Design Principles for Bispecific IgGs, Opportunities and Pitfalls of Artificial Disulfide Bonds




Abstract
1. Introduction
2. Materials and Methods
2.1. Construction of Expression Vectors
2.2. Production of Antibodies in E. coli
2.3. Transient Antibody Expression in HEK293 Cells
2.4. Stable Antibody Expression in CHO Cells
2.5. SDS-PAGE Electrophoresis, Immunoblotting and ELISA
2.6. Surface Plasmon Resonance Analysis
2.7. Thermal Stability Assay
2.8. MTT Cell Viability Assay
2.9. Animal Studies
2.10. Statistical Analysis
3. Results
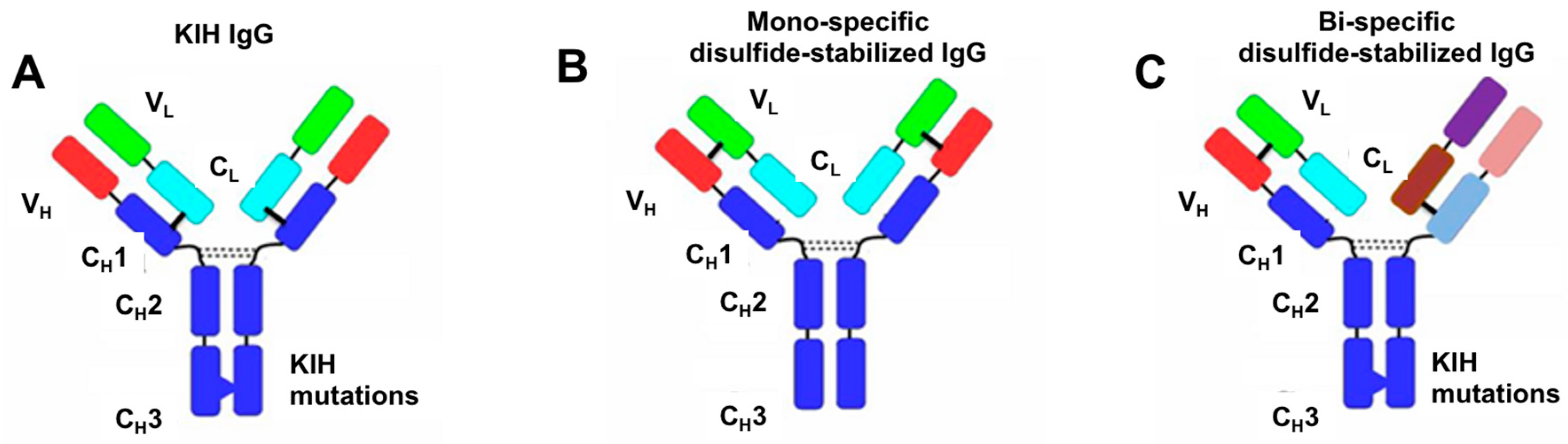
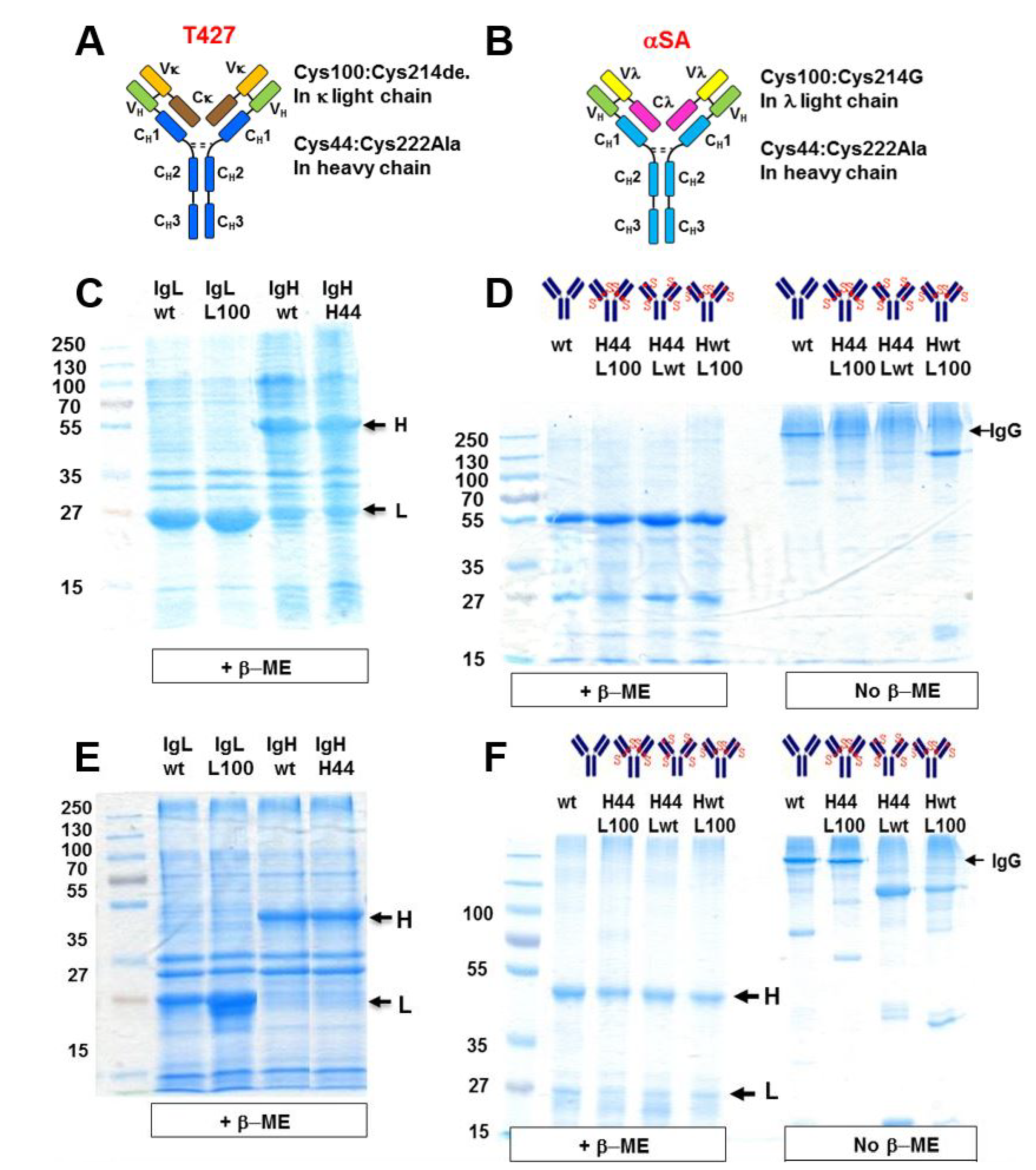
3.1. Design of Artificial Disulfide Bonds for H–L Chain Pairing
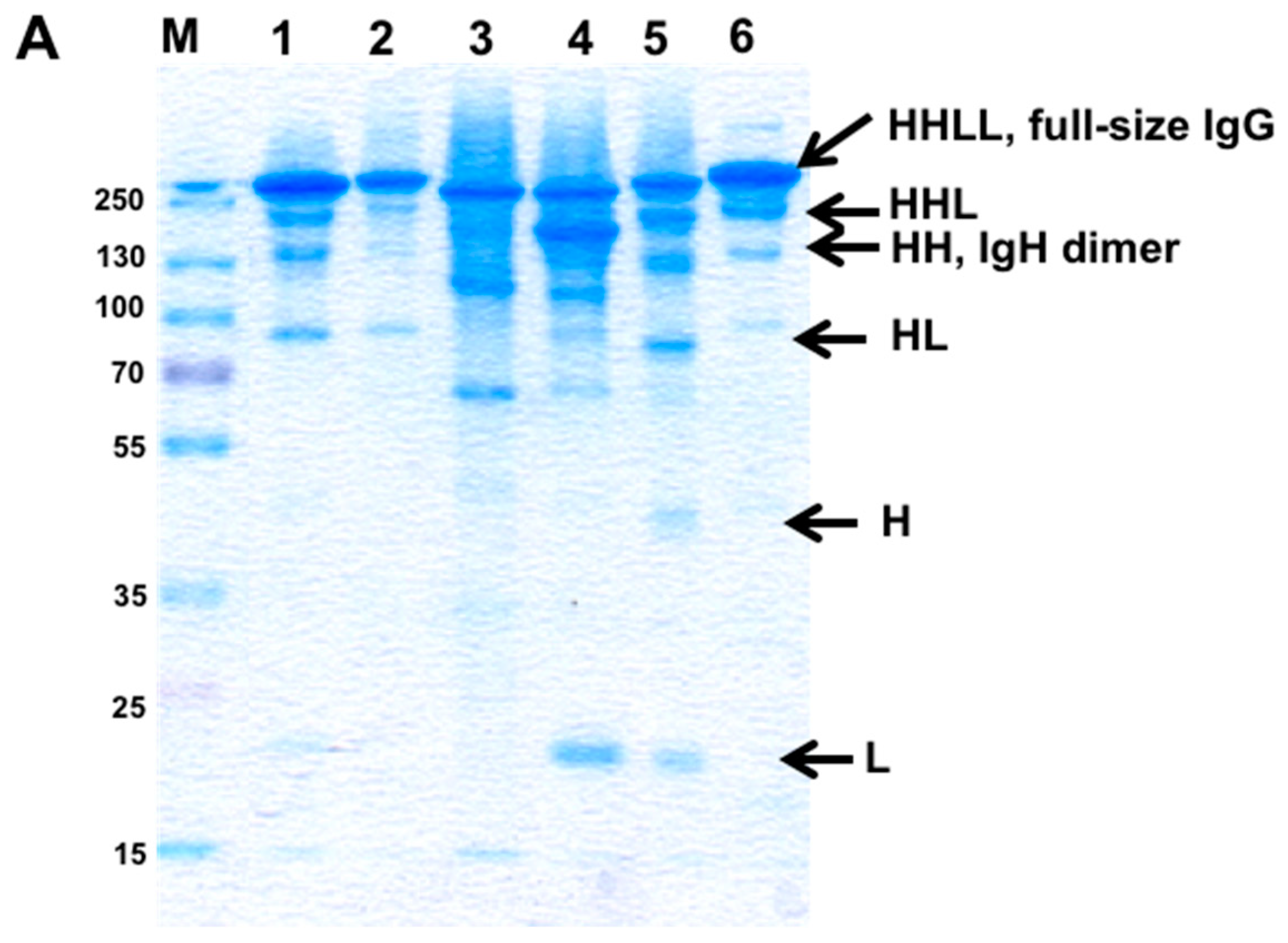
3.2. Evaluation of H–H Chain Heterodimerization in Refolded Bispecific IgGs
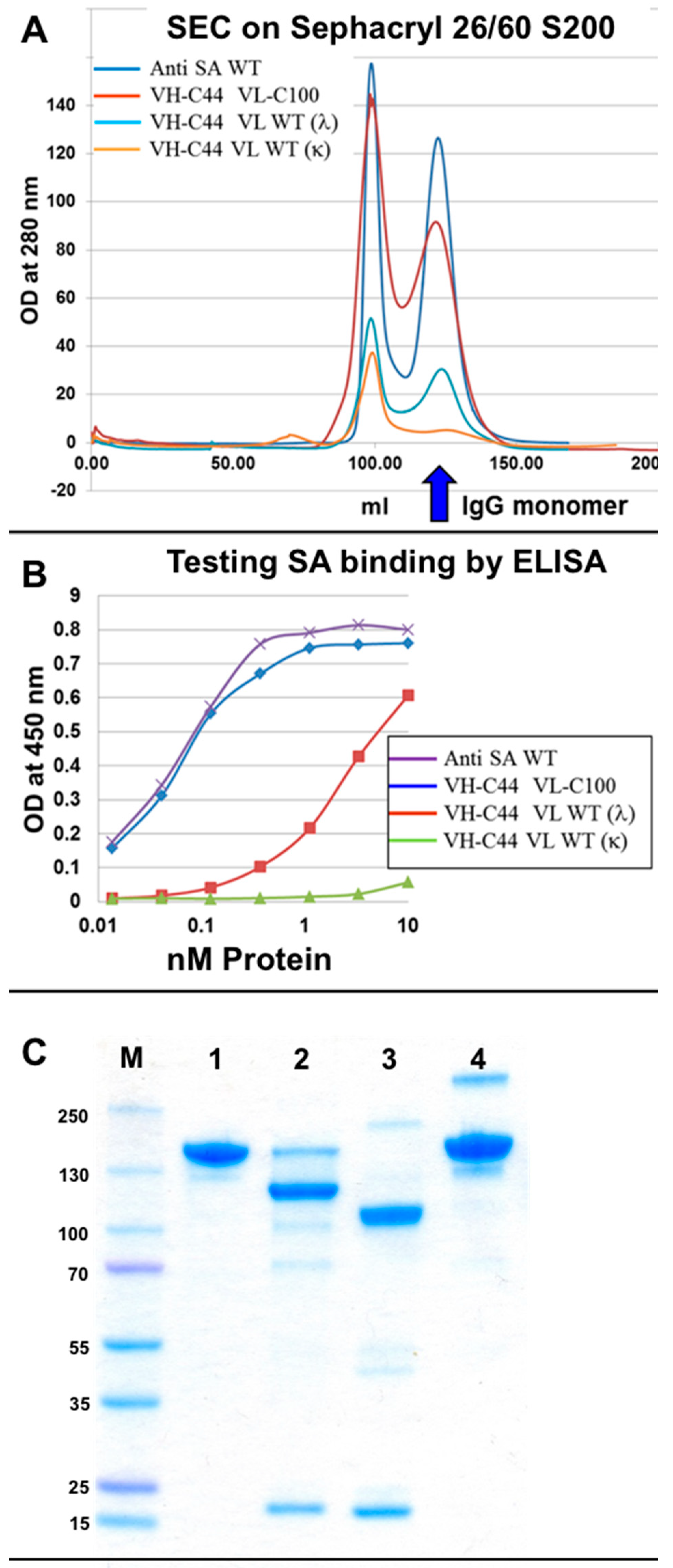
3.3. Evaluation of H–L Chain Heterodimerization in Refolded IgGs
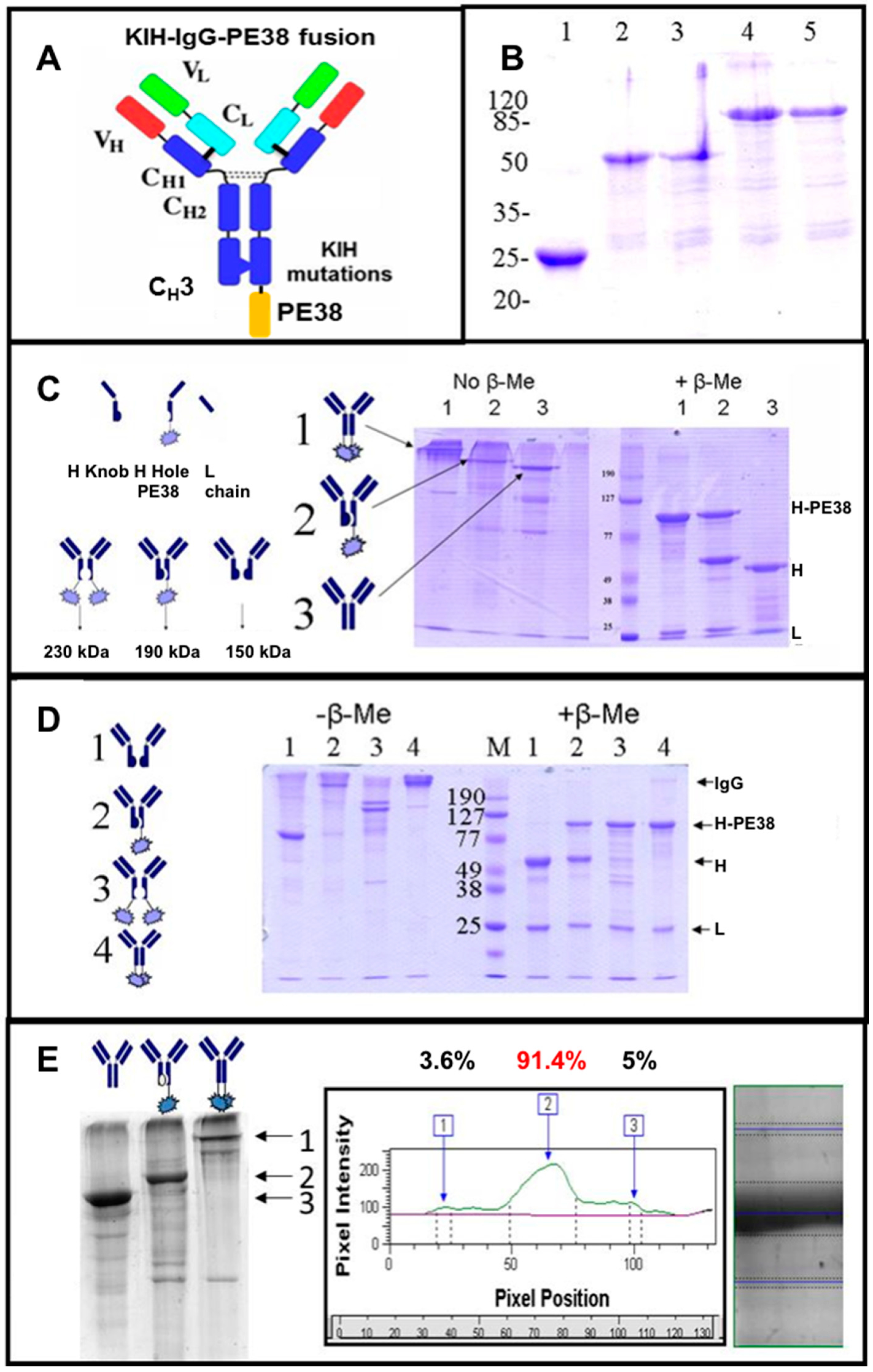
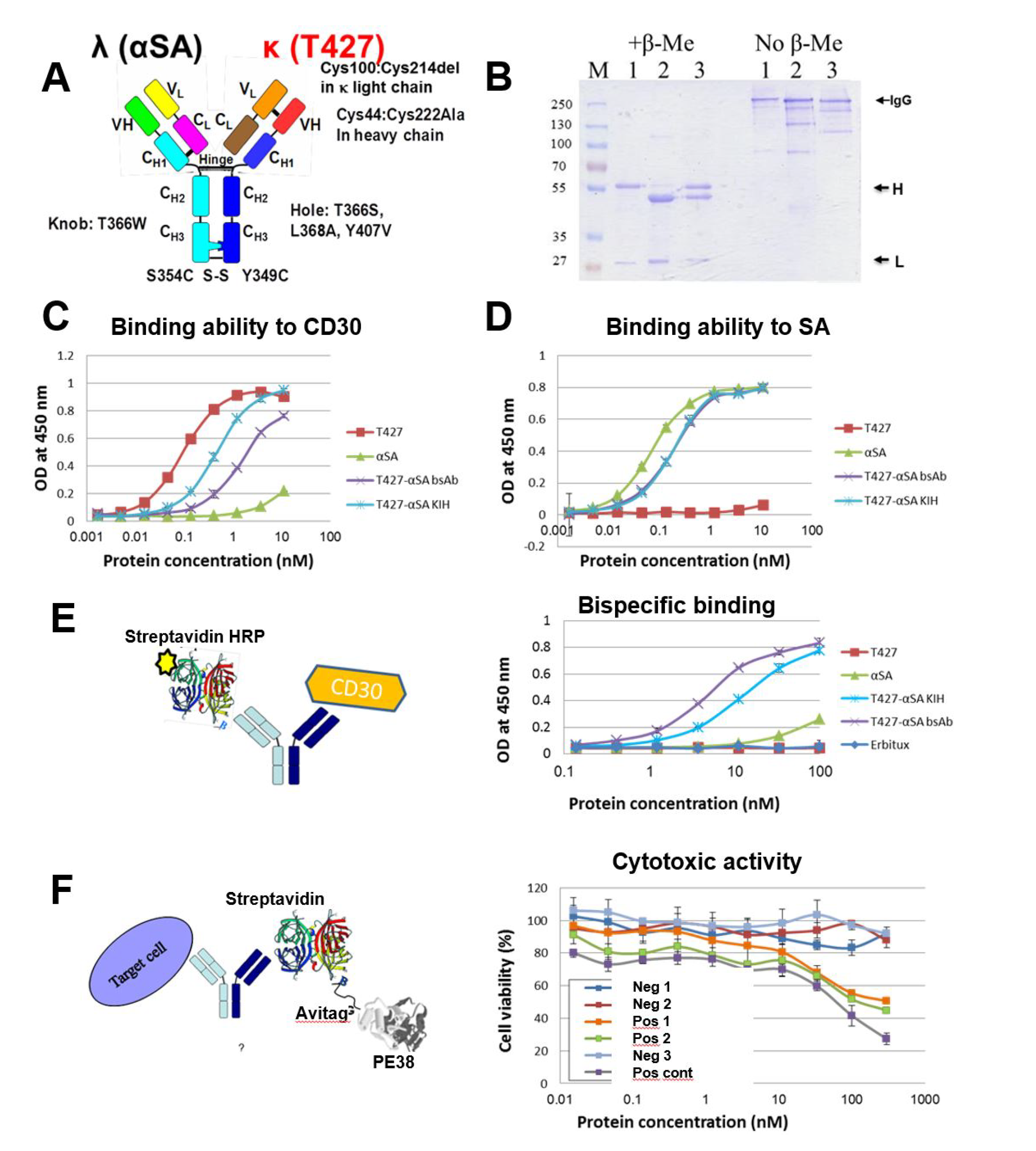
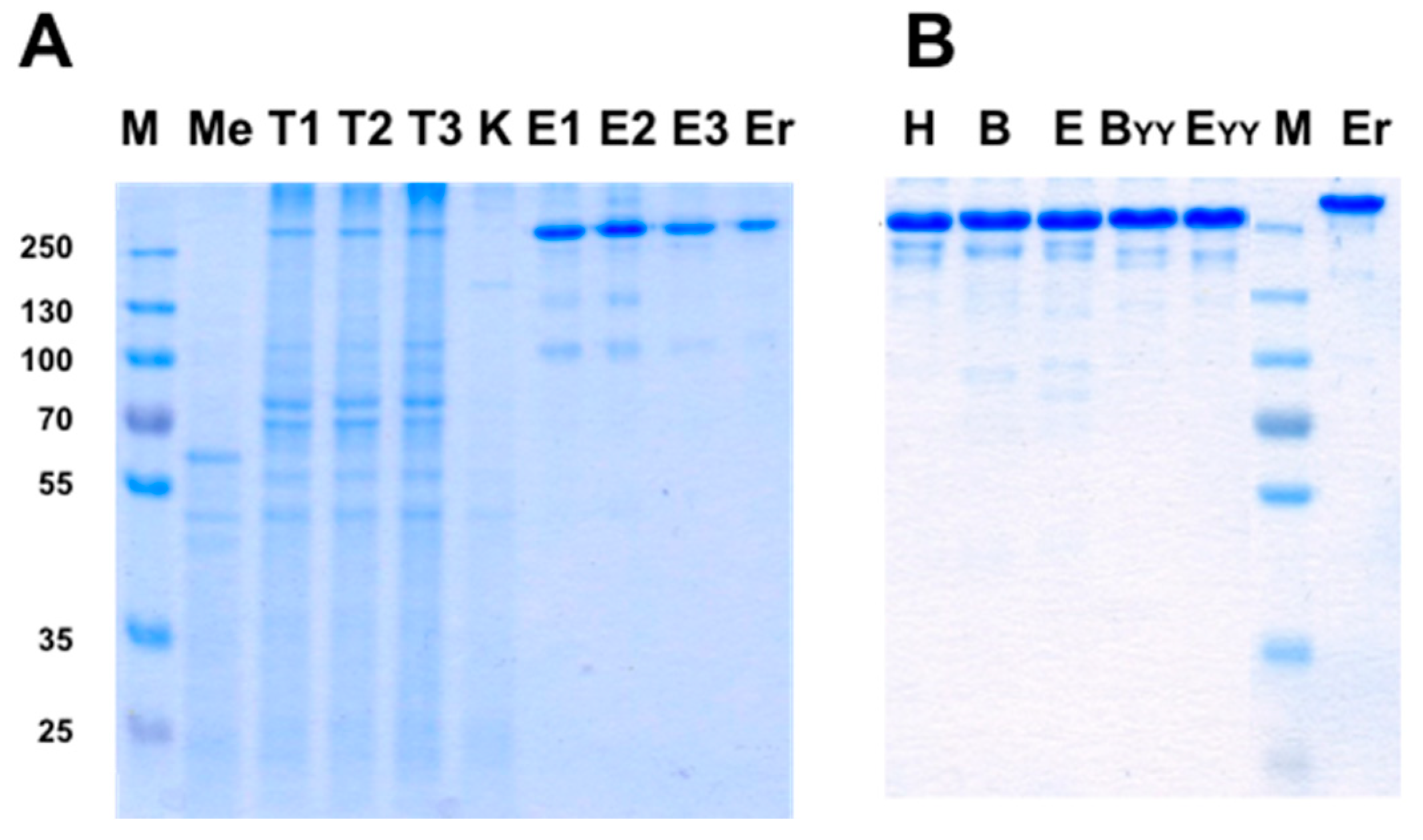
3.4. Production of bsAbs by Refolding and Their Functional Evaluation
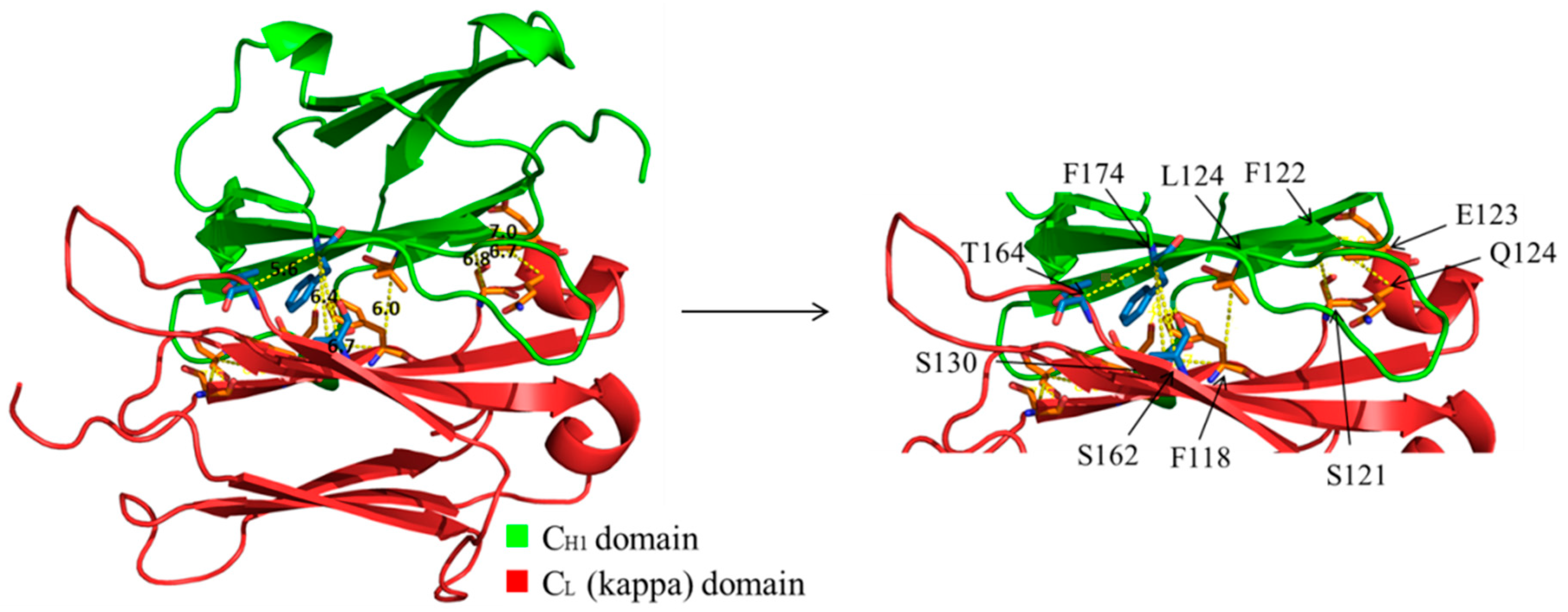
3.5. Evaluation of Additional Positions for Disulfide-Stabilization of the Fab Arm Interface of bsAbs
3.6. Evaluation of CH1-CL Positions for Disulfide-Stabilization of the Fab Arm Interface of bsAbs
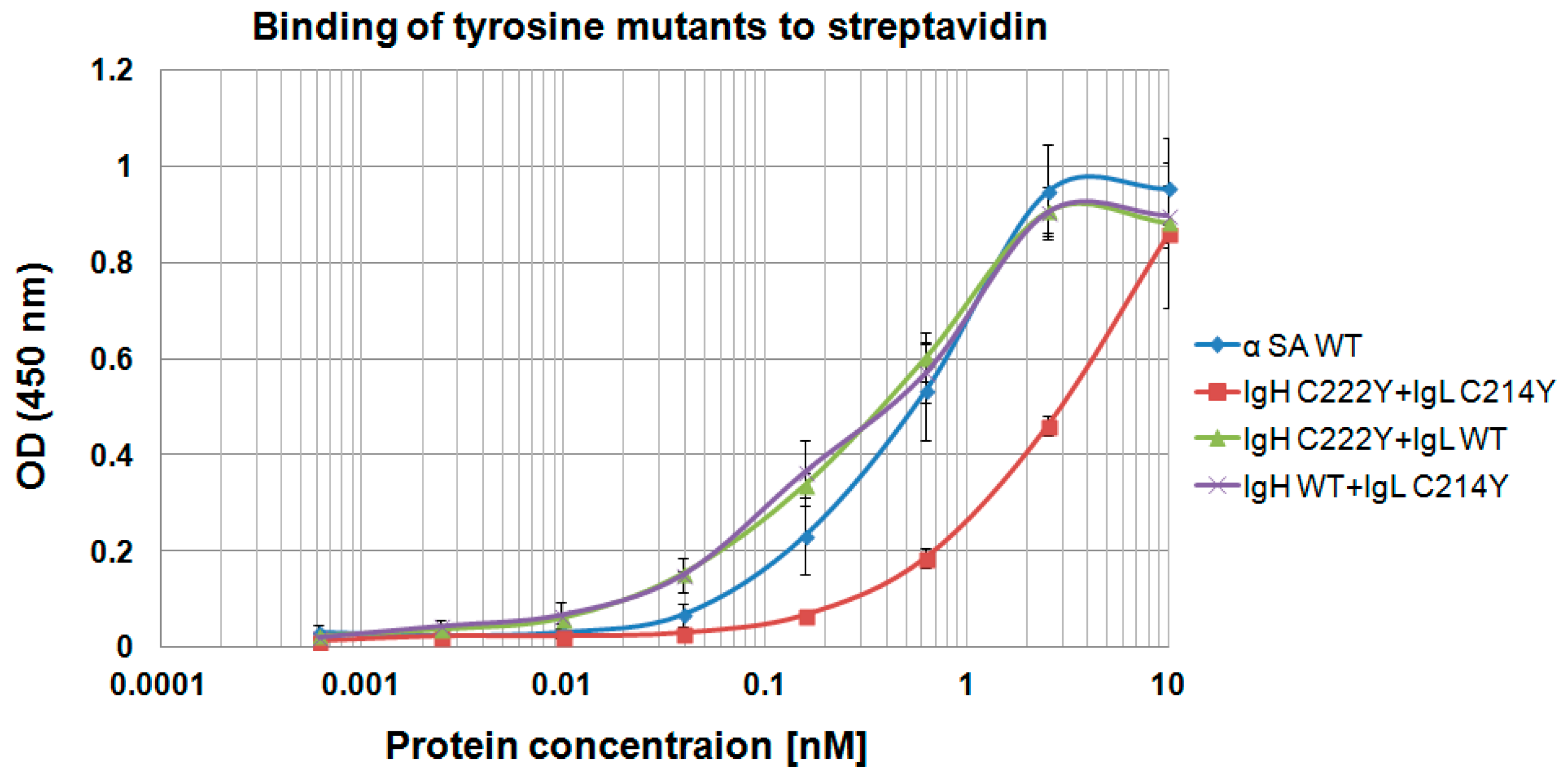
3.7. Using “Destructive Mutations” to Prevent “Illegitimate” H–L Chain Pairing
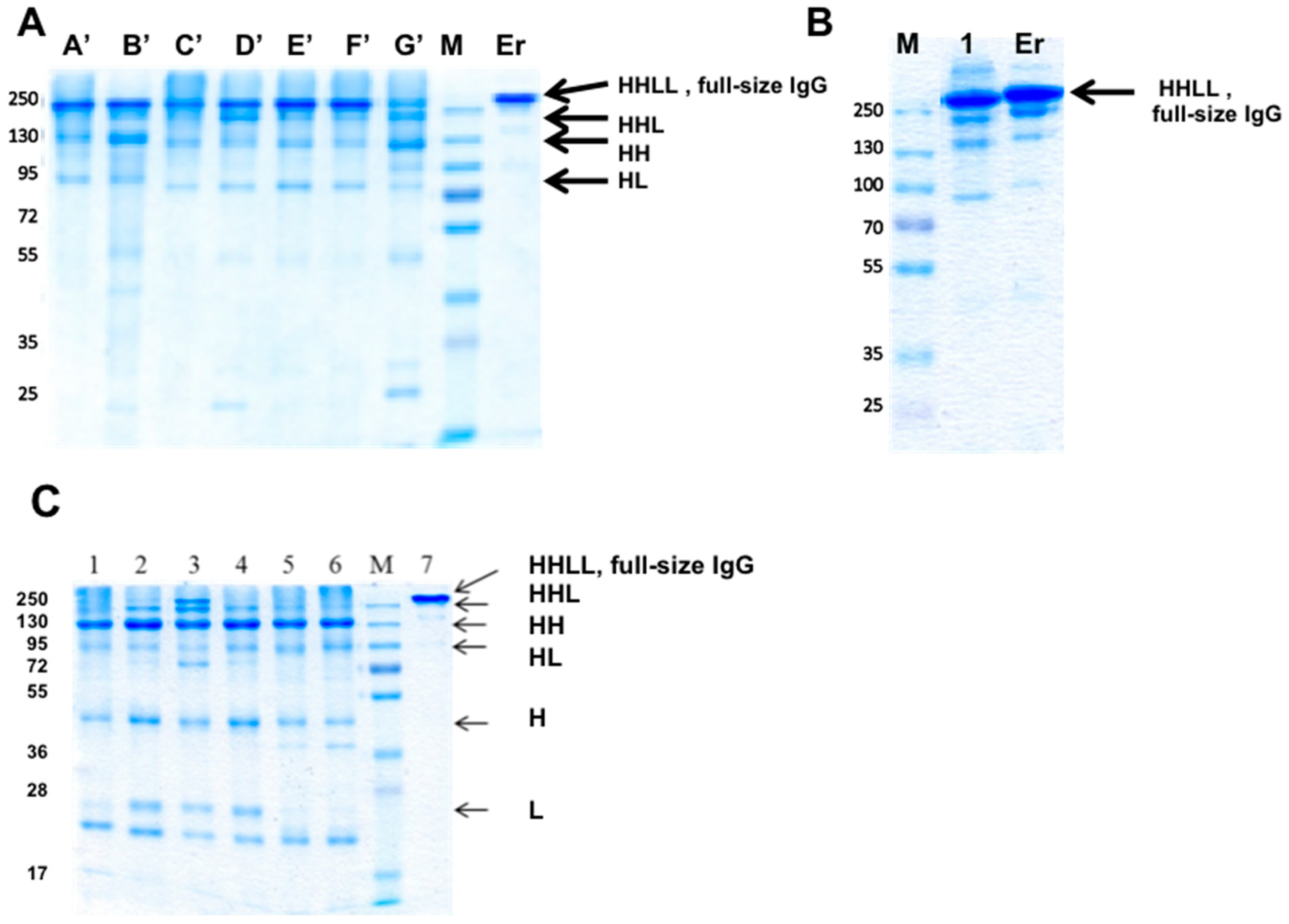
3.8. Production of bsAbs in Transfected Mammalian Cells
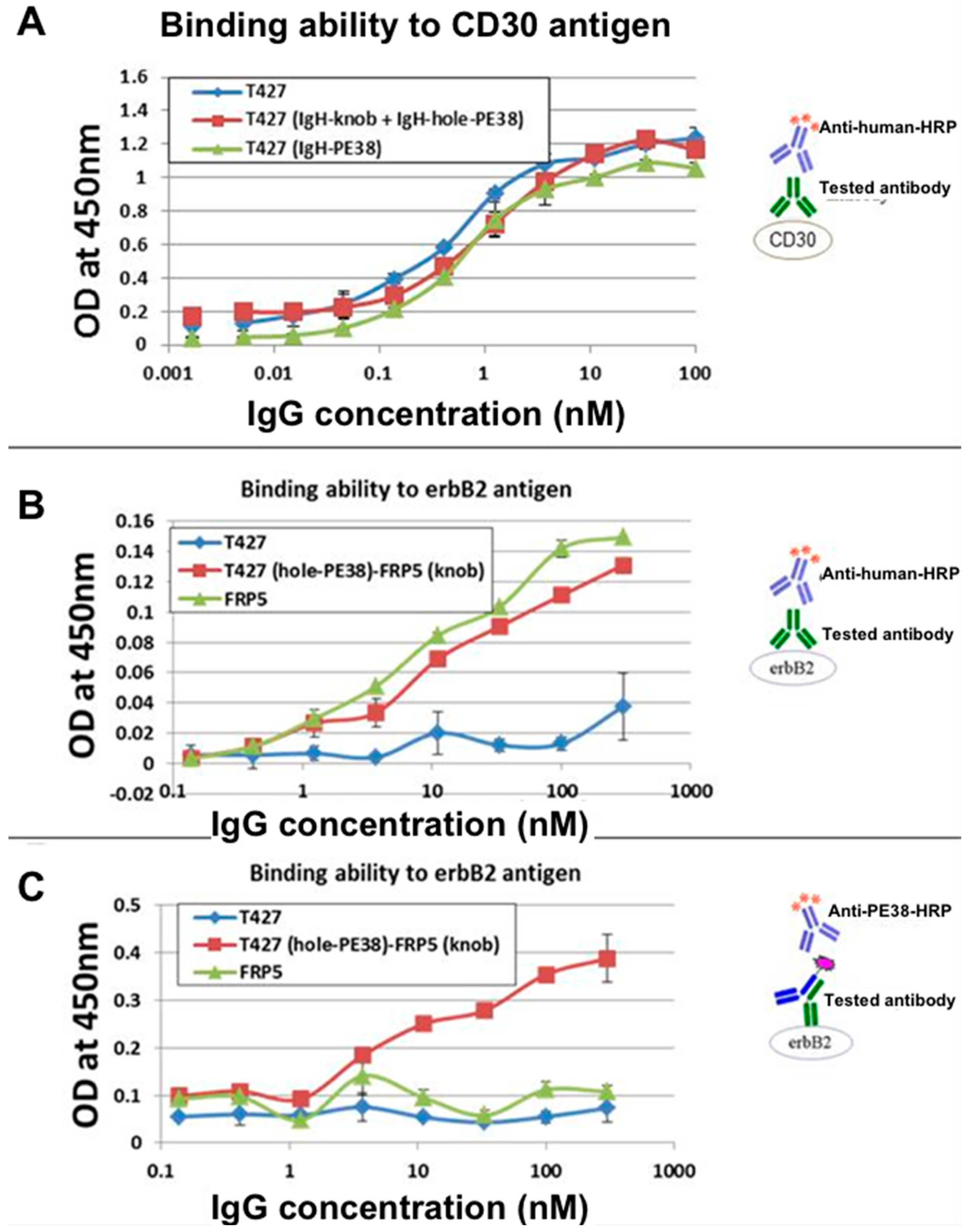
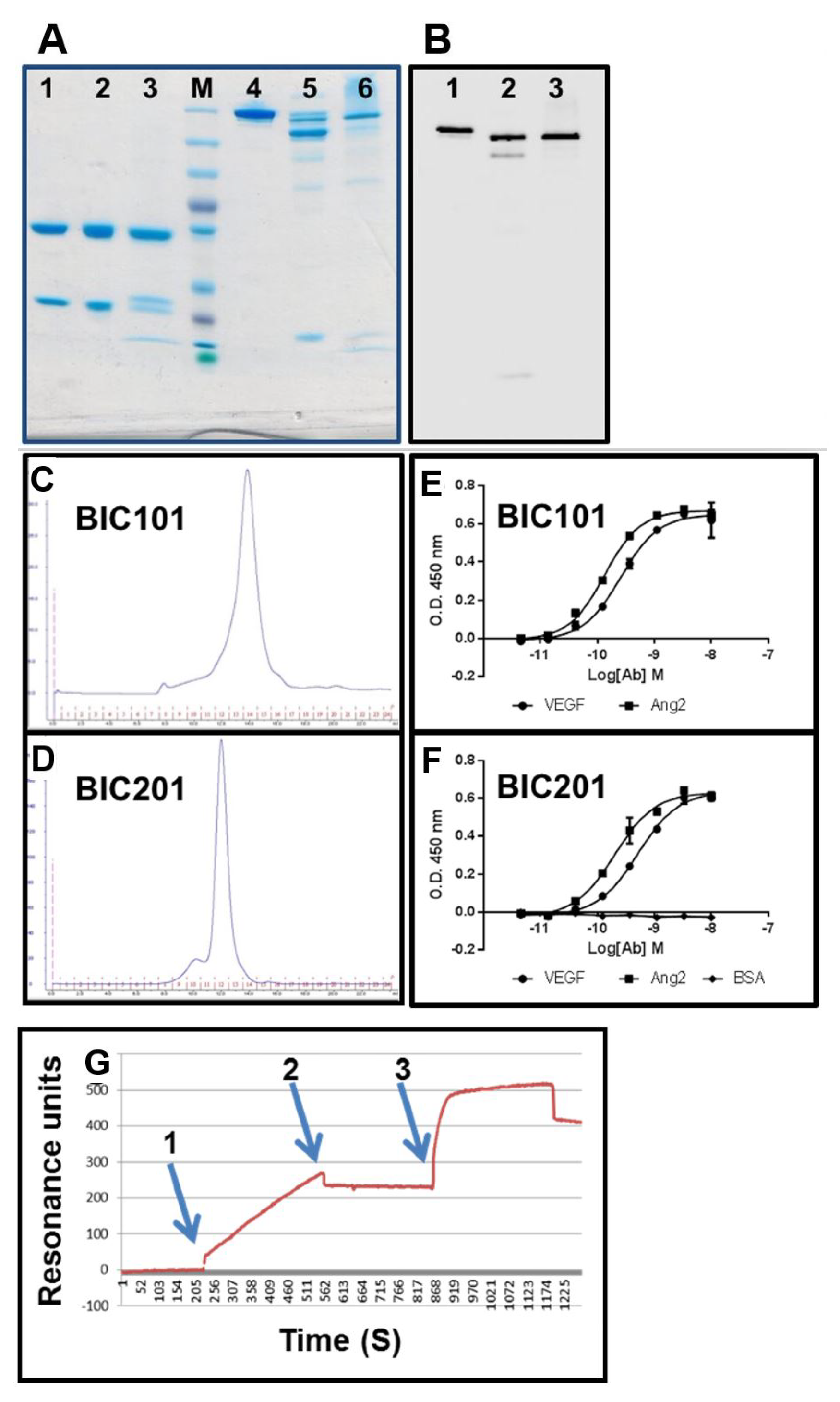
3.9. Evaluation of Binding Affinity by SPR
3.10. Large-Scale Production in CHO Cells
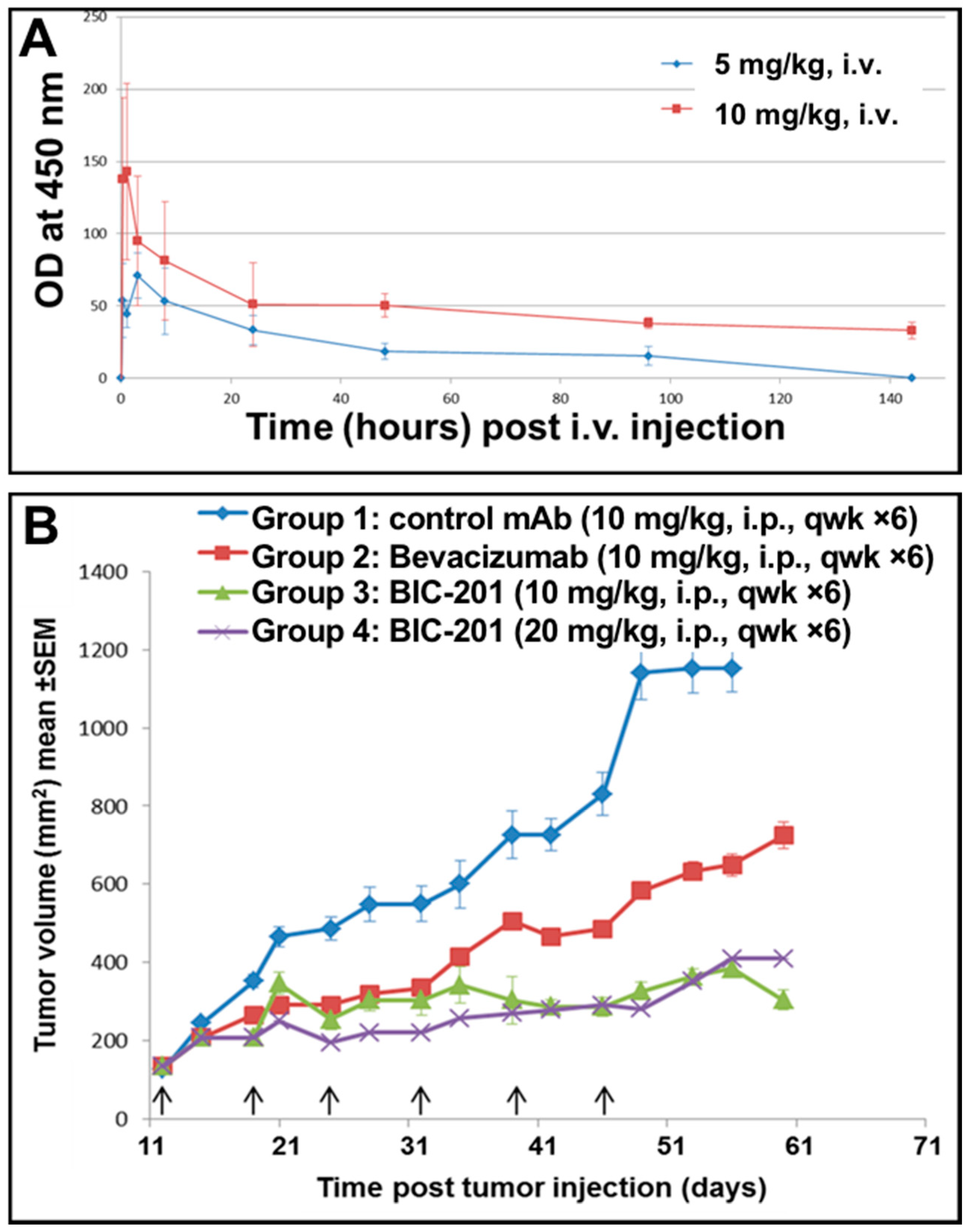
3.11. Study of the Blood Pharmacokinetics of BIC101 and BIC201 bsAbs Made in CHO Cells in Mice
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reichert, J.M. Antibodies to watch in 2014. mAbs 2014, 6, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R. Dual targeting strategies with bispecific antibodies. mAbs 2012, 4, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Riethmuller, G. Symmetry breaking: Bispecific antibodies, the beginnings, and 50 years on. Cancer Immun. 2012, 12, 12. [Google Scholar] [PubMed]
- Jost, C.; Pluckthun, A. Engineered proteins with desired specificity: Darpins, other alternative scaffolds and bispecific iggs. Curr. Opin. Struct. Biol. 2014, 27C, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. mAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed]
- Demarest, S.J.; Glaser, S.M. Antibody therapeutics, antibody engineering, and the merits of protein stability. Curr. Opin. Drug Discov. Dev. 2008, 11, 675–687. [Google Scholar]
- Dong, J.; Sereno, A.; Snyder, W.B.; Miller, B.R.; Tamraz, S.; Doern, A.; Favis, M.; Wu, X.; Tran, H.; Langley, E.; et al. Stable IgG-like bispecific antibodies directed toward the type I insulin-like growth factor receptor demonstrate enhanced ligand blockade and anti-tumor activity. J. Biol. Chem. 2011, 286, 4703–4717. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Sustmann, C.; Thomas, M.; Stubenrauch, K.; Croasdale, R.; Schanzer, J.; Brinkmann, U.; Kettenberger, H.; Regula, J.T.; Schaefer, W. Progress in overcoming the chain association issue in bispecific heterodimeric IgG antibodies. mAbs 2012, 4, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, W.; Regula, J.T.; Bahner, M.; Schanzer, J.; Croasdale, R.; Durr, H.; Gassner, C.; Georges, G.; Kettenberger, H.; Imhof-Jung, S.; et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific igg antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 11187–11192. [Google Scholar] [CrossRef] [PubMed]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An efficient route to human bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.M.; Wu, X.; Pustilnik, A.; Sereno, A.; Huang, F.; Rick, H.L.; Guntas, G.; Leaver-Fay, A.; Smith, E.M.; Ho, C.; et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat. Biotechnol. 2014, 32, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, J.B.; Presta, L.G.; Carter, P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996, 9, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.M.; Li, C.; Lei, M.; Lundin, V.; Lee, H.Y.; Ninonuevo, M.; Lin, K.; Han, G.; Sandoval, W.; Lei, D.; et al. Structural and functional characterization of a hole-hole homodimer variant in a “knob-into-hole” bispecific antibody. Anal. Chem. 2017, 89, 13494–13501. [Google Scholar] [CrossRef] [PubMed]
- Mazor, Y.; Oganesyan, V.; Yang, C.; Hansen, A.; Wang, J.; Liu, H.; Sachsenmeier, K.; Carlson, M.; Gadre, D.V.; Borrok, M.J.; et al. Improving target cell specificity using a novel monovalent bispecific IgG design. mAbs 2015, 7, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Kabat, E.A.; Wu, T.T.; Perry, H.M.; Gottesman, K.S.; Foeller, C. Sequences of Proteins of Immunological Interest, 5th ed.; US Department of Health and Human Services, Public Health Service, National Institutes of Health: Bethesda, MD, USA, 1991. [Google Scholar]
- Lefranc, M.P.; Giudicelli, V.; Kaas, Q.; Duprat, E.; Jabado-Michaloud, J.; Scaviner, D.; Ginestoux, C.; Clément, O.; Chaume, D.; Lefranc, G. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res. 2005, 33, D593–D597. [Google Scholar]
- Hakim, R.; Benhar, I. “Inclonals”: IgGs and IgG-enzyme fusion proteins produced in an E. coli expression-refolding system. mAbs 2009, 1, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, N.; Welschof, M.; Zewe, M.; Braunagel, M.; Dubel, S.; Breitling, F.; Little, M. Simultaneous mutagenesis of antibody CDR regions by overlap extension and PCR. BioTechniques 1994, 17, 310, 312, 314–315. [Google Scholar] [PubMed]
- GE Healthcare. Biacore™ Assay Handbook; General Electric Company: Boston, MA, USA, 2012. [Google Scholar]
- Senisterra, G.; Chau, I.; Vedadi, M. Thermal denaturation assays in chemical biology. Assay Drug Dev. Technol. 2012, 10, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U.; Reiter, Y.; Jung, S.H.; Lee, B.; Pastan, I. A recombinant immunotoxin containing a disulfide-stabilized Fv fragment. Proc. Natl. Acad. Sci. USA 1993, 90, 7538–7542. [Google Scholar] [CrossRef] [PubMed]
- Reiter, Y.; Brinkmann, U.; Jung, S.H.; Lee, B.; Kasprzyk, P.G.; King, C.R.; Pastan, I. Improved binding and antitumor activity of a recombinant anti-ErbB2 immunotoxin by disulfide stabilization of the fv fragment. J. Biol. Chem. 1994, 269, 18327–18331. [Google Scholar] [PubMed]
- Reiter, Y.; Brinkmann, U.; Kreitman, R.J.; Jung, S.H.; Lee, B.; Pastan, I. Stabilization of the Fv fragments in recombinant immunotoxins by disulfide bonds engineered into conserved framework regions. Biochemistry 1994, 33, 5451–5459. [Google Scholar] [CrossRef] [PubMed]
- Harwerth, I.M.; Wels, W.; Marte, B.M.; Hynes, N.E. Monoclonal antibodies against the extracellular domain of the ErbB-2 receptor function as partial ligand agonists. J. Biol. Chem. 1992, 267, 15160–15167. [Google Scholar] [PubMed]
- Nagata, S.; Numata, Y.; Onda, M.; Ise, T.; Hahn, Y.; Lee, B.; Pastan, I. Rapid grouping of monoclonal antibodies based on their topographical epitopes by a label-free competitive immunoassay. J. Immunol. Methods 2004, 292, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Ofir, K.; Berdichevsky, Y.; Benhar, I.; Azriel-Rosenfeld, R.; Lamed, R.; Barak, Y.; Bayer, E.A.; Morag, E. Versatile protein microarray based on carbohydrate-binding modules. Proteomics 2005, 5, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Azriel-Rosenfeld, R.; Valensi, M.; Benhar, I. A human synthetic combinatorial library of arrayable single-chain antibodies based on shuffling in vivo formed CDRs into general framework regions. J. Mol. Biol. 2004, 335, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Onda, M.; Numata, Y.; Santora, K.; Beers, R.; Kreitman, R.J.; Pastan, I. Novel anti-CD30 recombinant immunotoxins containing disulfide-stabilized Fv fragments. Clin. Cancer Res. 2002, 8, 2345–2355. [Google Scholar] [PubMed]
- Reiter, Y.; Brinkmann, U.; Jung, S.H.; Pastan, I.; Lee, B. Disulfide stabilization of antibody Fv: Computer predictions and experimental evaluation. Protein Eng. 1995, 8, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U.; Lee, B.K.; Pastan, I. Recombinant immunotoxins containing the VH or VL domain of monoclonal antibody B3 fused to Pseudomonas exotoxin. J. Immunol. 1993, 150, 2774–2782. [Google Scholar] [PubMed]
- Byrne, H.; Conroy, P.J.; Whisstock, J.C.; O’Kennedy, R.J. A tale of two specificities: Bispecific antibodies for therapeutic and diagnostic applications. Trends Biotechnol. 2013, 31, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.L.; Rossi, E.A.; Cardillo, T.M.; Goldenberg, D.M.; Chang, C.H. A new class of bispecific antibodies to redirect T cells for cancer immunotherapy. mAbs 2014, 6, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Jackman, J.; Chen, Y.; Huang, A.; Moffat, B.; Scheer, J.M.; Leong, S.R.; Lee, W.P.; Zhang, J.; Sharma, N.; Lu, Y.; et al. Development of a two-part strategy to identify a therapeutic human bispecific antibody that inhibits IgE receptor signaling. J. Biol. Chem. 2010, 285, 20850–20859. [Google Scholar] [CrossRef] [PubMed]
- Spiess, C.; Merchant, M.; Huang, A.; Zheng, Z.; Yang, N.Y.; Peng, J.; Ellerman, D.; Shatz, W.; Reilly, D.; Yansura, D.G.; et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat. Biotechnol. 2013, 31, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.H.; McCarthy, S.G.; Brosnan, K.; Goldberg, K.M.; Scallon, B.J. Correlations between pharmacokinetics of IgG antibodies in primates vs. FcRn-transgenic mice reveal a rodent model with predictive capabilities. mAbs 2013, 5, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.H.; Aperlo, C.; Li, Y.; Kurosawa, E.; Lan, Y.; Lo, K.M.; Huston, J.S. Seedbodies: Fusion proteins based on strand-exchange engineered domain (seed) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng. Des. Sel. 2010, 23, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, K.; Pentony, M.; Shen, M.; Garrett, L.; Forte, C.; Woodward, A.; Ng, S.B.; Born, T.; Retter, M.; Manchulenko, K.; et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: Applications to bispecific molecules and monovalent IgG. J. Biol. Chem. 2010, 285, 19637–19646. [Google Scholar] [CrossRef] [PubMed]
- Strop, P.; Ho, W.H.; Boustany, L.M.; Abdiche, Y.N.; Lindquist, K.C.; Farias, S.E.; Rickert, M.; Appah, C.T.; Pascua, E.; Radcliffe, T.; et al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J. Mol. Biol. 2012, 420, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Rispens, T.; Meesters, J.; Rose, R.J.; den Bleker, T.H.; Loverix, S.; van den Bremer, E.T.; Neijssen, J.; Vink, T.; Lasters, I.; et al. Species-specific determinants in the IgG CH3 domain enable Fab-arm exchange by affecting the noncovalent CH3-CH3 interaction strength. J. Immunol. 2011, 187, 3238–3246. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, G.; Haber, L.; Crocker, L.M.; Shia, S.; Shao, L.; Dowbenko, D.; Totpal, K.; Wong, A.; Lee, C.V.; Stawicki, S.; et al. A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell 2011, 20, 472–486. [Google Scholar] [CrossRef] [PubMed]
- Mazor, Y.; Hansen, A.; Yang, C.; Chowdhury, P.S.; Wang, J.; Stephens, G.; Wu, H.; Dall’Acqua, W.F. Insights into the molecular basis of a bispecific antibody’s target selectivity. mAbs 2015, 7, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.H.; Pastan, I.; Lee, B. Design of interchain disulfide bonds in the framework region of the Fv fragment of the monoclonal antibody B3. Proteins 1994, 19, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Reiter, Y.; Brinkmann, U.; Webber, K.O.; Jung, S.H.; Lee, B.; Pastan, I. Engineering interchain disulfide bonds into conserved framework regions of Fv fragments: Improved biochemical characteristics of recombinant immunotoxins containing disulfide-stabilized Fv. Protein Eng. 1994, 7, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Pastan, I.; Lee, B.; Jung, S.-H.; Brinkmann, U. Recombinant Disulfide-Stabilized Polypeptide Fragments Having Binding Specificity. U.S. Patent 5,747,654, 1998. [Google Scholar]
- Wozniak-Knopp, G.; Ruker, F. A C-terminal interdomain disulfide bond significantly stabilizes the Fc fragment of IgG. Arch. Biochem. Biophys. 2012, 526, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Wozniak-Knopp, G.; Stadlmann, J.; Ruker, F. Stabilisation of the Fc fragment of human IgG1 by engineered intradomain disulfide bonds. PLoS ONE 2012, 7, e30083. [Google Scholar] [CrossRef] [PubMed]
- Schachner, L.; Han, G.; Dillon, M.; Zhou, J.; McCarty, L.; Ellerman, D.; Yin, Y.; Spiess, C.; Lill, J.R.; Carter, P.J.; et al. Characterization of chain pairing variants of bispecific IgG expressed in a single host cell by high-resolution native and denaturing mass spectrometry. Anal. Chem. 2016, 88, 12122–12127. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Han, G.; Zhou, J.; Dillon, M.; McCarty, L.; Gavino, L.; Ellerman, D.; Spiess, C.; Sandoval, W.; Carter, P.J. Precise quantification of mixtures of bispecific IgG produced in single host cells by liquid chromatography-orbitrap high-resolution mass spectrometry. mAbs 2016, 8, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Leitner, A.; Walzthoeni, T.; Aebersold, R. Lysine-specific chemical cross-linking of protein complexes and identification of cross-linking sites using Lc-Ms/Ms and the xquest/xprophet software pipeline. Nat. Protoc. 2014, 9, 120–137. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.; Chen, Z.A.; Rappsilber, J. Quantitative cross-linking/mass spectrometry using isotope-labelled cross-linkers. J. Proteom. 2013, 88, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Slavin, M.; Kalisman, N. Structural analysis of protein complexes by cross-linking and mass spectrometry. Methods Mol. Biol. 2018, 1764, 173–183. [Google Scholar] [PubMed]
- Olson, G.; Fouque, N.; Depoisier, J.-F.; Fischer, N.; Magistl’elli, G. Methods of Purifying Antibodies. U.S. Patent 2013/0317200 a1, 2013. [Google Scholar]
- Kim, J.K.; Firan, M.; Radu, C.G.; Kim, C.H.; Ghetie, V.; Ward, E.S. Mapping the site on human IgG for binding of the MHC class I-related receptor, FcRn. Eur. J. Immunol. 1999, 29, 2819–2825. [Google Scholar] [CrossRef]
- Simmons, L.C.; Reilly, D.; Klimowski, L.; Raju, T.S.; Meng, G.; Sims, P.; Hong, K.; Shields, R.L.; Damico, L.A.; Rancatore, P.; et al. Expression of full-length immunoglobulins in Escherichia coli: Rapid and efficient production of aglycosylated antibodies. J. Immunol. Methods 2002, 263, 133–147. [Google Scholar] [CrossRef]
- Kontermann, R.E.; Brinkmann, U. Bispecific antibodies. Drug Discov. Today 2015, 20, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E.; Brinkmann, U. Corrigendum to “bispecific antibodies” [drug discov. Today 20 (July (7)) (2015) 838–847]. Drug Discov. Today 2018. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule Name | Residue in CH1 | Residue in CL | Cα-Cα Distance (Å) |
|---|---|---|---|
| A | F174 | S162 | 6.4 |
| B | F174 | T164 | 5.6 |
| C | L124 | F118 | 6 |
| D | F122 | S121 | 6.8 |
| E | F122 | E123 | 7 |
| F | F122 | Q124 | 6.7 |
| G | S130 | F118 | 6.7 |
| Molecule Name | Heavy Chain | Light Chain | EC50 (nM) |
|---|---|---|---|
| α SA WT | α SA WT | α SA WT | 0.15 |
| 1 | α SA WT | C214A | 0.05 |
| 2 | C222A | α SA WT | 0.15 |
| 3 | α SA WT | C214R | 0.09 |
| 4 | C222R | α SA WT | 0.1 |
| 5 | C222R | C214R | 0.2 |
| 6 | C222A | C214A | 0.05 |
| 7 | C222R | C214A | 0.25 |
| 8 | C222A | C214R | 0.08 |
| Molecule Name and Mutations | Tm1 (°C) | Tm2 (°C) |
|---|---|---|
| H (Heavy chain C44/Light chain C100) | 67.0 | 76.8 |
| B (Heavy chain C174/Light chain C164) | 67.3 | 77.1 |
| E (Heavy chain C122/Light chain C123) | 67.7 | 77.5 |
| BYY (B with Heavy chain Y222/Light chain Y214) | 67.6 | 75.2 |
| EYY (E with Heavy chain Y222/Light chain Y214) | 68.0 | 76.1 |
| T427 WT | 67.0 | 81.3 |
| αSA WT | 70.2 | 78.2 |
| Molecule Name | Yield (µg/30 mL Transfection) | Total Calculated Yield (mg/L) |
|---|---|---|
| H | 230 | 7.7 |
| B | 525 | 17.5 |
| E | 360 | 12 |
| BYY | 610 | 20.3 |
| EYY | 215 | 7.2 |
| T427 WT | 2.6 mg * | 86 |
| αSA WT | 10 mg * | 330 |
| Antibody | Binding to CD30 | Binding to SA | ||||
|---|---|---|---|---|---|---|
| Ka (1/Ms) | Kd (1/s) | KD (Nm) | Ka (M-1s-1) | Kd (1/s) | KD (nM) | |
| H | 2.04 × 105 | 2.97 × 10−3 | 14.6 | 5.26 × 104 | 1.93 × 10−3 | 36.6 |
| B | 1.67 × 105 | 1.99 × 10−3 | 11.9 | 1.29 × 105 | 2.35 × 10−3 | 18.2 |
| E | 1.34 × 105 | 1.80 × 10−3 | 13.5 | 1.28 × 105 | 2.48 × 10−3 | 19.3 |
| BYY | 2.00 × 105 | 2.29 × 10−3 | 11.5 | 4.30 × 104 | 2.65 × 10−3 | 61.7 |
| EYY | 1.21 × 105 | 1.94 × 10−3 | 16 | 2.20 × 104 | 3.18 × 10−3 | 145 |
| T427 WT | 4.69 × 105 | 4.22 × 10−4 | 0.899 | - | - | - |
| αSA WT | - | - | - | 3.68 × 105 | 1.41 × 10−4 | 0.384 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaks, L.; Litvak-Greenfeld, D.; Dror, S.; Shefet-Carasso, L.; Matatov, G.; Nahary, L.; Shapira, S.; Hakim, R.; Alroy, I.; Benhar, I. Design Principles for Bispecific IgGs, Opportunities and Pitfalls of Artificial Disulfide Bonds. Antibodies 2018, 7, 27. https://doi.org/10.3390/antib7030027
Vaks L, Litvak-Greenfeld D, Dror S, Shefet-Carasso L, Matatov G, Nahary L, Shapira S, Hakim R, Alroy I, Benhar I. Design Principles for Bispecific IgGs, Opportunities and Pitfalls of Artificial Disulfide Bonds. Antibodies. 2018; 7(3):27. https://doi.org/10.3390/antib7030027
Chicago/Turabian StyleVaks, Lilach, Dana Litvak-Greenfeld, Stav Dror, LeeRon Shefet-Carasso, Galia Matatov, Limor Nahary, Shiran Shapira, Rahely Hakim, Iris Alroy, and Itai Benhar. 2018. "Design Principles for Bispecific IgGs, Opportunities and Pitfalls of Artificial Disulfide Bonds" Antibodies 7, no. 3: 27. https://doi.org/10.3390/antib7030027
APA StyleVaks, L., Litvak-Greenfeld, D., Dror, S., Shefet-Carasso, L., Matatov, G., Nahary, L., Shapira, S., Hakim, R., Alroy, I., & Benhar, I. (2018). Design Principles for Bispecific IgGs, Opportunities and Pitfalls of Artificial Disulfide Bonds. Antibodies, 7(3), 27. https://doi.org/10.3390/antib7030027