A Strategy to Optimize the Generation of Stable Chromobody Cell Lines for Visualization and Quantification of Endogenous Proteins in Living Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
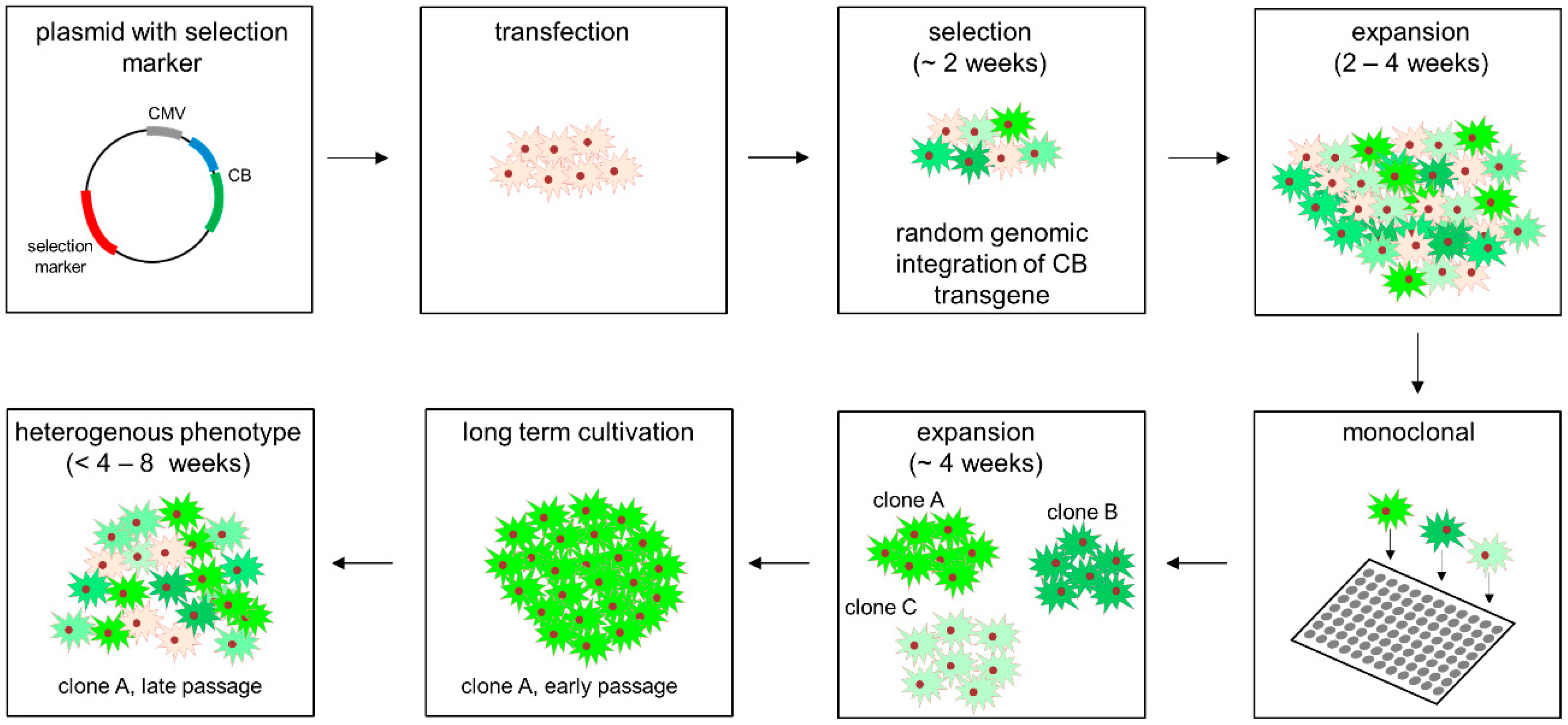
2. Material and Methods
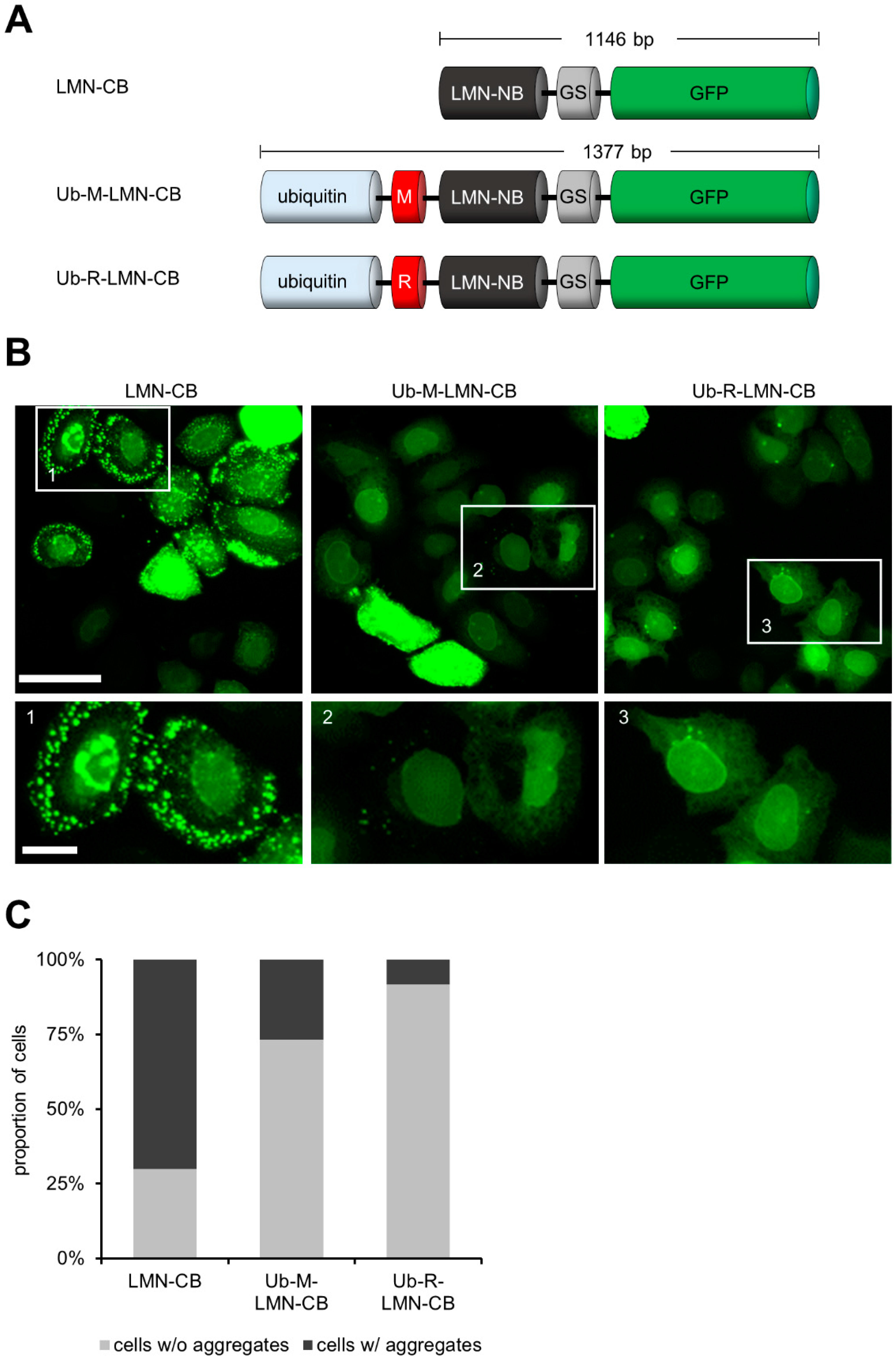
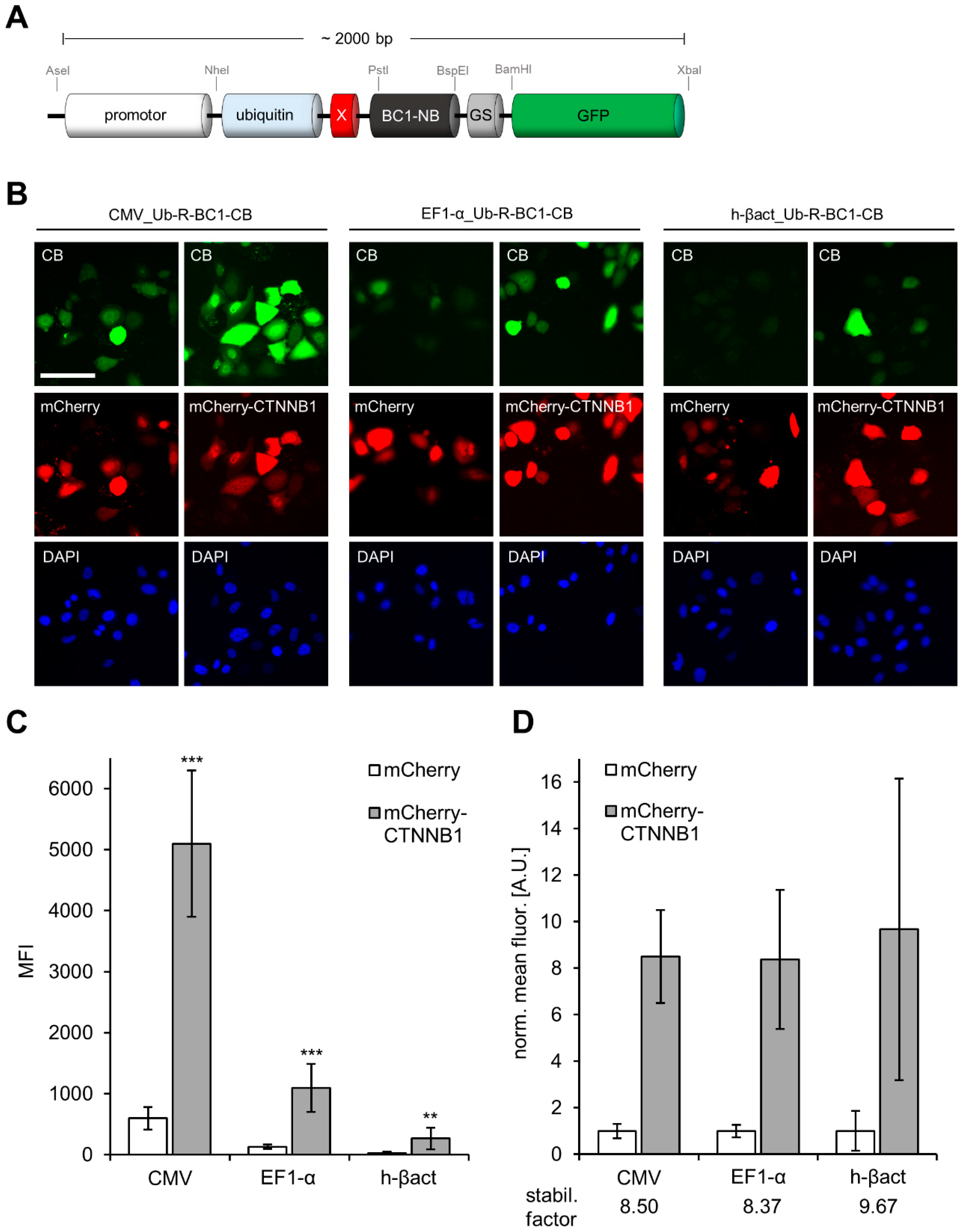
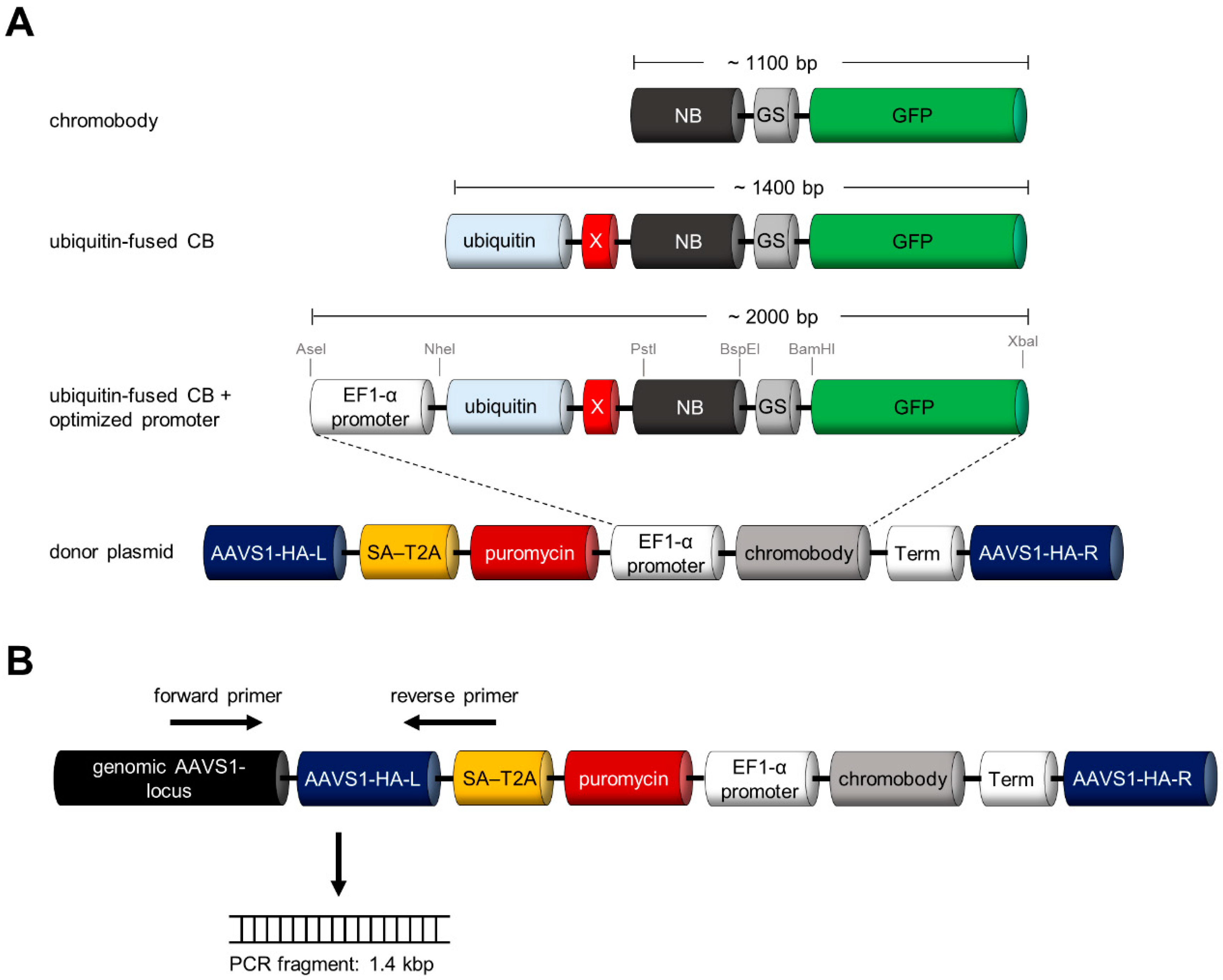
2.1. Expression Constructs
2.2. Cell Culture, Transfection, Stable Cell Line Generation and Compound Treatment
2.3. Fluorescence Imaging, Image Segmentation and Analysis
2.4. Western Blot
3. Results
3.1. Expression of CBs as Ubiquitin Fusions Reduces Intracellular Aggregation
3.2. Comparison of CB Expression Driven by the CMV-, EF1-α or β-actin Promoter to Monitor Changes in Antigen Concentration
3.3. Design and Construction of AAVS1 Donor Vector for Site-Directed Stable Integration of Turnover-Accelerated CBs
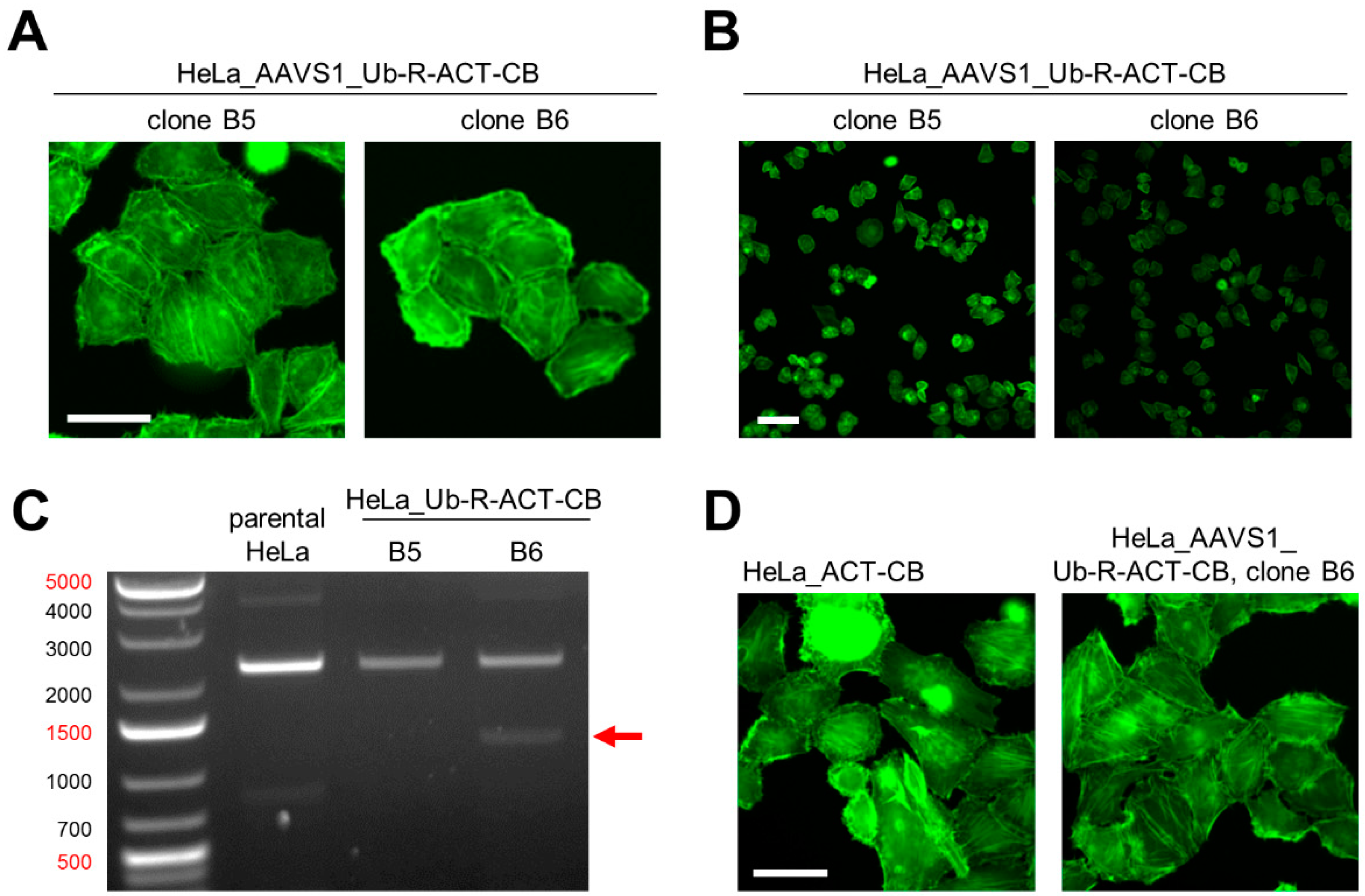
3.4. Site-Directed Integration of the Turnover-Accelerated Actin-Specific CB into the AAVS1 Locus
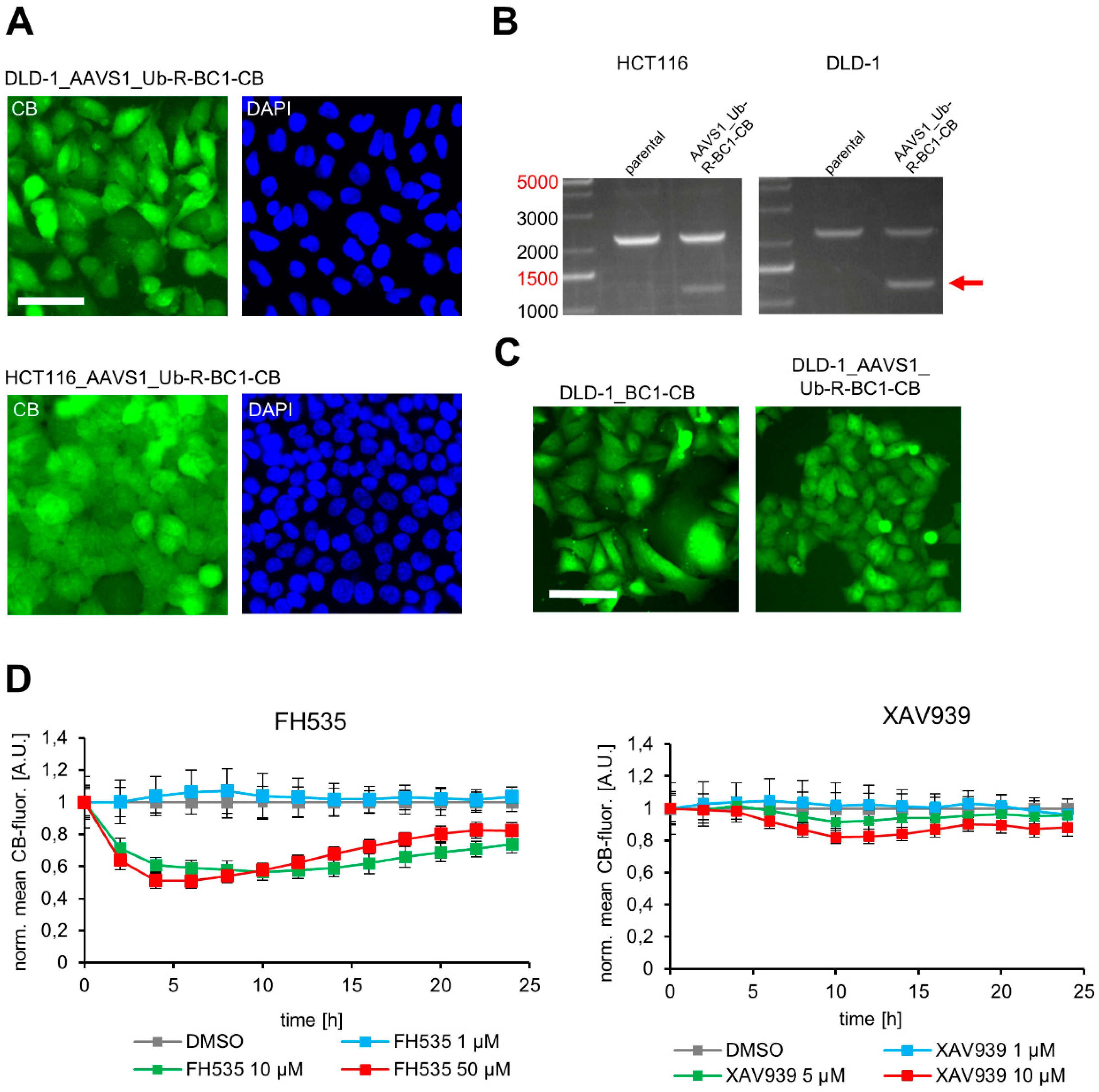
3.5. Site-Directed Integration of the Turnover-Accelerated BC1-CB into the AAVS1 Locus of Human CRC Cell Lines
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clemons, P.A. Complex phenotypic assays in high-throughput screening. Curr. Opin. Chem. Boil. 2004, 8, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Zock, J.M. Applications of high content screening in life science research. Comb. Chem. High Throughput Screen 2009, 12, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Bickle, M. The beautiful cell: High-content screening in drug discovery. Anal. Bioanal. Chem. 2010, 398, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Hosein, R.E.; Williams, S.A.; Haye, K.; Gavin, R.H. Expression of gfp-actin leads to failure of nuclear elongation and cytokinesis in tetrahymena thermophila. J. Eukaryot. Microbiol. 2003, 50, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [PubMed]
- Stadler, C.; Rexhepaj, E.; Singan, V.R.; Murphy, R.F.; Pepperkok, R.; Uhlen, M.; Simpson, J.C.; Lundberg, E. Immunofluorescence and fluorescent-protein tagging show high correlation for protein localization in mammalian cells. Nat. Methods 2013, 10, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Snapp, E.L. Fluorescent proteins: A cell biologist’s user guide. Trends Cell Boil. 2009, 19, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Virant, D.; Traenkle, B.; Maier, J.; Kaiser, P.D.; Bodenhofer, M.; Schmees, C.; Vojnovic, I.; Pisak-Lukats, B.; Endesfelder, U.; Rothbauer, U. A peptide tag-specific nanobody enables high-quality labelling for dstorm imaging. Nat. Commun. 2018, 9, 930. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, P.D.; Maier, J.; Traenkle, B.; Emele, F.; Rothbauer, U. Recent progress in generating intracellular functional antibody fragments to target and trace cellular components in living cells. Biochim. Biophys. Acta 2014, 1844, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Helma, J.; Cardoso, M.C.; Muyldermans, S.; Leonhardt, H. Nanobodies and recombinant binders in cell biology. J. Cell Boil. 2015, 209, 633–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutel, S.; Bery, N.; Bernard, V.; Keller, L.; Lemesre, E.; de Marco, A.; Ligat, L.; Rain, J.C.; Favre, G.; Olichon, A.; et al. NaLi-H1: A universal synthetic library of humanized nanobodies providing highly functional antibodies and intrabodies. eLife 2016, 5, e16228. [Google Scholar] [CrossRef] [PubMed]
- Rothbauer, U.; Zolghadr, K.; Tillib, S.; Nowak, D.; Schermelleh, L.; Gahl, A.; Backmann, N.; Conrath, K.; Muyldermans, S.; Cardoso, M.C.; et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat. Methods 2006, 3, 887–889. [Google Scholar] [CrossRef] [PubMed]
- Irannejad, R.; Tomshine, J.C.; Tomshine, J.R.; Chevalier, M.; Mahoney, J.P.; Steyaert, J.; Rasmussen, S.G.; Sunahara, R.K.; El-Samad, H.; Huang, B.; et al. Conformational biosensors reveal GPCR signalling from endosomes. Nature 2013, 495, 534–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, A.; Lorca, T.; Castro, A. Quantitative live imaging of endogenous DNA replication in mammalian cells. PLoS ONE 2012, 7, e45726. [Google Scholar] [CrossRef] [PubMed]
- Panza, P.; Maier, J.; Schmees, C.; Rothbauer, U.; Sollner, C. Live imaging of endogenous protein dynamics in zebrafish using chromobodies. Development 2015, 142, 1879–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traenkle, B.; Emele, F.; Anton, R.; Poetz, O.; Haeussler, R.S.; Maier, J.; Kaiser, P.D.; Scholz, A.M.; Nueske, S.; Buchfellner, A.; et al. Monitoring interactions and dynamics of endogenous beta-catenin with intracellular nanobodies in living cells. Mol. Cell. Proteom. 2015, 14, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.; Traenkle, B.; Rothbauer, U. Real-time analysis of epithelial-mesenchymal transition using fluorescent single-domain antibodies. Sci. Rep. 2015, 5, 13402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schorpp, K.; Rothenaigner, I.; Maier, J.; Traenkle, B.; Rothbauer, U.; Hadian, K. A multiplexed high-content screening approach using the chromobody technology to identify cell cycle modulators in living cells. J. Biomol. Screen. 2016, 21, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Halff, E.F.; Versteeg, M.; Brondijk, T.H.; Huizinga, E.G. When less becomes more: Optimization of protein expression in HEK293-EBNA1 cells using plasmid titration—A case study for NLRs. Protein Expr. Purif. 2014, 99, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Vavouri, T.; Semple, J.I.; Garcia-Verdugo, R.; Lehner, B. Intrinsic protein disorder and interaction promiscuity are widely associated with dosage sensitivity. Cell 2009, 138, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, W.; de Jong, J.; Pindyurin, A.V.; Pagie, L.; Meuleman, W.; de Ridder, J.; Berns, A.; Wessels, L.F.; van Lohuizen, M.; van Steensel, B. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell 2013, 154, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Bringmann, P.; McClary, J.; Jones, P.P.; Manzana, W.; Zhu, Y.; Wang, S.; Liu, Y.; Harvey, S.; Madlansacay, M.R.; et al. High levels of protein expression using different mammalian cmv promoters in several cell lines. Protein Expr. Purif. 2006, 45, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Zolghadr, K.; Gregor, J.; Leonhardt, H.; Rothbauer, U. Case study on live cell apoptosis-assay using lamin-chromobody cell-lines for high-content analysis. Methods Mol. Boil. 2012, 911, 569–575. [Google Scholar]
- Keller, B.M.; Maier, J.; Secker, K.A.; Egetemaier, S.M.; Parfyonova, Y.; Rothbauer, U.; Traenkle, B. Chromobodies to quantify changes of endogenous protein concentration in living cells. Mol. Cell. Proteom. 2018, 17, 2518–2533. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A., III; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Damdindorj, L.; Karnan, S.; Ota, A.; Takahashi, M.; Konishi, Y.; Hossain, E.; Hosokawa, Y.; Konishi, H. Assessment of the long-term transcriptional activity of a 550-bp-long human β-actin promoter region. Plasmid 2012, 68, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Oceguera-Yanez, F.; Kim, S.I.; Matsumoto, T.; Tan, G.W.; Xiang, L.; Hatani, T.; Kondo, T.; Ikeya, M.; Yoshida, Y.; Inoue, H.; et al. Engineering the aavs1 locus for consistent and scalable transgene expression in human ipscs and their differentiated derivatives. Methods 2016, 101, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. Ubiquitin fusion technique and its descendants. Methods Enzymol. 2000, 327, 578–593. [Google Scholar] [PubMed]
- Varshavsky, A. The n-end rule pathway and regulation by proteolysis. Protein Sci. 2011, 20, 1298–1345. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. Ubiquitin fusion technique and related methods. Methods Enzymol. 2005, 399, 777–799. [Google Scholar] [PubMed]
- Schmidthals, K.; Helma, J.; Zolghadr, K.; Rothbauer, U.; Leonhardt, H. Novel antibody derivatives for proteome and high-content analysis. Anal. Bioanal. Chem. 2010, 397, 3203–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, M.; Park, J.M.; Cao, F.; Wang, D.; Paulmurugan, R.; Tseng, J.R.; Gonzalgo, M.L.; Gambhir, S.S.; Wu, J.C. Effects of epigenetic modulation on reporter gene expression: Implications for stem cell imaging. FASEB J. 2006, 20, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Mariati; Chusainow, J.; Yap, M.G. DNA methylation contributes to loss in productivity of monoclonal antibody-producing CHO cell lines. J. Biotechnol. 2010, 147, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Paredes, V.; Park, J.S.; Jeong, Y.; Yoon, J.; Baek, K. Unstable expression of recombinant antibody during long-term culture of cho cells is accompanied by histone H3 hypoacetylation. Biotechnol. Lett. 2013, 35, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Li, H.P.; Hung, Y.H.; Leu, Y.W.; Wu, W.H.; Wang, F.S.; Lee, K.D.; Chang, P.J.; Wu, C.S.; Lu, Y.J.; et al. Targeted methylation of cmv and e1a viral promoters. Biochem. Biophys. Res. Commun. 2010, 402, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.Y.; Zhang, L.; Clift, K.L.; Hulur, I.; Xiang, A.P.; Ren, B.Z.; Lahn, B.T. Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PLoS ONE 2010, 5, e10611. [Google Scholar] [CrossRef] [PubMed]
- Norrman, K.; Fischer, Y.; Bonnamy, B.; Wolfhagen Sand, F.; Ravassard, P.; Semb, H. Quantitative comparison of constitutive promoters in human ES cells. PLoS ONE 2010, 5, e12413. [Google Scholar] [CrossRef]
- Wurm, F.M. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat. Biotechnol. 2004, 22, 1393–1398. [Google Scholar] [CrossRef]
- Rodolosse, A.; Barbat, A.; Chantret, I.; Lacasa, M.; Brot-Laroche, E.; Zweibaum, A.; Rousset, M. Selecting agent hygromycin B alters expression of glucose-regulated genes in transfected Caco-2 cells. Am. J. Physiol. 1998, 274, G931–G938. [Google Scholar] [CrossRef]
- Valera, A.; Perales, J.C.; Hatzoglou, M.; Bosch, F. Expression of the neomycin-resistance (neo) gene induces alterations in gene expression and metabolism. Hum. Gene Ther. 1994, 5, 449–456. [Google Scholar] [CrossRef]
- McDaniel, L.D.; Schultz, R.A. Elevation of sister chromatid exchange frequency in transformed human fibroblasts following exposure to widely used aminoglycosides. Environ. Mol. Mutagen. 1993, 21, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Kotin, R.M.; Linden, R.M.; Berns, K.I. Characterization of a preferred site on human chromosome 19q for integration of adeno-associated virus DNA by non-homologous recombination. EMBO J. 1992, 11, 5071–5078. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Liu, C.; Cerbini, T.; San, H.; Lin, Y.; Chen, G.; Rao, M.S.; Zou, J. Stable enhanced green fluorescent protein expression after differentiation and transplantation of reporter human induced pluripotent stem cells generated by aavs1 transcription activator-like effector nucleases. Stem Cells Transl. Med. 2014, 3, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Sekine, K.; Takebe, T.; Taniguchi, H. Fluorescent labeling and visualization of human induced pluripotent stem cells with the use of transcription activator-like effector nucleases. Transplant. Proc. 2014, 46, 1205–1207. [Google Scholar] [CrossRef]
- Zhang, P.W.; Haidet-Phillips, A.M.; Pham, J.T.; Lee, Y.; Huo, Y.; Tienari, P.J.; Maragakis, N.J.; Sattler, R.; Rothstein, J.D. Generation of gfap::Gfp astrocyte reporter lines from human adult fibroblast-derived i PS cells using zinc-finger nuclease technology. Glia 2016, 64, 63–75. [Google Scholar] [CrossRef]
- Sadelain, M.; Papapetrou, E.P.; Bushman, F.D. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer 2011, 12, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.R.; Maguire, S.; Davis, L.A.; Alexander, M.; Yang, F.; Chandran, S.; ffrench-Constant, C.; Pedersen, R.A. Robust, persistent transgene expression in human embryonic stem cells is achieved with AAVS1-targeted integration. Stem Cells 2008, 26, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknaes, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef]
- Handeli, S.; Simon, J.A. A small-molecule inhibitor of Tcf/β-catenin signaling down-regulates PPARγ and PPARδ activities. Mol. Cancer Ther. 2008, 7, 521–529. [Google Scholar] [CrossRef]
- Yao, H.; Ashihara, E.; Maekawa, T. Targeting the Wnt/β-catenin signaling pathway in human cancers. Expert Opin. Ther. Targets 2011, 15, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Boutros, M.; Heigwer, F.; Laufer, C. Microscopy-based high-content screening. Cell 2015, 163, 1314–1325. [Google Scholar] [CrossRef] [PubMed]
- Traenkle, B.; Rothbauer, U. Under the microscope: Single-domain antibodies for live-cell imaging and super-resolution microscopy. Front. Immunol. 2017, 8, 1030. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.T.; Smith, S.A.; Marano, R.; McKee, J.; Board, P.G. Protein expression using cotranslational fusion and cleavage of ubiquitin. Mutagenesis of the glutathione-binding site of human Pi class glutathione S-transferase. J. Boil. Chem. 1994, 269, 25381–25386. [Google Scholar]
- Papapetrou, E.P.; Schambach, A. Gene insertion into genomic safe harbors for human gene therapy. Mol. Ther. 2016, 24, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Frattini, A.; Fabbri, M.; Valli, R.; De Paoli, E.; Montalbano, G.; Gribaldo, L.; Pasquali, F.; Maserati, E. High variability of genomic instability and gene expression profiling in different hela clones. Sci. Rep. 2015, 5, 15377. [Google Scholar] [CrossRef]
- Koch, B.; Nijmeijer, B.; Kueblbeck, M.; Cai, Y.; Walther, N.; Ellenberg, J. Generation and validation of homozygous fluorescent knock-in cells using CRISPR-Cas9 genome editing. Nat. Protoc. 2018, 13, 1465–1487. [Google Scholar] [CrossRef]
- Ratz, M.; Testa, I.; Hell, S.W.; Jakobs, S. CRISPR/Cas9-mediated endogenous protein tagging for resolft super-resolution microscopy of living human cells. Sci. Rep. 2015, 5, 9592. [Google Scholar] [CrossRef]
- Zerjatke, T.; Gak, I.A.; Kirova, D.; Fuhrmann, M.; Daniel, K.; Gonciarz, M.; Muller, D.; Glauche, I.; Mansfeld, J. Quantitative cell cycle analysis based on an endogenous all-in-one reporter for cell tracking and classification. Cell Rep. 2017, 19, 1953–1966. [Google Scholar] [CrossRef]
- Lackner, D.H.; Carre, A.; Guzzardo, P.M.; Banning, C.; Mangena, R.; Henley, T.; Oberndorfer, S.; Gapp, B.V.; Nijman, S.M.; Brummelkamp, T.R.; et al. A generic strategy for CRISPR-Cas9-mediated gene tagging. Nat. Commun. 2015, 6, 10237. [Google Scholar] [CrossRef]
- Specht, E.A.; Braselmann, E.; Palmer, A.E. A critical and comparative review of fluorescent tools for live-cell imaging. Annu. Rev. Physiol. 2017, 79, 93–117. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keller, B.-M.; Maier, J.; Weldle, M.; Segan, S.; Traenkle, B.; Rothbauer, U. A Strategy to Optimize the Generation of Stable Chromobody Cell Lines for Visualization and Quantification of Endogenous Proteins in Living Cells. Antibodies 2019, 8, 10. https://doi.org/10.3390/antib8010010
Keller B-M, Maier J, Weldle M, Segan S, Traenkle B, Rothbauer U. A Strategy to Optimize the Generation of Stable Chromobody Cell Lines for Visualization and Quantification of Endogenous Proteins in Living Cells. Antibodies. 2019; 8(1):10. https://doi.org/10.3390/antib8010010
Chicago/Turabian StyleKeller, Bettina-Maria, Julia Maier, Melissa Weldle, Soeren Segan, Bjoern Traenkle, and Ulrich Rothbauer. 2019. "A Strategy to Optimize the Generation of Stable Chromobody Cell Lines for Visualization and Quantification of Endogenous Proteins in Living Cells" Antibodies 8, no. 1: 10. https://doi.org/10.3390/antib8010010
APA StyleKeller, B.-M., Maier, J., Weldle, M., Segan, S., Traenkle, B., & Rothbauer, U. (2019). A Strategy to Optimize the Generation of Stable Chromobody Cell Lines for Visualization and Quantification of Endogenous Proteins in Living Cells. Antibodies, 8(1), 10. https://doi.org/10.3390/antib8010010