google Scholar] [CrossRef]
- Li, J.; Zhou, C.; Dong, B.; Zhong, H.; Chen, S.; Li, Q.; Wang, Z. Single domain antibody-based bispecific antibody induces potent specific anti-tumor activity. Cancer Biol. Ther. 2016, 17, 1231–1239. [Google Scholar] [CrossRef]
- Li, A.; Xing, J.; Li, L.; Zhou, C.; Dong, B.; He, P.; Li, Q.; Wang, Z. A single-domain antibody-linked Fab bispecific antibody Her2-S-Fab has potent cytotoxicity against Her2-expressing tumor cells. AMB Express 2016, 6, 32. [Google Scholar] [CrossRef]
- Deng, W.; Liu, J.; Pan, H.; Li, L.; Zhou, C.; Wang, X.; Shu, R.; Dong, B.; Cao, D.; Li, Q.; et al. A Bispecific Antibody Based on Pertuzumab Fab Has Potent Antitumor Activity. J. Immunother. 2018, 41, 1–8. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Pan, H.; Xing, J.; Wu, X.; Li, Q.; Wang, Z. A GPC3-targeting Bispecific Antibody, GPC3-S-Fab, with Potent Cytotoxicity. J. Vis. Exp. 2018, 137, e57588. [Google Scholar] [CrossRef]
- Gray, M.A.; Tao, R.N.; DePorter, S.M.; Spiegel, D.A.; McNaughton, B.R. A Nanobody Activation Immunotherapeutic that Selectively Destroys HER2-Positive Breast Cancer Cells. Chembiochem 2016, 17, 155–158. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef]
- Böttcher, J.P.; e Sousa, C.R. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef]
- Sockolosky, J.T.; Dougan, M.; Ingram, J.R.; Ho, C.C.M.; Kauke, M.J.; Almo, S.C.; Ploegh, H.L.; Garcia, K.C. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc. Natl. Acad. Sci. USA 2016, 113, E2646–E2654. [Google Scholar] [CrossRef]
- Ingram, J.R.; Blomberg, O.S.; Sockolosky, J.T.; Ali, L.; Schmidt, F.I.; Pishesha, N.; Espinosa, C.; Dougan, S.K.; Garcia, K.C.; Ploegh, H.L.; et al. Localized CD47 blockade enhances immunotherapy for murine melanoma. Proc. Natl. Acad. Sci. USA 2017, 114, 10184–10189. [Google Scholar] [CrossRef]
- Tang, C.; Wang, X.; Li, Z.; Yue, Q.; Yang, Z.; Fan, K.; Hoon, D.; Hua, W. A Systemic Review of Clinical Trials on Dendritic-Cells Based Vaccine Against Malignant Glioma. J. Carcinog. Mutagen 2015, 6. [Google Scholar] [CrossRef]
- Garg, A.D.; Perez, M.V.; Schaaf, M.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Dendritic cell-based anticancer immunotherapy. OncoImmunology 2017, 6, e1328341. [Google Scholar] [CrossRef]
- Duarte, J.N.; Cragnolini, J.J.; Swee, L.K.; Bilate, A.M.; Bader, J.; Ingram, J.R.; Rashidfarrokhi, A.; Fang, T.; Schiepers, A.; Hanke, L.; et al. Generation of Immunity against Pathogens via Single-Domain Antibody–Antigen Constructs. J. Immunol. 2016, 197, 4838–4847. [Google Scholar] [CrossRef]
- Fang, T.; Van Elssen, C.H.M.J.; Duarte, J.N.; Guzman, J.S.; Chahal, J.S.; Ling, J.; Ploegh, H.L. Targeted antigen delivery by an anti-class II MHC VHH elicits focused αMUC1(Tn) immunity †Electronic supplementary information (ESI) available. Chem. Sci. 2017, 8, 5591–5597. [Google Scholar] [CrossRef]
- Kwon, S.; Duarte, J.N.; Li, Z.; Ling, J.J.; Cheneval, O.; Durek, T.; Schroeder, C.I.; Craik, D.J.; Ploegh, H.L. Targeted Delivery of Cyclotides via Conjugation to a Nanobody. ACS Chem. Biol. 2018, 13, 2973–2980. [Google Scholar] [CrossRef]
- Sun, X.; Gao, D.; Gao, L.; Zhang, C.; Yu, X.; Jia, B.; Wang, F.; Liu, Z. Molecular imaging of tumor-infiltrating macrophages in a preclinical mouse model of breast cancer. Theranostics 2015, 5, 597–608. [Google Scholar] [CrossRef]
- Dong, P.; Ma, L.; Liu, L.; Zhao, G.; Zhang, S.; Dong, L.; Xue, R.; Chen, S. CD86+/CD206+, Diametrically Polarized Tumor-Associated Macrophages, Predict Hepatocellular Carcinoma Patient Prognosis. Int. J. Mol. Sci. 2016, 17, 320. [Google Scholar] [CrossRef]
- Nuhn, L.; Bolli, E.; Massa, S.; Vandenberghe, I.; Movahedi, K.; Devreese, B.; Van Ginderachter, J.A.; De Geest, B.G. Targeting Protumoral Tumor-Associated Macrophages with Nanobody-Functionalized Nanogels through Strain Promoted Azide Alkyne Cycloaddition Ligation. Bioconjug. Chem. 2018, 29, 2394–2405. [Google Scholar] [CrossRef]
- Fang, T.; Duarte, J.N.; Ling, J.; Li, Z.; Guzman, J.S.; Ploegh, H.L. Structurally Defined αMHC-II Nanobody–Drug Conjugates: A Therapeutic and Imaging System for B-Cell Lymphoma. Angew. Chem. Int. Ed. 2016, 55, 2416–2420. [Google Scholar] [CrossRef]
- Georgoudaki, A.-M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Östling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef]
- Tariq, M.; Zhang, J.; Liang, G.; He, Q.; Ding, L.; Yang, B. Gefitinib inhibits M2-like polarization of tumor-associated macrophages in Lewis lung cancer by targeting the STAT6 signaling pathway. Acta Pharmacol. Sin. 2017, 38, 1501–1511. [Google Scholar] [CrossRef]
- Alupei, M.C.; Licarete, E.; Patras, L.; Banciu, M. Liposomal simvastatin inhibits tumor growth via targeting tumor-associated macrophages-mediated oxidative stress. Cancer Lett. 2015, 356, 946–952. [Google Scholar] [CrossRef]
- Yin, W.; Yu, X.; Kang, X.; Zhao, Y.; Zhao, P.; Jin, H.; Fu, X.; Wan, Y.; Peng, C.; Huang, Y. Remodeling Tumor-Associated Macrophages and Neovascularization Overcomes EGFRT790M-Associated Drug Resistance by PD-L1 Nanobody-Mediated Codelivery. Small 2018, 14, 1802372. [Google Scholar] [CrossRef]
- McEwen-Smith, R.M.; Salio, M.; Cerundolo, V. The Regulatory Role of Invariant NKT Cells in Tumor Immunity. Cancer Immunol. Res. 2015, 3, 425–435. [Google Scholar] [CrossRef]
- Lameris, R.; de Bruin, R.C.; van Bergen en Henegouwen, P.M.; Verheul, H.M.; Zweegman, S.; de Gruijl, T.D.; van der Vliet, H.J. Generation and characterization of CD1d-specific single-domain antibodies with distinct functional features. Immunology 2016, 149, 111–121. [Google Scholar] [CrossRef]
- Goyvaerts, C.; De Groeve, K.; Dingemans, J.; Van Lint, S.; Robays, L.; Heirman, C.; Reiser, J.; Zhang, X.-Y.; Thielemans, K.; De Baetselier, P.; et al. Development of the Nanobody display technology to target lentiviral vectors to antigen-presenting cells. Gene Ther. 2012, 19, 1133–1140. [Google Scholar] [CrossRef]
- Goyvaerts, C.; De Vlaeminck, Y.; Escors, D.; Lienenklaus, S.; Keyaerts, M.; Raes, G.; Breckpot, K. Antigen-presenting cell-targeted lentiviral vectors do not support the development of productive T-cell effector responses: Implications for in vivo targeted vaccine delivery. Gene Ther. 2017, 24, 370–375. [Google Scholar] [CrossRef]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef]
- Zhao, X.; Rong, L.; Zhao, X.; Li, X.; Liu, X.; Deng, J.; Wu, H.; Xu, X.; Erben, U.; Wu, P.; et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J. Clin. Investig. 2012, 122, 4094–4104. [Google Scholar] [CrossRef]
- Okubo, Y.; Mera, T.; Wang, L.; Faustman, D.L. Homogeneous Expansion of Human T-Regulatory Cells Via Tumor Necrosis Factor Receptor 2. Sci. Rep. 2013, 3, 3153. [Google Scholar] [CrossRef]
- Bertrand, F.; Montfort, A.; Marcheteau, E.; Imbert, C.; Gilhodes, J.; Filleron, T.; Rochaix, P.; Andrieu-Abadie, N.; Levade, T.; Meyer, N.; et al. TNFα blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat. Commun. 2017, 8, 2256. [Google Scholar] [CrossRef]
- Ji, X.; Peng, Z.; Li, X.; Yan, Z.; Yang, Y.; Qiao, Z.; Liu, Y. Neutralization of TNFα in tumor with a novel nanobody potentiates paclitaxel-therapy and inhibits metastasis in breast cancer. Cancer Lett. 2017, 386, 24–34. [Google Scholar] [CrossRef]
- Bakherad, H.; Gargari, S.L.M.; Sepehrizadeh, Z.; Aghamollaei, H.; Taheri, R.A.; Torshabi, M.; Yazdi, M.T.; Ebrahimizadeh, W.; Setayesh, N. Identification and in vitro characterization of novel nanobodies against human granulocyte colony-stimulating factor receptor to provide inhibition of G-CSF function. Biomed. Pharmacother. 2017, 93, 245–254. [Google Scholar] [CrossRef]
- Fan, Z.; Li, Y.; Zhao, Q.; Fan, L.; Tan, B.; Zuo, J.; Hua, K.; Ji, Q. Highly Expressed Granulocyte Colony-Stimulating Factor (G-CSF) and Granulocyte Colony-Stimulating Factor Receptor (G-CSFR) in Human Gastric Cancer Leads to Poor Survival. Med. Sci. Monit. 2018, 24, 1701–1711. [Google Scholar] [CrossRef]
- Morris, K.T.; Khan, H.; Ahmad, A.; Weston, L.L.; Nofchissey, R.A.; Pinchuk, I.V.; Beswick, E.J. G-CSF and G-CSFR are highly expressed in human gastric and colon cancers and promote carcinoma cell proliferation and migration. Br. J. Cancer 2014, 110, 1211–1220. [Google Scholar] [CrossRef]
- Agarwal, S.; Lakoma, A.; Chen, Z.; Hicks, J.; Metelitsa, L.S.; Kim, E.S.; Shohet, J.M. G-CSF promotes neuroblastoma tumorigenicity and metastasis via STAT3-dependent cancer stem cell activation. Cancer Res. 2015, 75, 2566–2579. [Google Scholar] [CrossRef]
- Sackstein, R.; Schatton, T.; Barthel, S.R. T-lymphocyte homing: An underappreciated yet critical hurdle for successful cancer immunotherapy. Lab. Investig. 2017, 97, 669–697. [Google Scholar] [CrossRef]
- Liao, Y.X.; Zhou, C.H.; Zeng, H.; Zuo, D.Q.; Wang, Z.Y.; Yin, F.; Hua, Y.Q.; Cai, Z.D. The role of the CXCL12-CXCR4/CXCR7 axis in the progression and metastasis of bone sarcomas (Review). Int. J. Mol. Med. 2013, 32, 1239–1246. [Google Scholar] [CrossRef]
- Krikun, G. The CXL12/CXCR4/CXCR7 axis in female reproductive tract disease: Review. Am. J. Reprod. Immunol. 2018, 80, e13028. [Google Scholar] [CrossRef]
- Brennecke, P.; Arlt, M.J.E.; Campanile, C.; Husmann, K.; Gvozdenovic, A.; Apuzzo, T.; Thelen, M.; Born, W.; Fuchs, B. CXCR4 antibody treatment suppresses metastatic spread to the lung of intratibial human osteosarcoma xenografts in mice. Clin. Exp. Metastasis 2014, 31, 339–349. [Google Scholar] [CrossRef]
- Benedicto, A.; Romayor, I.; Arteta, B. CXCR4 receptor blockage reduces the contribution of tumor and stromal cells to the metastatic growth in the liver. Oncol. Rep. 2018, 39, 2022–2030. [Google Scholar] [CrossRef]
- Lefort, S.; Thuleau, A.; Kieffer, Y.; Sirven, P.; Bieche, I.; Marangoni, E.; Vincent-Salomon, A.; Mechta-Grigoriou, F. CXCR4 inhibitors could benefit to HER2 but not to triple-negative breast cancer patients. Oncogene 2017, 36, 1211–1222. [Google Scholar] [CrossRef]
- Maussang, D.; Mujić-Delić, A.; Descamps, F.J.; Stortelers, C.; Vanlandschoot, P.; Walsum, M.S.; Vischer, H.F.; van Roy, M.; Vosjan, M.; Gonzalez-Pajuelo, M.; et al. Llama-derived Single Variable Domains (Nanobodies) Directed against Chemokine Receptor CXCR7 Reduce Head and Neck Cancer Cell Growth In Vivo. J. Biol. Chem. 2013, 288, 29562–29572. [Google Scholar] [CrossRef]
- Zheng, N.; Liu, W.; Chen, J.; Li, B.; Liu, J.; Wang, J.; Gao, Y.; Shao, J.; Jia, L. CXCR7 is not obligatory for CXCL12-CXCR4-induced epithelial-mesenchymal transition in human ovarian cancer. Mol. Carcinog. 2019, 58, 144–155. [Google Scholar] [CrossRef]
- Duda, D.G.; Kozin, S.V.; Kirkpatrick, N.D.; Xu, L.; Fukumura, D.; Jain, R.K. CXCL12 (SDF1α)-CXCR4/CXCR7 Pathway Inhibition: An Emerging Sensitizer for Anticancer Therapies? Clin. Cancer Res. 2011, 17, 2074–2080. [Google Scholar] [CrossRef]
- Van Hout, A.; Klarenbeek, A.; Bobkov, V.; Doijen, J.; Arimont, M.; Zhao, C.; Heukers, R.; Rimkunas, R.; de Graaf, C.; Verrips, T.; et al. CXCR4-targeting nanobodies differentially inhibit CXCR4 function and HIV entry. Biochem. Pharmacol. 2018, 158, 402–412. [Google Scholar] [CrossRef]
- Jähnichen, S.; Blanchetot, C.; Maussang, D.; Gonzalez-Pajuelo, M.; Chow, K.Y.; Bosch, L.; Vrieze, S.D.; Serruys, B.; Ulrichts, H.; Vandevelde, W.; et al. CXCR4 nanobodies (VHH-based single variable domains) potently inhibit chemotaxis and HIV-1 replication and mobilize stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 20565–20570. [Google Scholar] [CrossRef]
- Bobkov, V.; Zarca, A.M.; Van Hout, A.; Arimont, M.; Doijen, J.; Bialkowska, M.; Toffoli, E.; Klarenbeek, A.; van der Woning, B.; van der Vliet, H.J.; et al. Nanobody-Fc constructs targeting chemokine receptor CXCR4 potently inhibit signaling and CXCR4-mediated HIV-entry and induce antibody effector functions. Biochem. Pharmacol. 2018, 158, 413–424. [Google Scholar] [CrossRef]
- Rosenberg, S.A. IL-2: The First Effective Immunotherapy for Human Cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef]
- Dougan, M.; Ingram, J.R.; Jeong, H.-J.; Mosaheb, M.M.; Bruck, P.T.; Ali, L.; Pishesha, N.; Blomberg, O.; Tyler, P.M.; Servos, M.M.; et al. Targeting Cytokine Therapy to the Pancreatic Tumor Microenvironment Using PD-L1–Specific VHHs. Cancer Immunol. Res. 2018, 6, 389–401. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Xing, J.; Li, Y.; Liu, J.; Wang, Z. A novel multifunctional anti-CEA-IL15 molecule displays potent antitumor activities. Drug Des. Dev. Ther. 2018, 12, 2645–2654. [Google Scholar] [CrossRef]
- Rubinstein, M.P.; Kovar, M.; Purton, J.F.; Cho, J.-H.; Boyman, O.; Surh, C.D.; Sprent, J. Converting IL-15 to a superagonist by binding to soluble IL-15Rα. Proc. Natl. Acad. Sci. USA 2006, 103, 9166–9171. [Google Scholar] [CrossRef]
- Schoonooghe, S.; Laoui, D.; Van Ginderachter, J.A.; Devoogdt, N.; Lahoutte, T.; De Baetselier, P.; Raes, G. Novel applications of nanobodies for in vivo bio-imaging of inflamed tissues in inflammatory diseases and cancer. Immunobiology 2012, 217, 1266–1272. [Google Scholar] [CrossRef]
- Fang, T.; Lu, X.; Berger, D.; Gmeiner, C.; Cho, J.; Schalek, R.; Ploegh, H.; Lichtman, J. Nanobody immunostaining for correlated light and electron microscopy with preservation of ultrastructure. Nat. Methods 2018, 15, 1029–1032. [Google Scholar] [CrossRef]
- Witte, M.D.; Wu, T.; Guimaraes, C.P.; Theile, C.S.; Blom, A.E.M.; Ingram, J.R.; Li, Z.; Kundrat, L.; Goldberg, S.D.; Ploegh, H.L. Site-specific protein modification using immobilized sortase in batch and continuous-flow systems. Nat. Protoc. 2015, 10, 508–516. [Google Scholar] [CrossRef]
- Massa, S.; Vikani, N.; Betti, C.; Ballet, S.; Vanderhaegen, S.; Steyaert, J.; Descamps, B.; Vanhove, C.; Bunschoten, A.; van Leeuwen, F.W.B.; et al. Sortase A-mediated site-specific labeling of camelid single-domain antibody-fragments: A versatile strategy for multiple molecular imaging modalities. Contrast Media Mol. Imaging 2016, 11, 328–339. [Google Scholar] [CrossRef]
- Xavier, C.; Devoogdt, N.; Hernot, S.; Vaneycken, I.; D’Huyvetter, M.; De Vos, J.; Massa, S.; Lahoutte, T.; Caveliers, V. Site-Specific Labeling of His-Tagged Nanobodies with 99mTc: A Practical Guide. In Single Domain Antibodies: Methods and Protocols; Saerens, D., Muyldermans, S., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; pp. 485–490. ISBN 978-1-61779-968-6. [Google Scholar]
- Broos, K.; Keyaerts, M.; Lecocq, Q.; Renmans, D.; Nguyen, T.; Escors, D.; Liston, A.; Raes, G.; Breckpot, K.; Devoogdt, N.; et al. Non-invasive assessment of murine PD-L1 levels in syngeneic tumor models by nuclear imaging with nanobody tracers. Oncotarget 2017, 8, 41932–41946. [Google Scholar] [CrossRef]
- Keyaerts, M.; Xavier, C.; Heemskerk, J.; Devoogdt, N.; Everaert, H.; Ackaert, C.; Vanhoeij, M.; Duhoux, F.P.; Gevaert, T.; Simon, P.; et al. Phase I Study of 68Ga-HER2-Nanobody for PET/CT Assessment of HER2 Expression in Breast Carcinoma. J. Nucl. Med. 2016, 57, 27–33. [Google Scholar] [CrossRef]
- Krasniqi, A.; D’Huyvetter, M.; Xavier, C.; der Jeught, K.V.; Muyldermans, S.; Heyden, J.V.D.; Lahoutte, T.; Tavernier, J.; Devoogdt, N. Theranostic Radiolabeled Anti-CD20 sdAb for Targeted Radionuclide Therapy of Non-Hodgkin Lymphoma. Mol. Cancer Ther. 2017, 16, 2828–2839. [Google Scholar] [CrossRef]
- Balhuizen, A.; Massa, S.; Mathijs, I.; Turatsinze, J.-V.; Vos, J.D.; Demine, S.; Xavier, C.; Villate, O.; Millard, I.; Egrise, D.; et al. A nanobody-based tracer targeting DPP6 for non-invasive imaging of human pancreatic endocrine cells. Sci. Rep. 2017, 7, 15130. [Google Scholar] [CrossRef]
- Warnders, F.J.; van Scheltinga, A.G.T.T.; Knuehl, C.; van Roy, M.; de Vries, E.F.J.; Kosterink, J.G.W.; de Vries, E.G.E.; Hooge, M.N.L. Human Epidermal Growth Factor Receptor 3–Specific Tumor Uptake and Biodistribution of 89Zr-MSB0010853 Visualized by Real-Time and Noninvasive PET Imaging. J. Nucl. Med. 2017, 58, 1210–1215. [Google Scholar] [CrossRef]
- Bannas, P.; Well, L.; Lenz, A.; Rissiek, B.; Haag, F.; Schmid, J.; Hochgräfe, K.; Trepel, M.; Adam, G.; Ittrich, H.; et al. In vivo near-infrared fluorescence targeting of T cells: Comparison of nanobodies and conventional monoclonal antibodies. Contrast Media Mol. Imaging 2014, 9, 135–142. [Google Scholar] [CrossRef]
- Rashidian, M.; Ingram, J.R.; Dougan, M.; Dongre, A.; Whang, K.A.; LeGall, C.; Cragnolini, J.J.; Bierie, B.; Gostissa, M.; Gorman, J.; et al. Predicting the response to CTLA-4 blockade by longitudinal noninvasive monitoring of CD8 T cells. J. Exp. Med. 2017, 214, 2243–2255. [Google Scholar] [CrossRef]
- Movahedi, K.; Schoonooghe, S.; Laoui, D.; Houbracken, I.; Waelput, W.; Breckpot, K.; Bouwens, L.; Lahoutte, T.; Baetselier, P.D.; Raes, G.; et al. Nanobody-Based Targeting of the Macrophage Mannose Receptor for Effective In Vivo Imaging of Tumor-Associated Macrophages. Cancer Res. 2012, 72, 4165–4177. [Google Scholar] [CrossRef]
- Blykers, A.; Schoonooghe, S.; Xavier, C.; D’hoe, K.; Laoui, D.; D’Huyvetter, M.; Vaneycken, I.; Cleeren, F.; Bormans, G.; Heemskerk, J.; et al. PET Imaging of Macrophage Mannose Receptor–Expressing Macrophages in Tumor Stroma Using 18F-Radiolabeled Camelid Single-Domain Antibody Fragments. J. Nucl. Med. 2015, 56, 1265–1271. [Google Scholar] [CrossRef]
- Van Elssen, C.H.M.J.; Rashidian, M.; Vrbanac, V.; Wucherpfennig, K.W.; el Habre, Z.; Sticht, J.; Freund, C.; Jacobsen, J.T.; Cragnolini, J.; Ingram, J.; et al. Noninvasive Imaging of Human Immune Responses in a Human Xenograft Model of Graft-Versus-Host Disease. J. Nucl. Med. 2017, 58, 1003–1008. [Google Scholar] [CrossRef]
- Rashidian, M.; Keliher, E.J.; Dougan, M.; Juras, P.K.; Cavallari, M.; Wojtkiewicz, G.R.; Jacobsen, J.T.; Edens, J.G.; Tas, J.M.J.; Victora, G.; et al. Use of 18F-2-Fluorodeoxyglucose to Label Antibody Fragments for Immuno-Positron Emission Tomography of Pancreatic Cancer. ACS Cent. Sci. 2015, 1, 142–147. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chanier, T.; Chames, P. Nanobody Engineering: Toward Next Generation Immunotherapies and Immunoimaging of Cancer. Antibodies 2019, 8, 13. https://doi.org/10.3390/antib8010013
Chanier T, Chames P. Nanobody Engineering: Toward Next Generation Immunotherapies and Immunoimaging of Cancer. Antibodies. 2019; 8(1):13. https://doi.org/10.3390/antib8010013
Chicago/Turabian StyleChanier, Timothée, and Patrick Chames. 2019. "Nanobody Engineering: Toward Next Generation Immunotherapies and Immunoimaging of Cancer" Antibodies 8, no. 1: 13. https://doi.org/10.3390/antib8010013
APA StyleChanier, T., & Chames, P. (2019). Nanobody Engineering: Toward Next Generation Immunotherapies and Immunoimaging of Cancer. Antibodies, 8(1), 13. https://doi.org/10.3390/antib8010013