Characterization of Monoclonal Antibody–Protein Antigen Complexes Using Small-Angle Scattering and Molecular Modeling


Abstract
:1. Introduction
2. Results
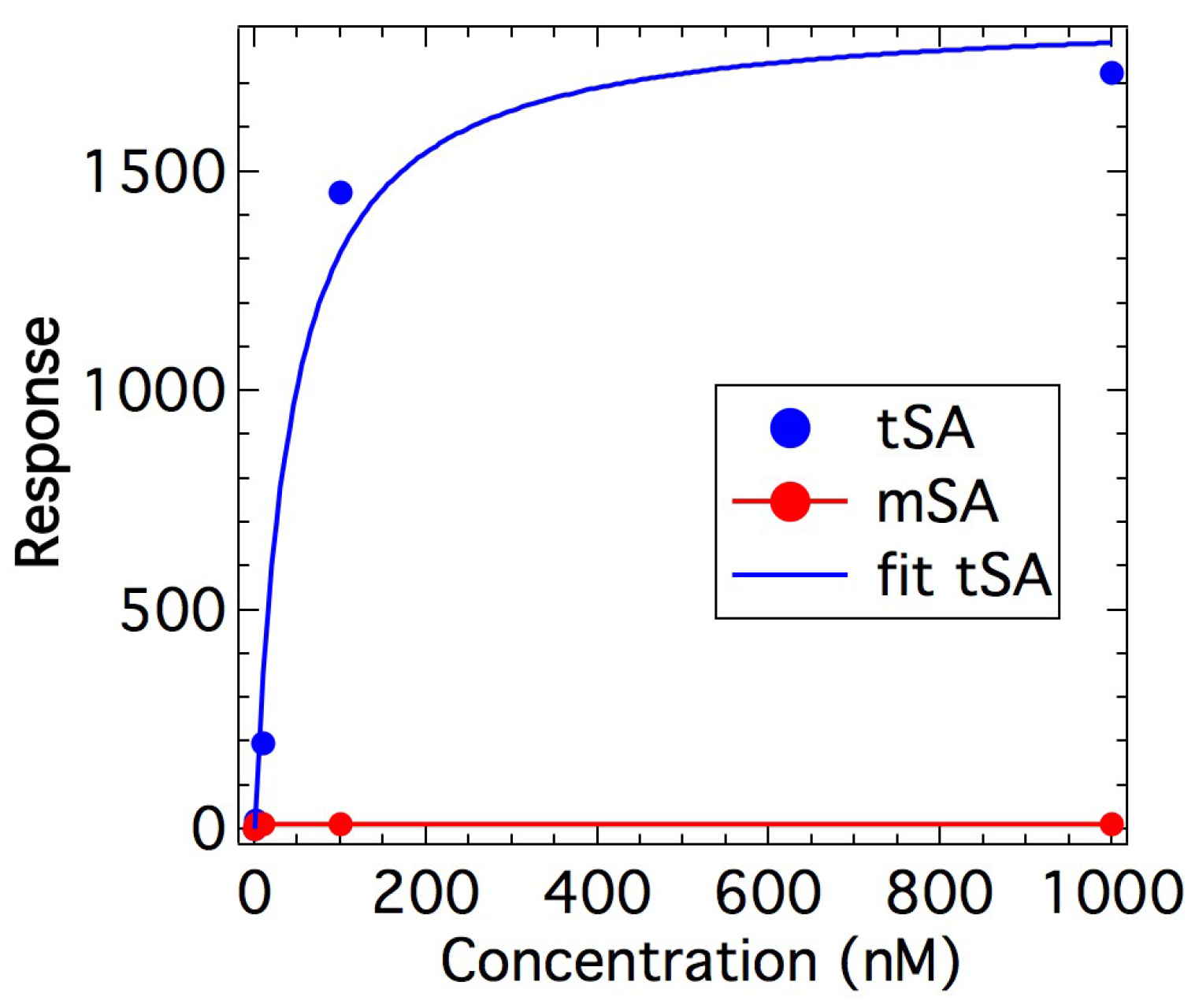
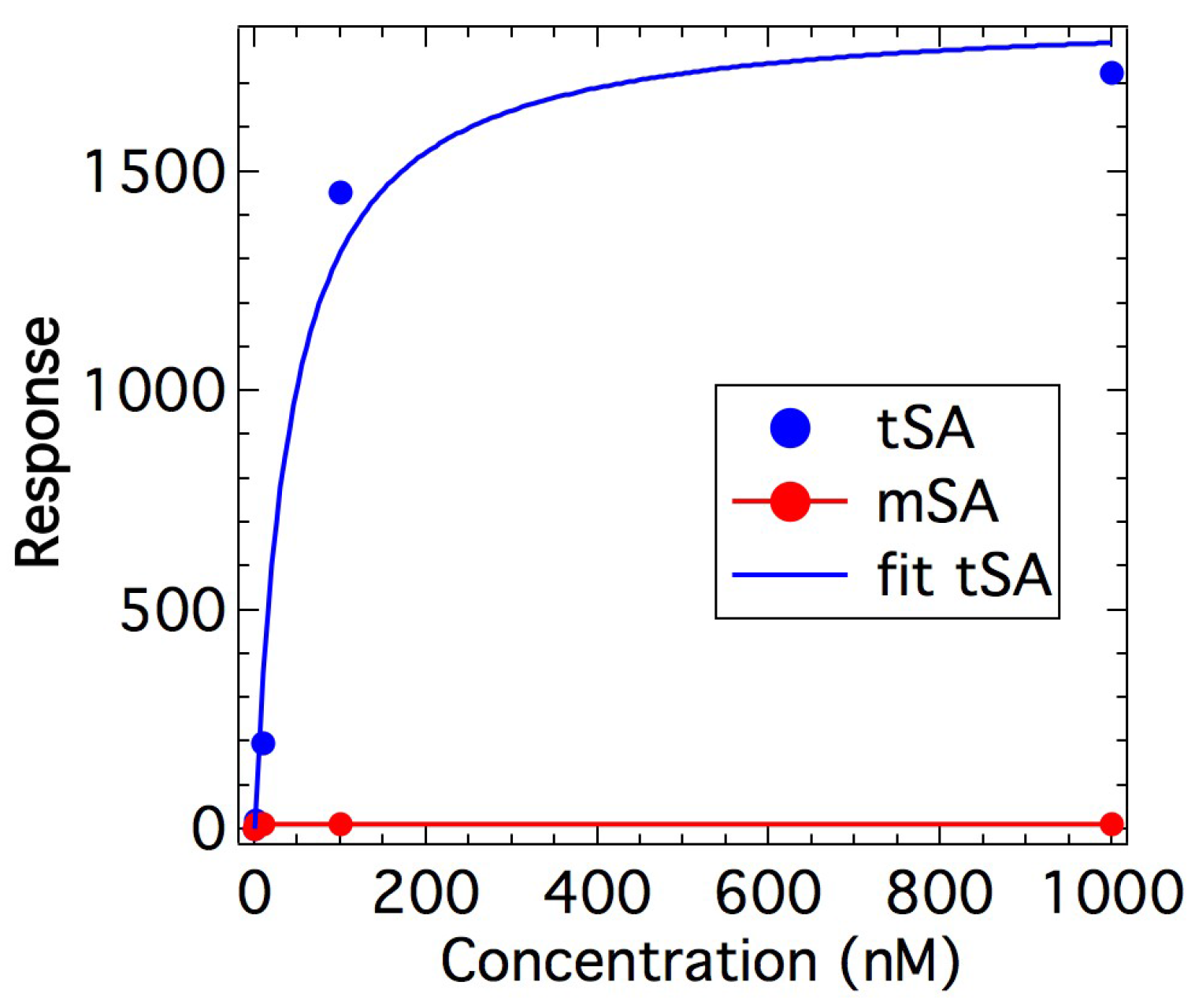
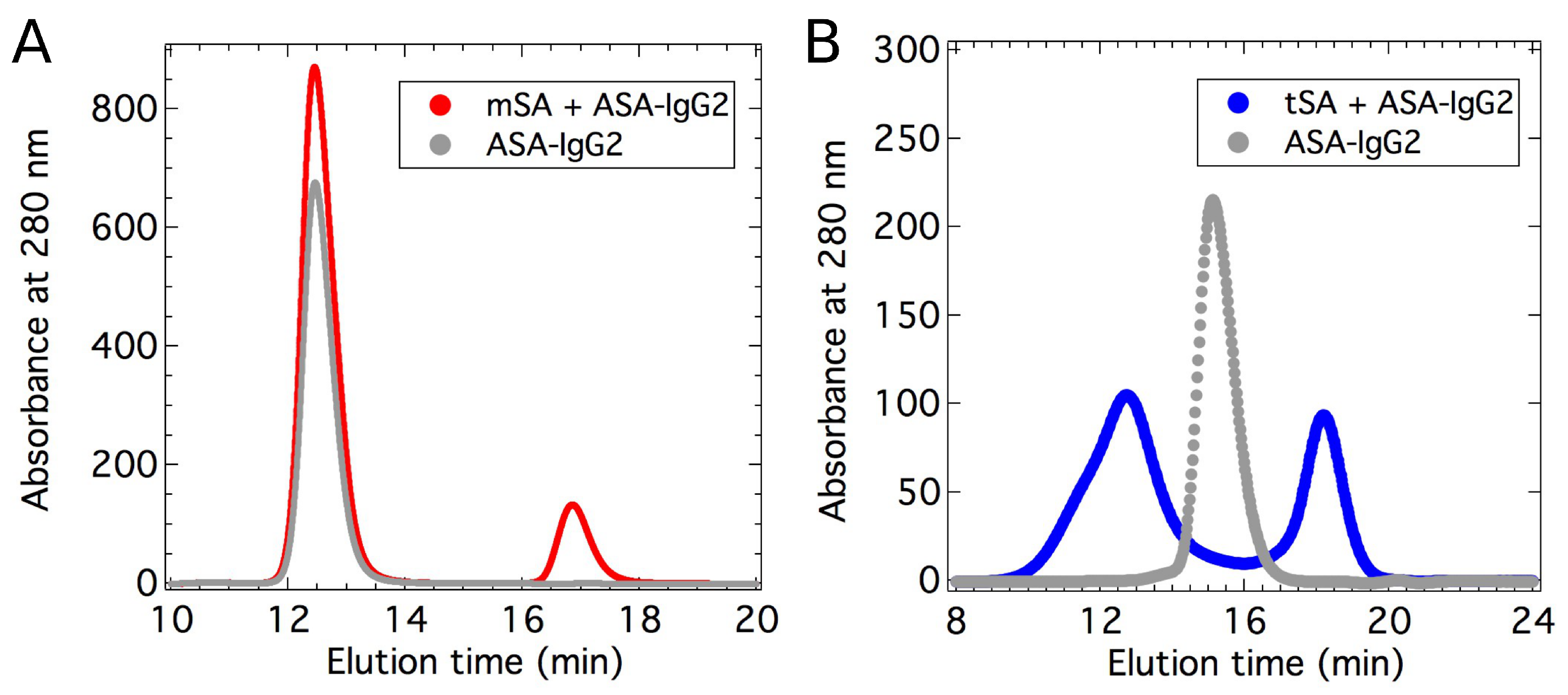
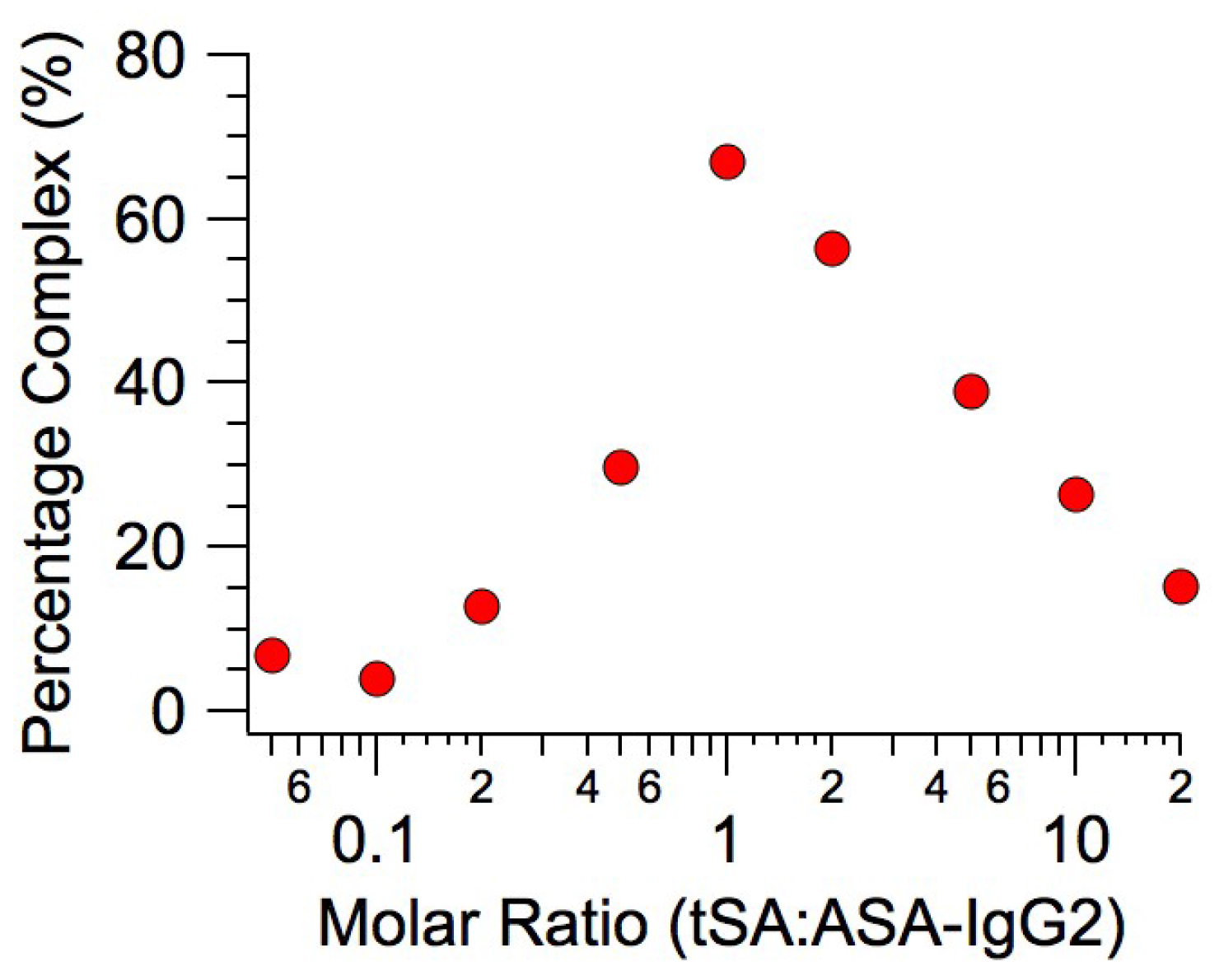
2.1. Binding Affinity Measurements
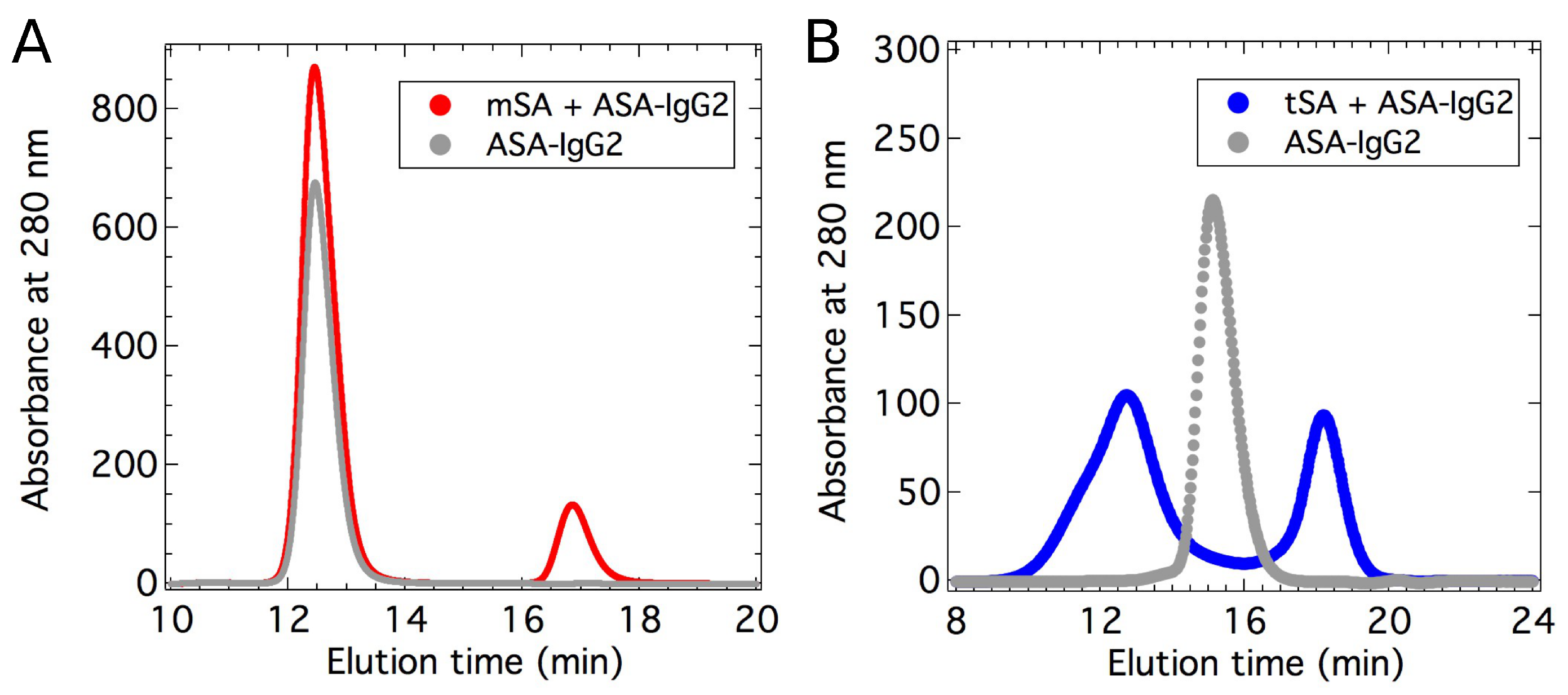
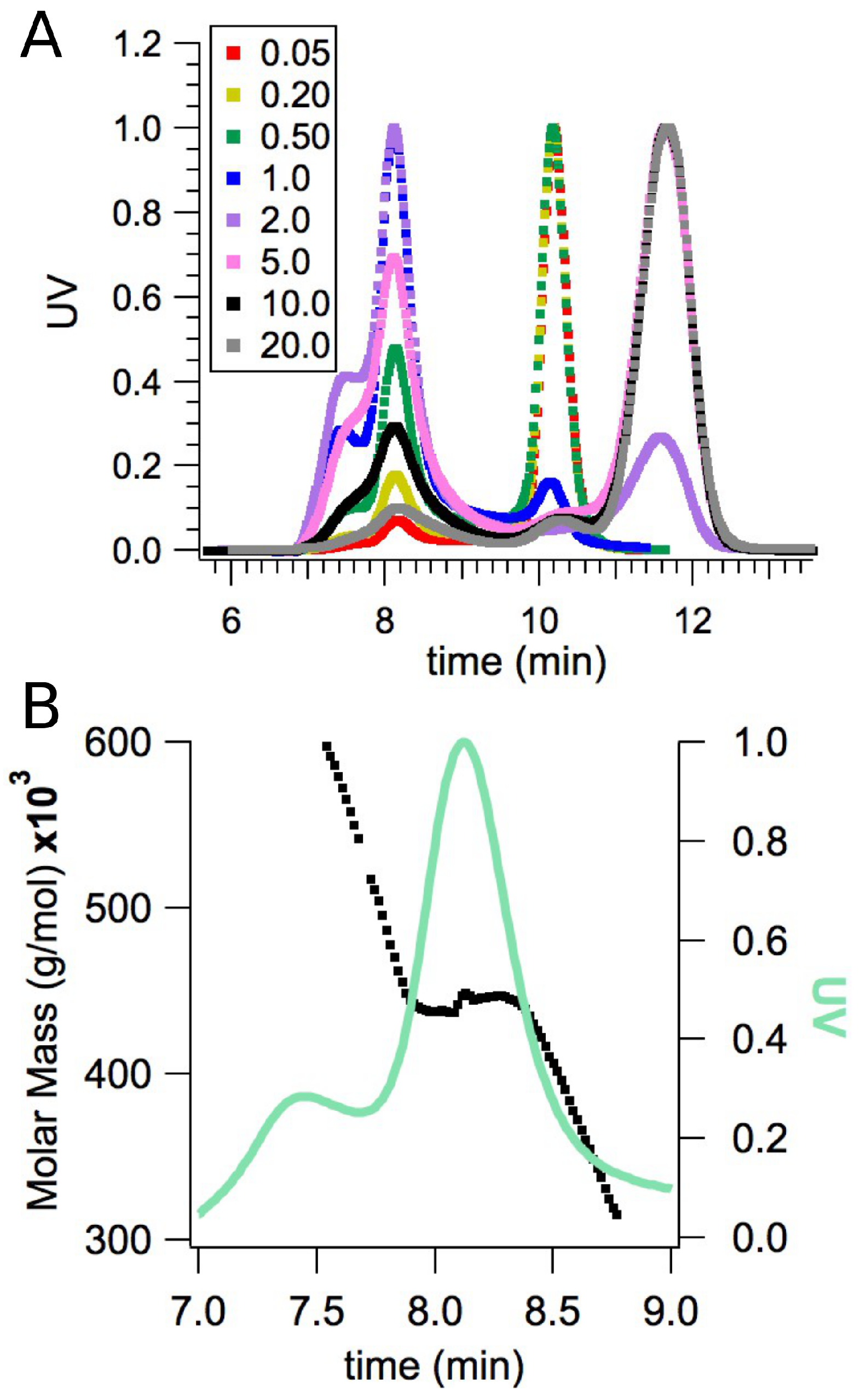
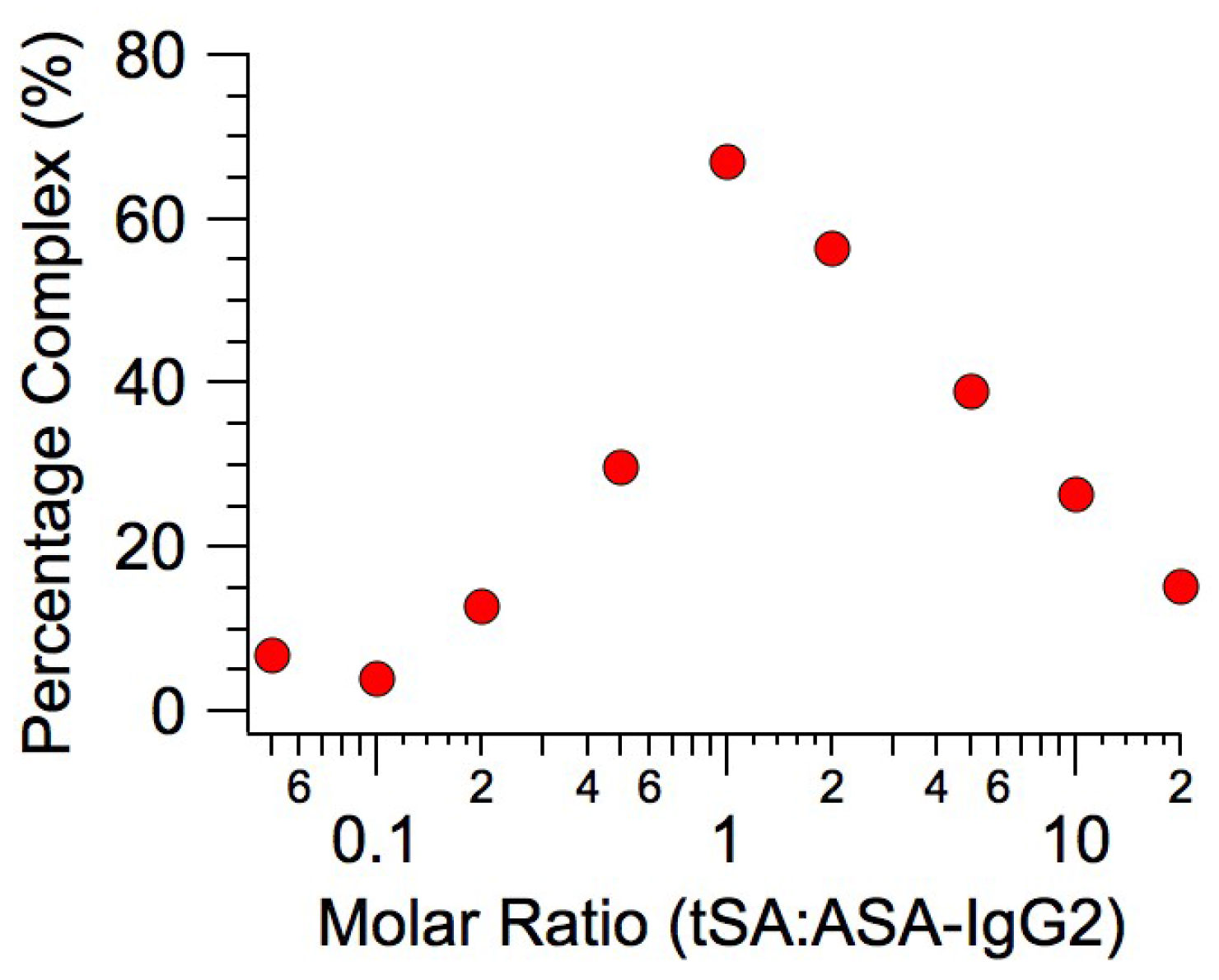
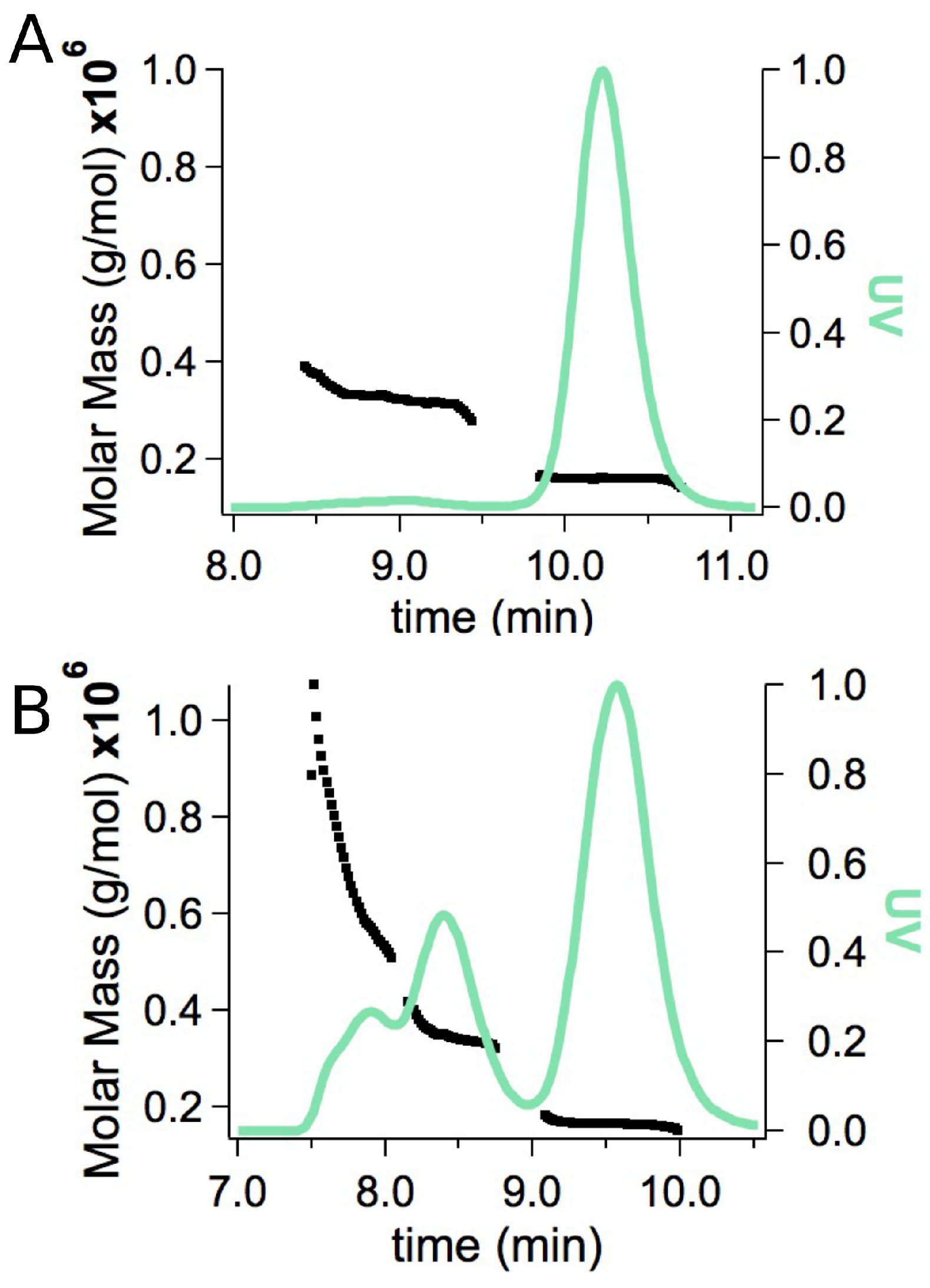
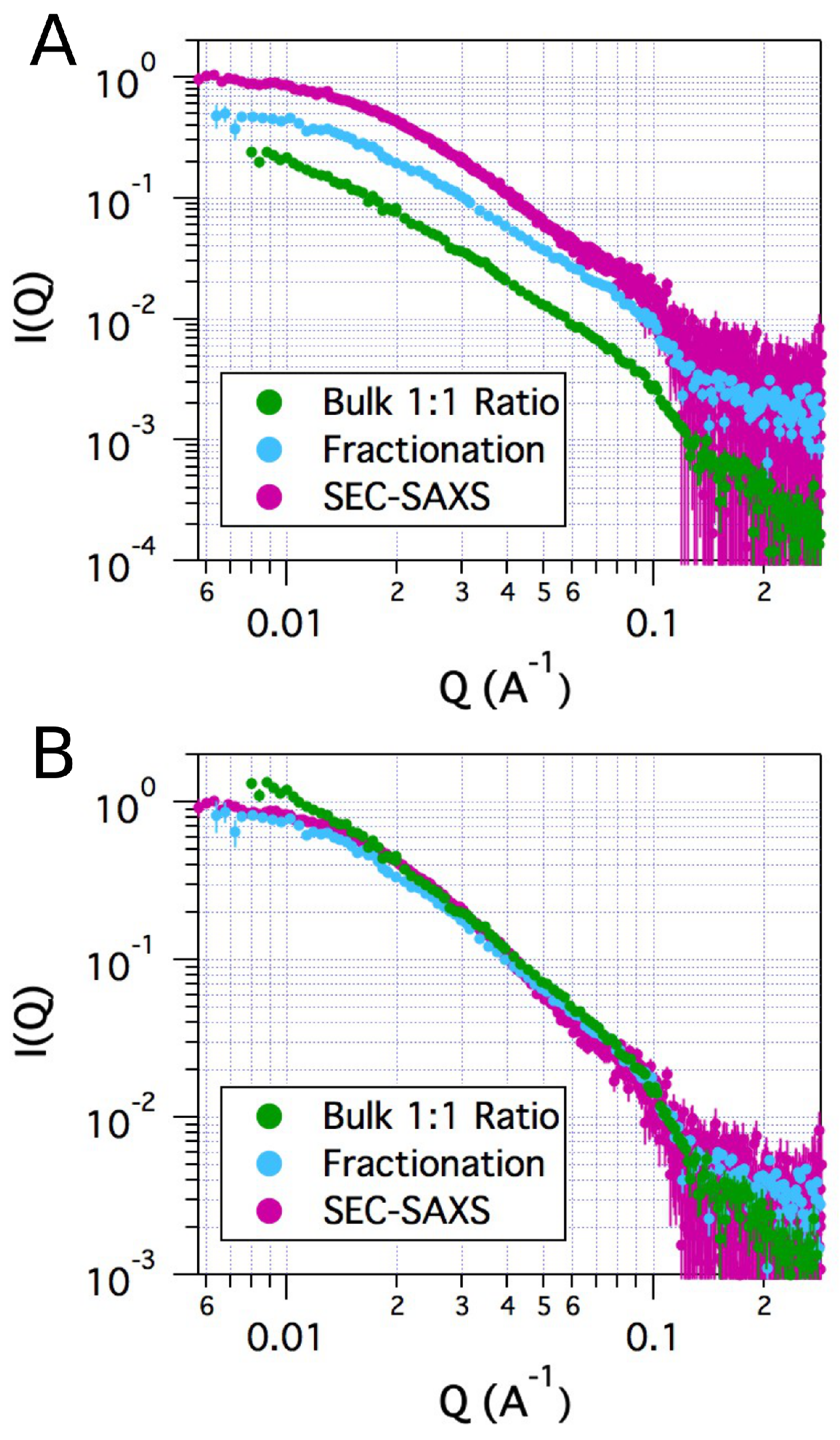
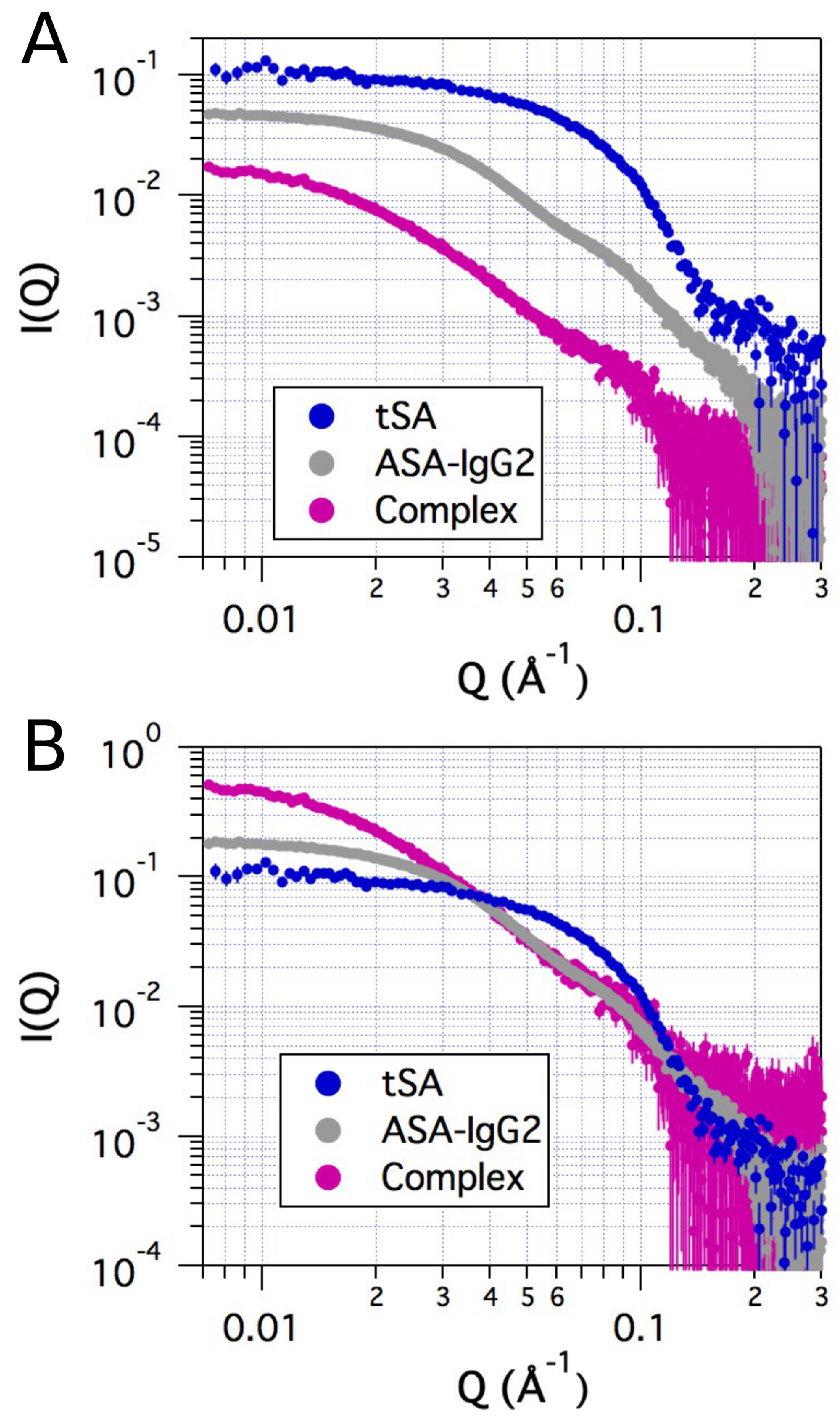
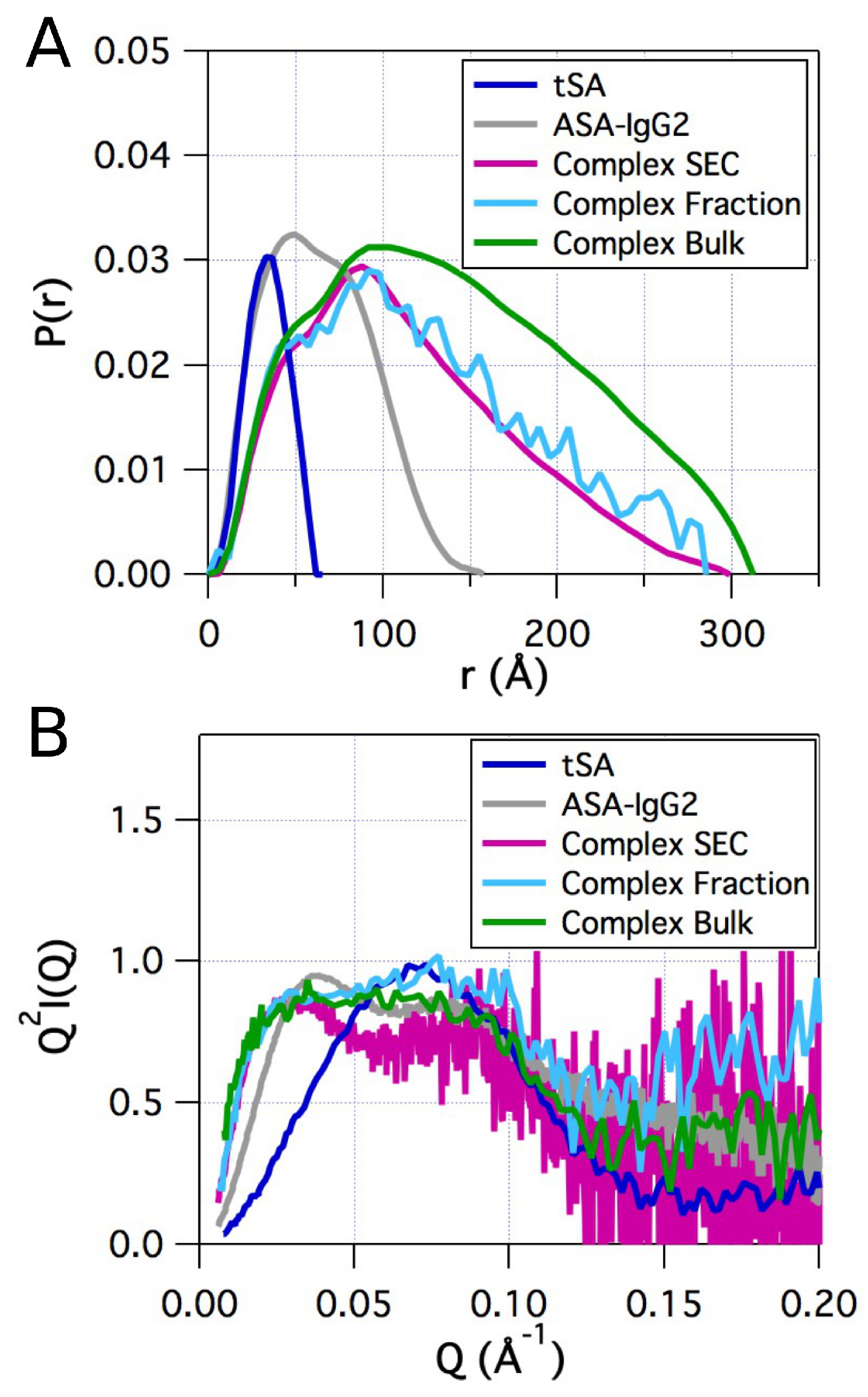
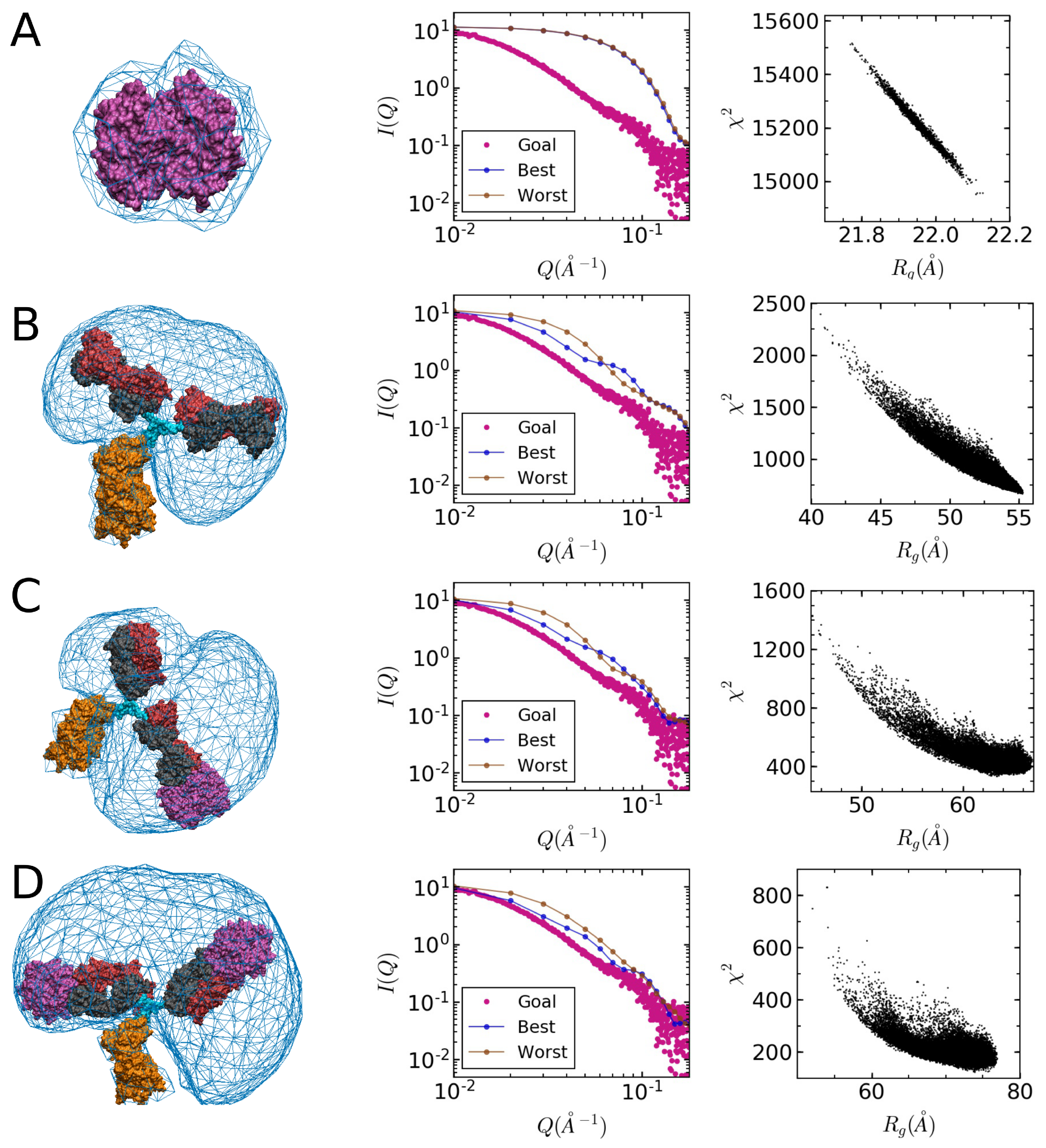
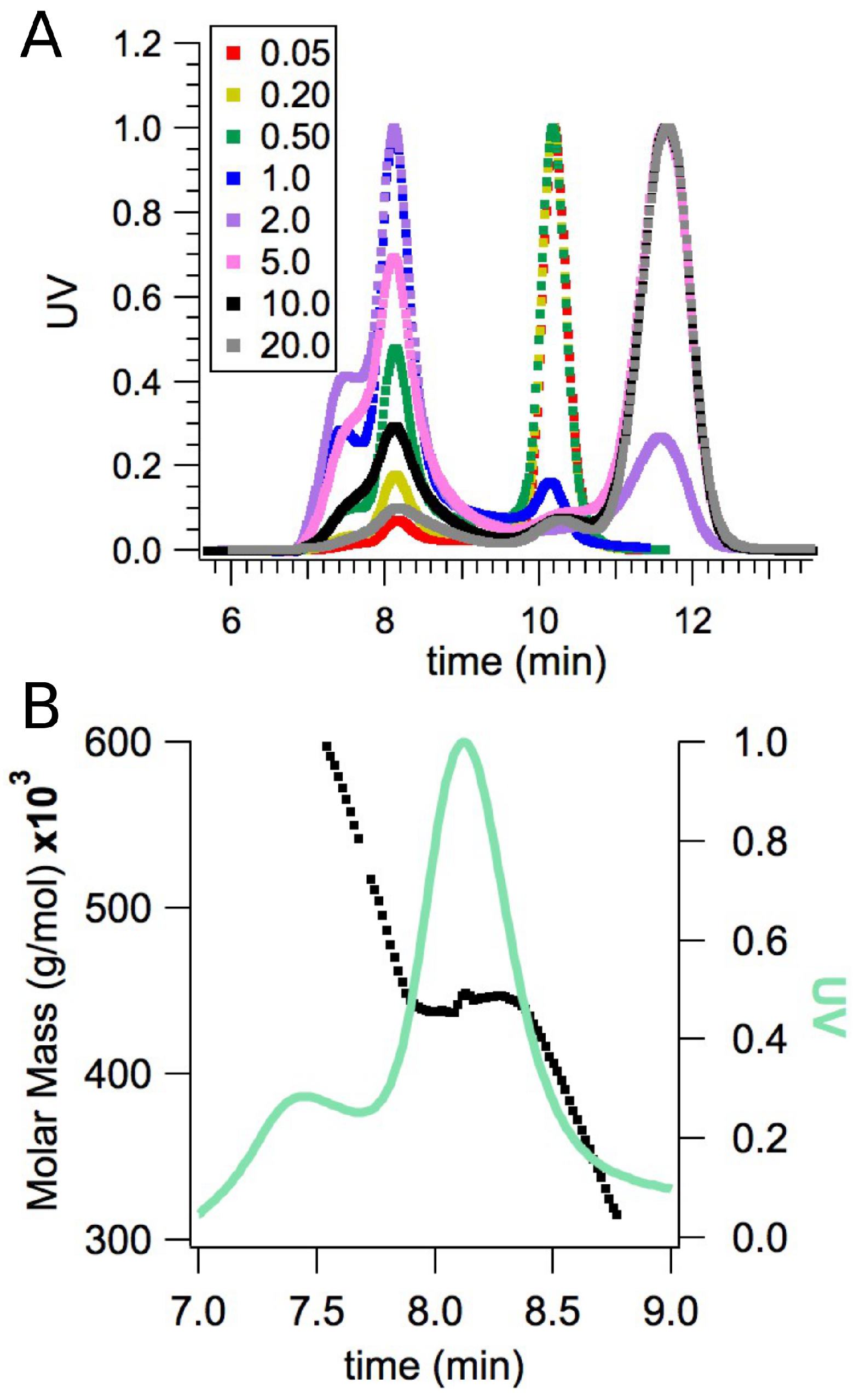
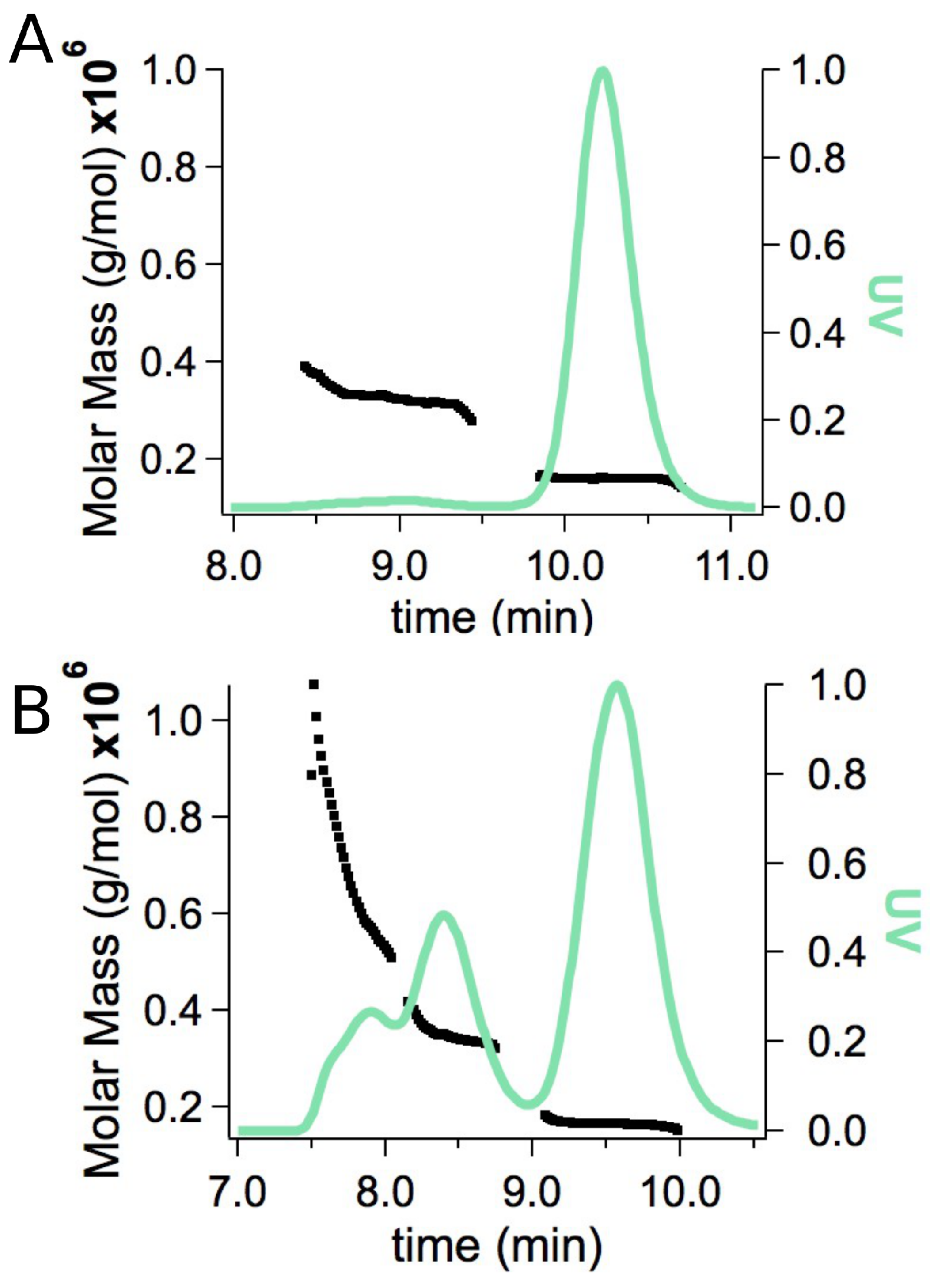
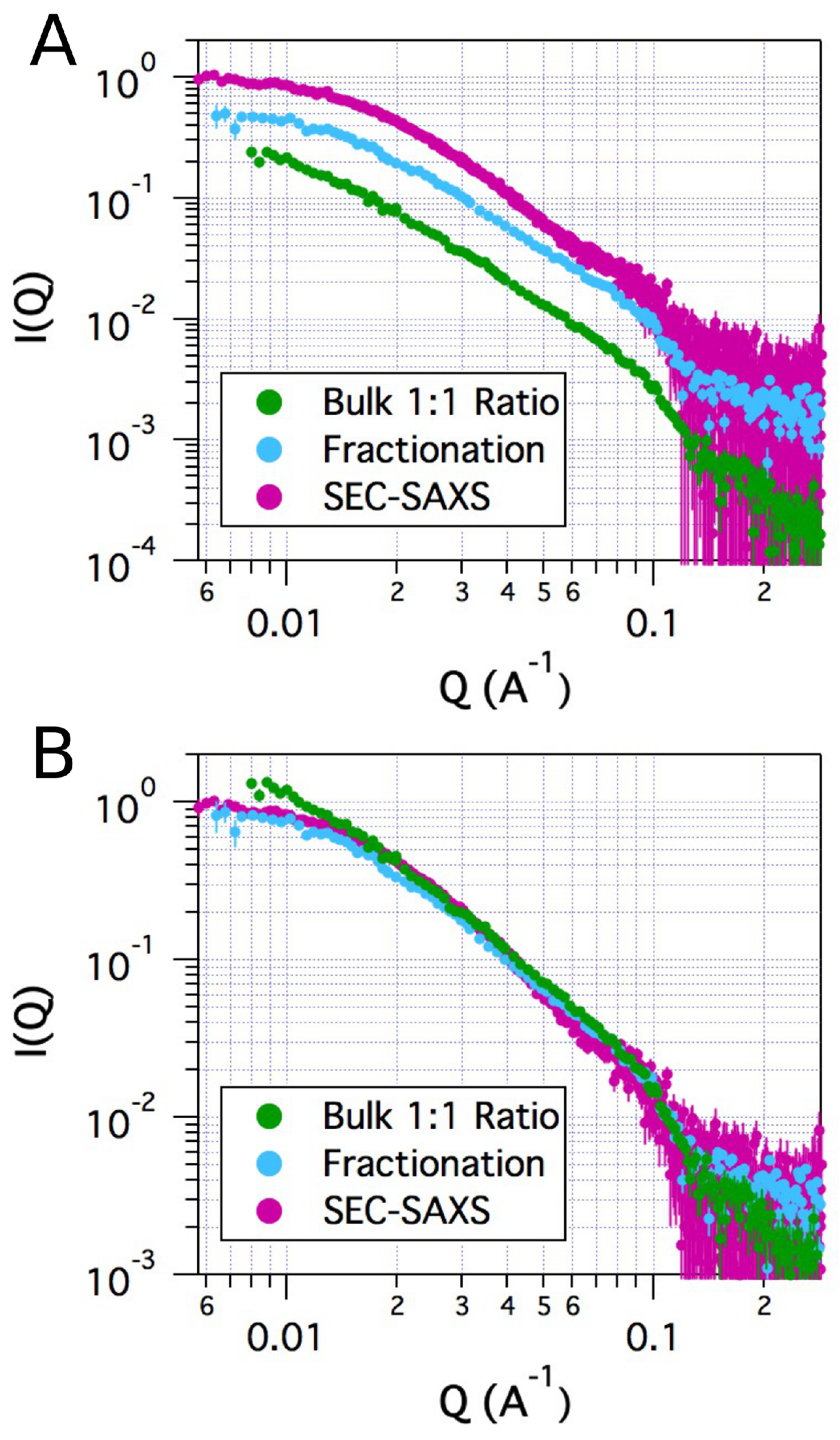
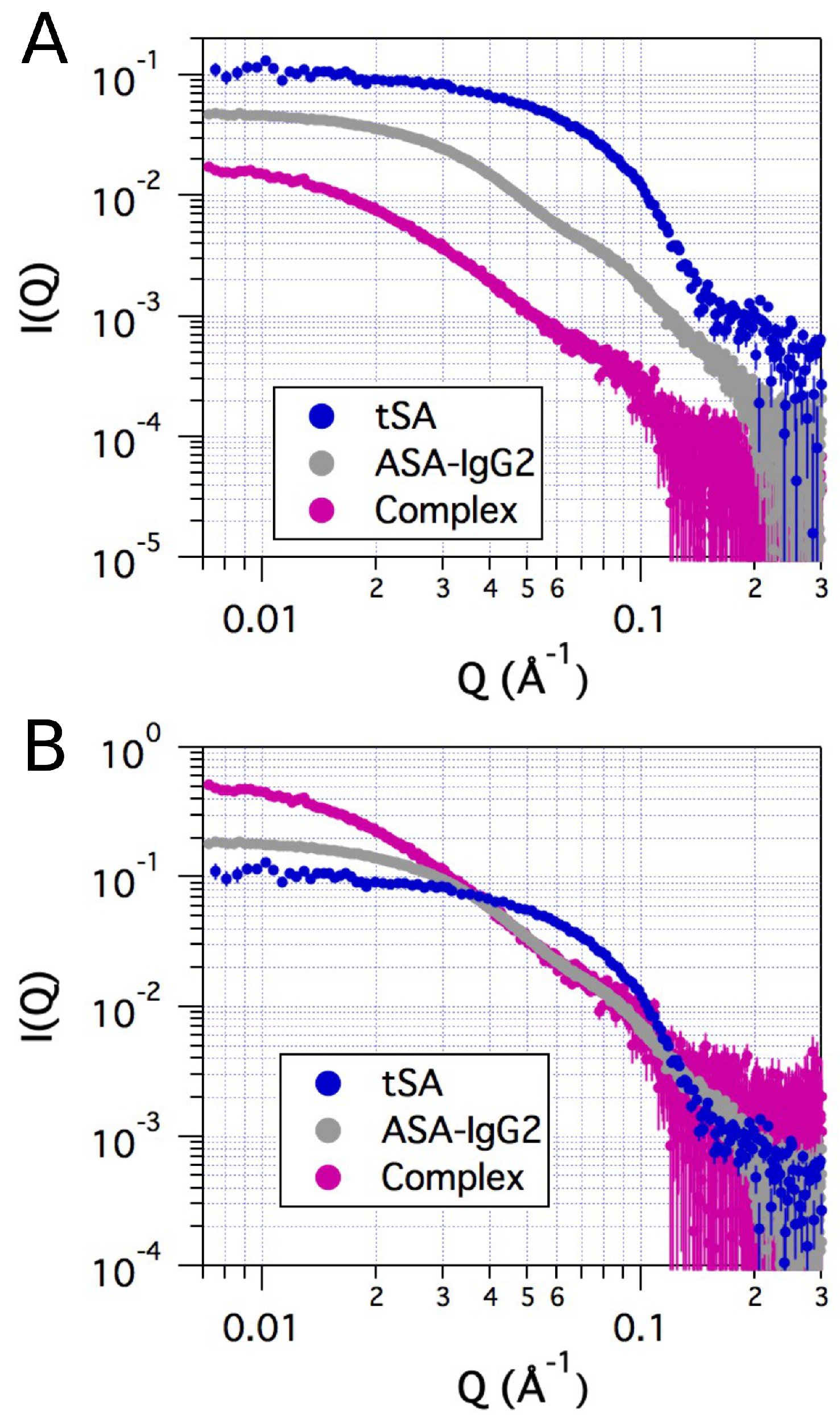
2.2. Small-Angle X-ray Scattering
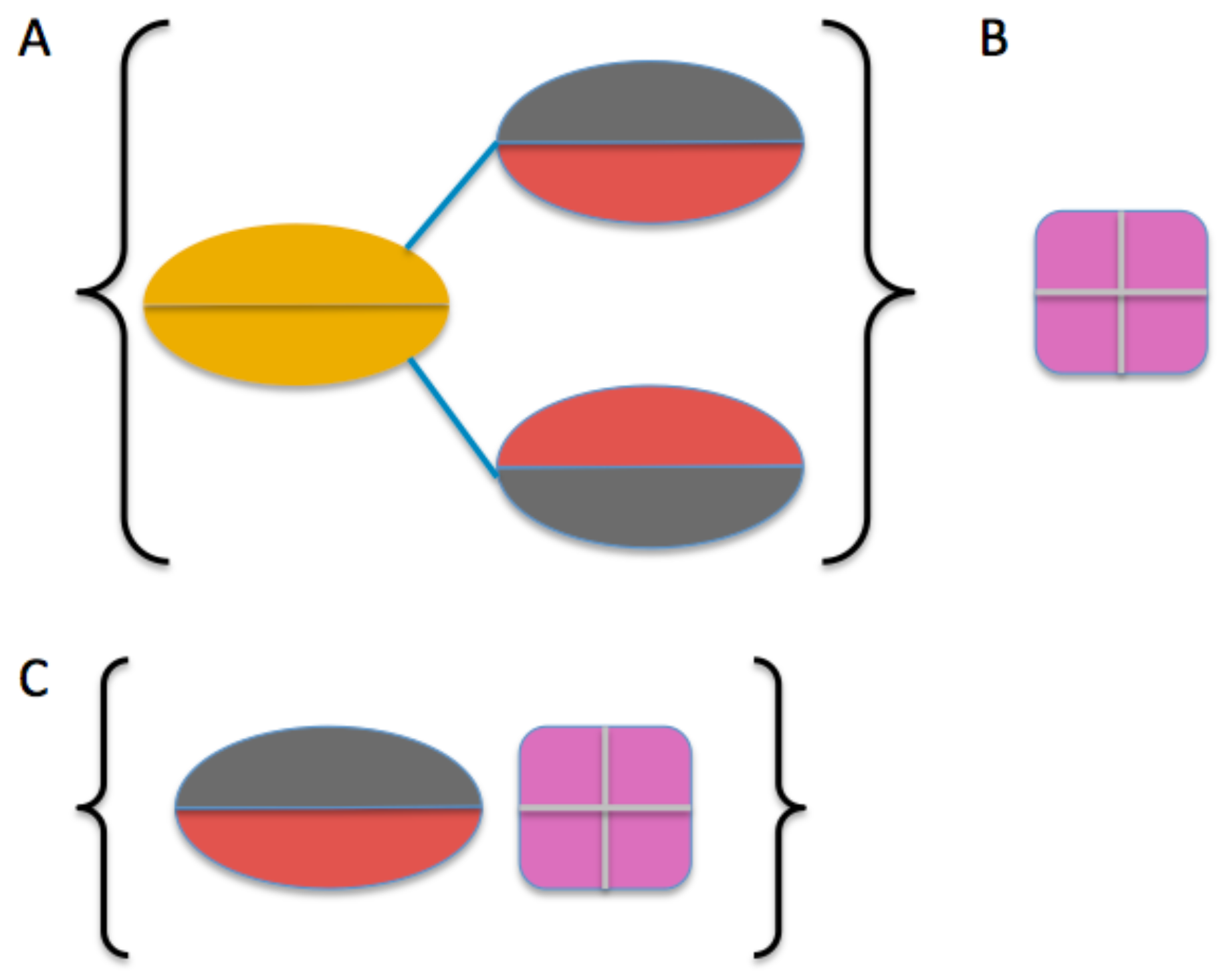
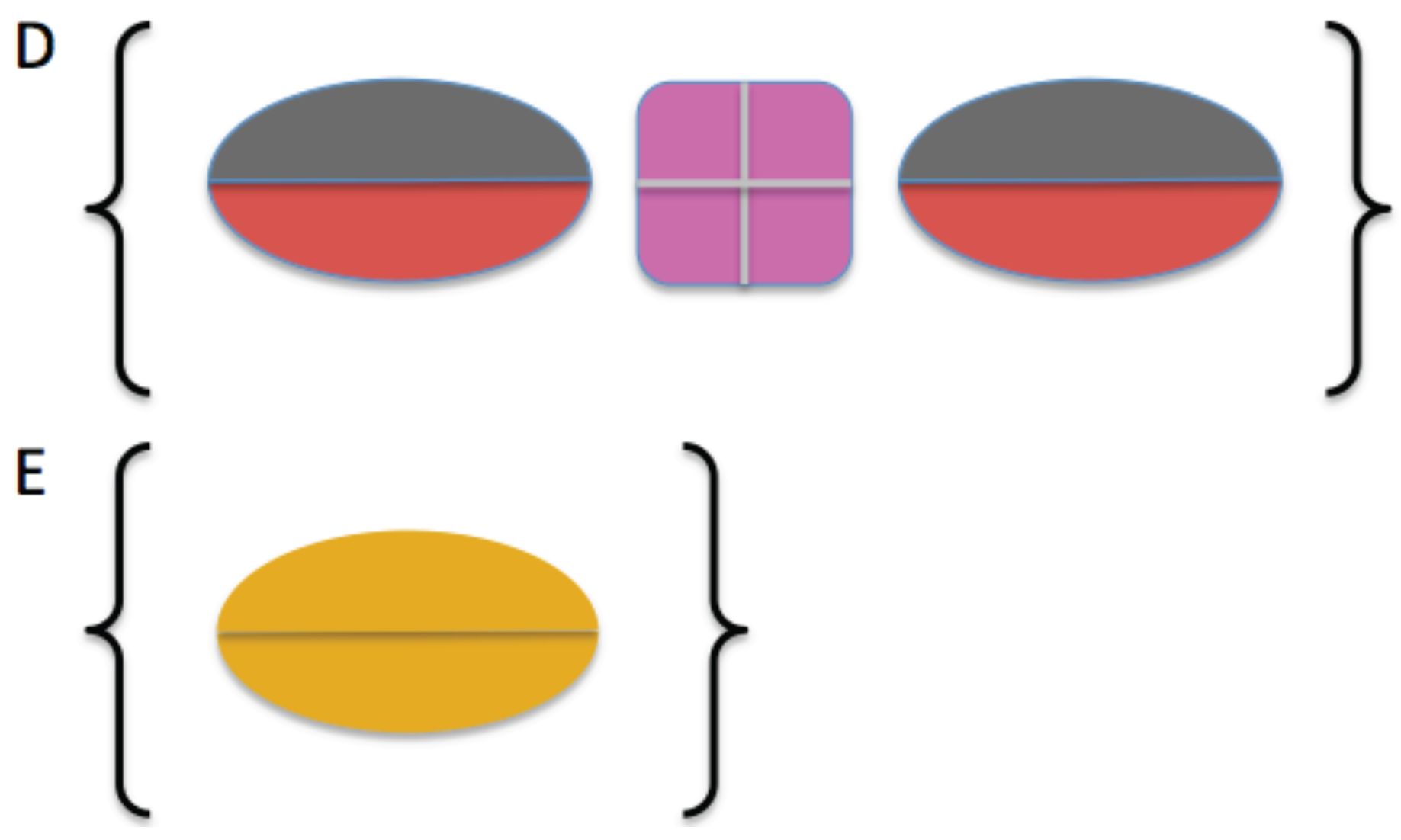
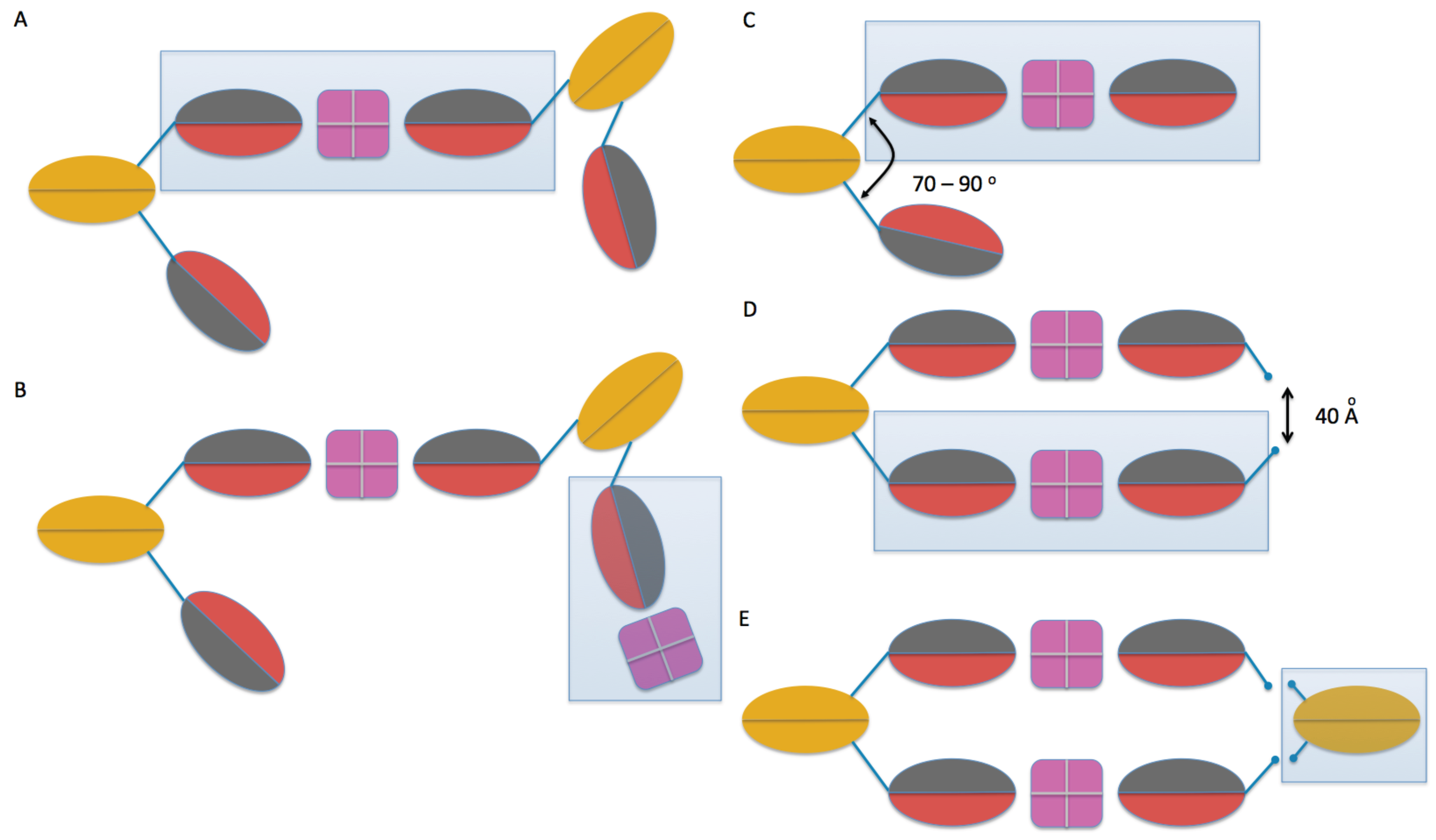
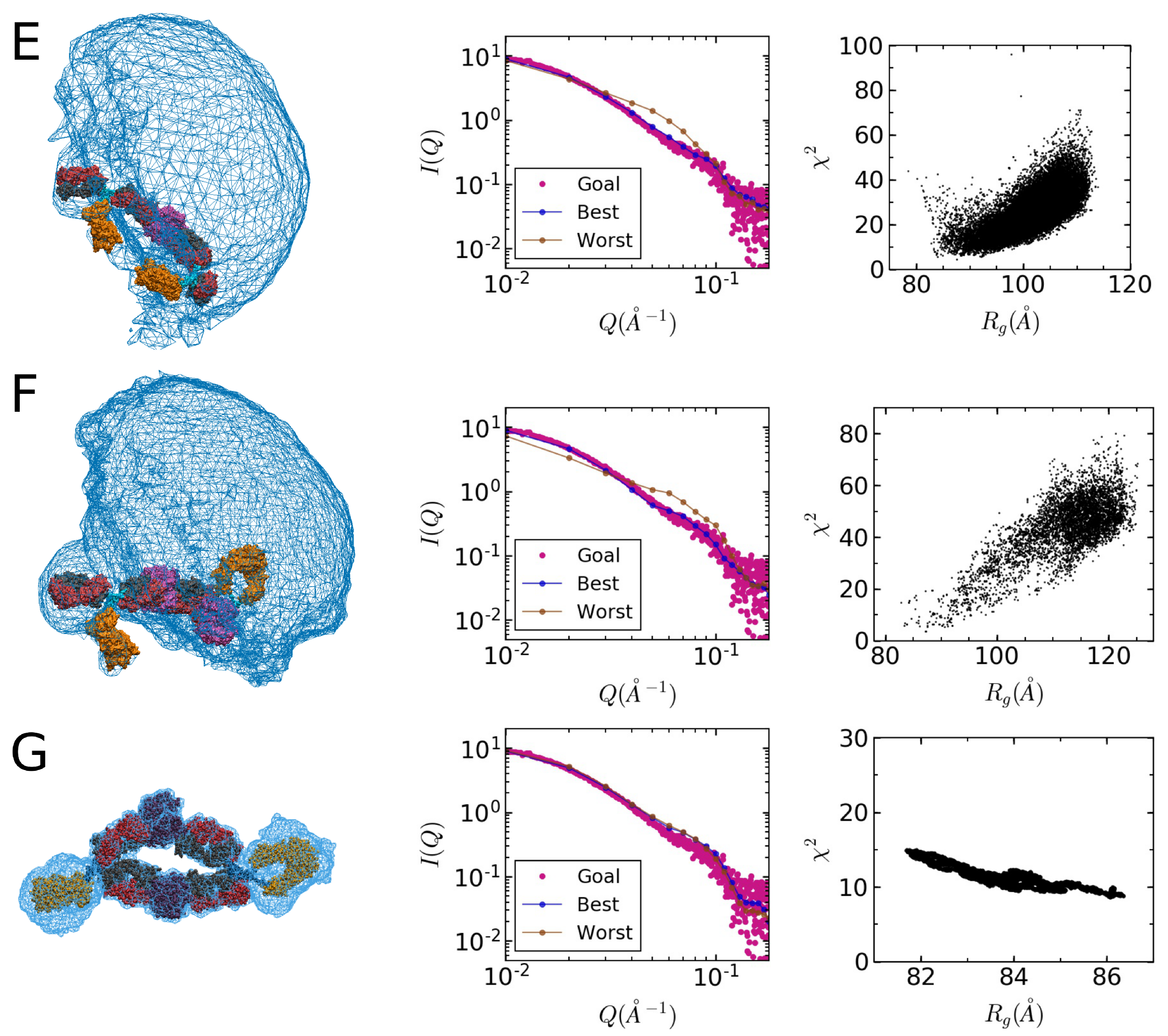
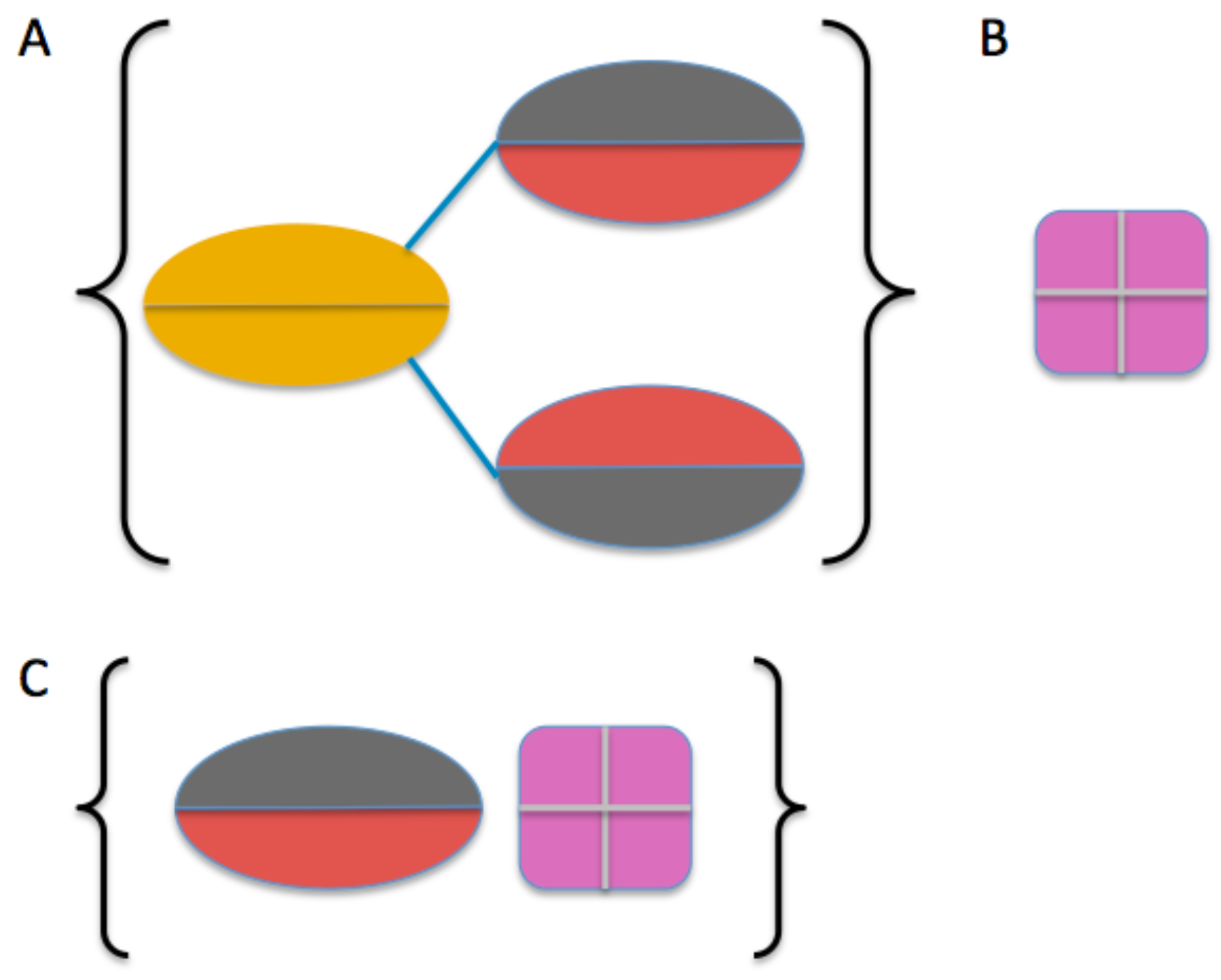
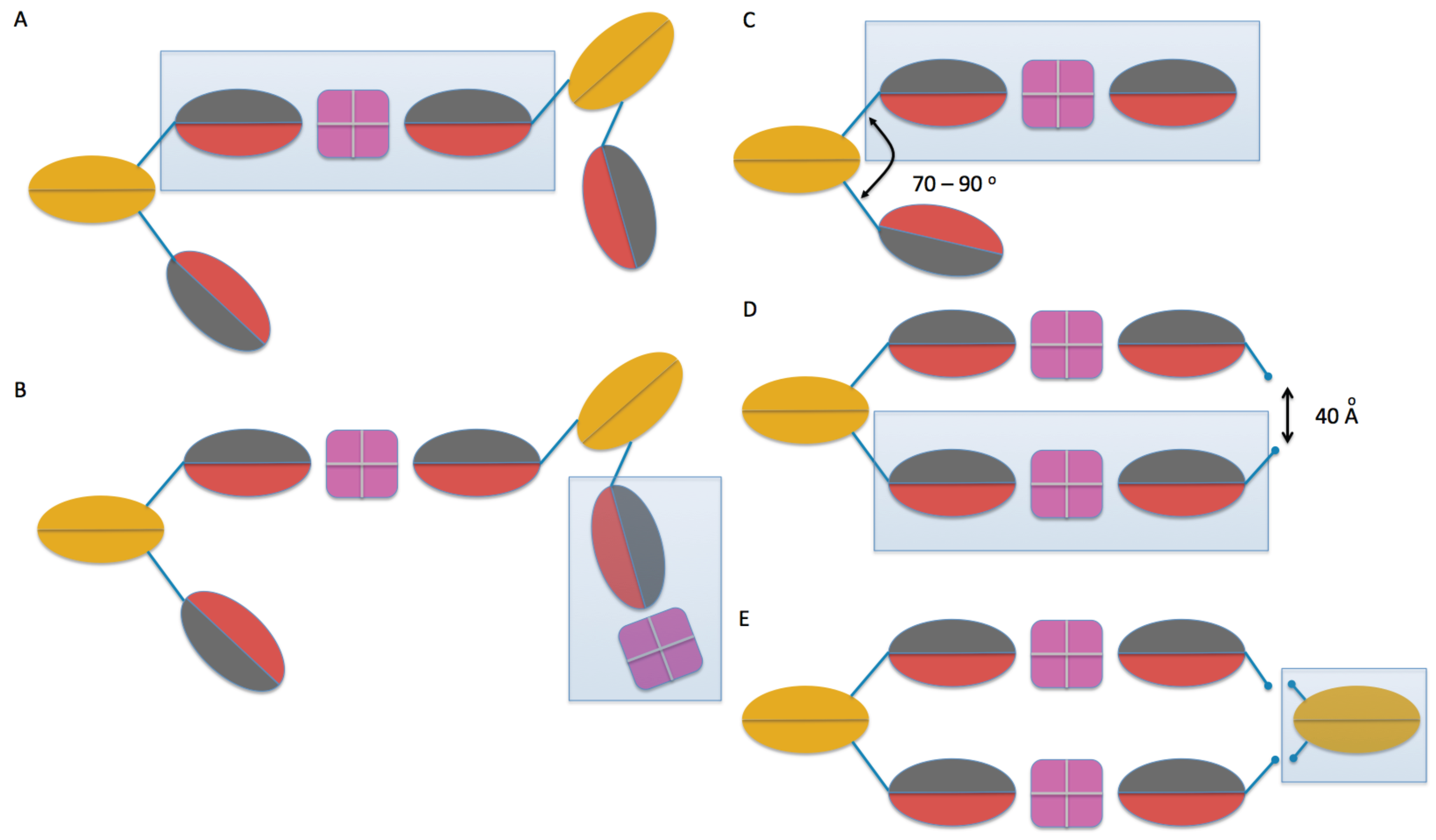
2.3. Model Building
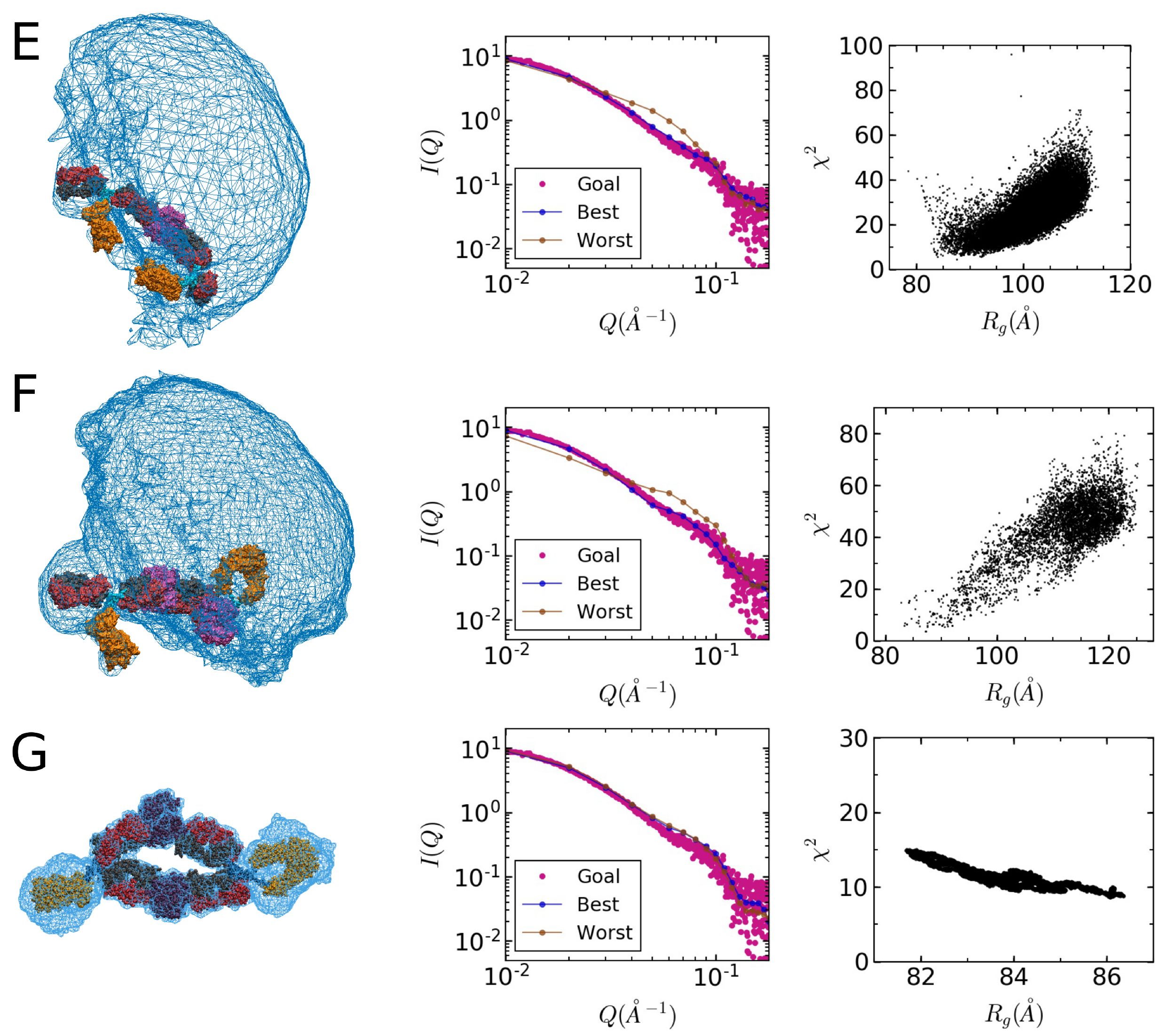
2.4. Comparison of Models to SAXS Data
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. Binding Measurements
4.3. Small-Angle X-ray Scattering Measurements
4.4. Molecular Modeling
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| SAXS | small-angle X-ray scattering |
| SANS | small-angle neutron scattering |
| SAS | small-angle scattering |
| SEC | size-exclusion chromatography |
| SEC-SAXS | size-exclusion chromatography coupled with small-angle X-ray scattering |
| SEC-MALS | size-exclusion chromatography coupled with multi-angle light scattering |
| SPR | surface plasmon resonance |
| MD | molecular dynamics |
| MC | Monte Carlo |
| Fab | antigen-binding region |
| Fc | fragment crystallizable region |
| IgG | immunoglobulin |
| SA | streptavidin |
References
- Egli, M. Diffraction Techniques in Structural Biology. In Current Protocols in Nucleic Acid Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001; Volume 65, pp. 7.13.1–7.13.41. [Google Scholar]
- Shi, Y. A Glimpse of Structural Biology through X-ray Crystallography. Cell 2014, 159, 995–1014. [Google Scholar] [CrossRef] [PubMed]
- Markwick, P.R.L.; Malliavin, T.; Nilges, M. Structural Biology by NMR: Structure, Dynamics, and Interactions. PLoS Comput. Biol. 2008, 4, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Marion, D. An Introduction to Biological NMR Spectroscopy. Mol. Cell. Proteom. 2013, 12, 3006–3025. [Google Scholar] [CrossRef] [PubMed]
- Stuhrmann, H.B.; Miller, A. Small-angle scattering of biological structures. J. Appl. Crystallogr. 1978, 11, 325–345. [Google Scholar] [CrossRef]
- Svergun, D.I.; Koch, M.H.J. Small-angle scattering studies of biological macromolecules in solution. Rep. Prog. Phys. 2003, 66, 1735–1782. [Google Scholar] [CrossRef]
- Jacques, D.A.; Trewhella, J. Small-angle scattering for structural biology—Expanding the frontier while avoiding the pitfalls. Protein Sci. 2010, 19, 642–657. [Google Scholar] [CrossRef] [PubMed]
- Stuhrmann, H.B. Small-angle scattering and its interplay with crystallography, contrast variation in SAXS and SANS. Acta Crystallogr. Sect. A 2008, 64, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Whitten, A.E.; Trewhella, J. Small-Angle Scattering and Neutron Contrast Variation for Studying Bio-Molecular Complexes. In Micro and Nano Technologies in Bioanalysis: Methods and Protocols; Foote, S.R., Lee, W.J., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 307–323. [Google Scholar]
- Krueger, S. Designing and Performing Biologicial Solution Small-Angle Neutron Scattering Contrast Variation Experiments on Multi-component Assemblies. In Biological Small Angle Scattering: Techniques, Strategies and Tips; Chaudhuri, B., Muñoz, I.G., Urban, V., Qian, S., Eds.; Springer: New York, NY, USA, 2017. [Google Scholar]
- Castellanos, M.M.; McAuley, A.; Curtis, J.E. Investigating Structure and Dynamics of Proteins in Amorphous Phases Using Neutron Scattering. Comput. Struct. Biotechnol. J. 2017, 15, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Perkins, S.J.; Bonner, A. Structure determinations of human and chimaeric antibodies by solution scattering and constrained molecular modelling. Biochem. Soc. Trans. 2008, 36, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Perkins, S.J.; Okemefuna, A.I.; Nan, R.; Li, K.; Bonner, A. Constrained solution scattering modelling of human antibodies and complement proteins reveals novel biological insights. J. R. Soc. Interface 2009, 6, S679–S696. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Gor, J.; Bracewell, D.G.; Perkins, S.J.; Dalby, P.A. Masking of the Fc region in human IgG4 by constrained X-ray scattering modelling: Implications for antibody function and therapy. Biochem. J. 2010, 432, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Ashish; Solanki, A.K.; Boone, C.D.; Krueger, J.K. Global structure of HIV-1 neutralizing antibody IgG1 b12 is asymmetric. Biochem. Biophys. Res. Commun. 2010, 391, 947–951. [Google Scholar] [CrossRef] [PubMed]
- Mosbæk, C.R.; Konarev, P.V.; Svergun, D.I.; Rischel, C.; Vestergaard, B. High concentration formulation studies of an IgG2 antibody using small angle X-ray scattering. Pharm. Res. 2012, 29, 2225–2235. [Google Scholar] [CrossRef] [PubMed]
- Lilyestrom, W.G.; Shire, S.J.; Scherer, T.M. Influence of the cosolute environment on IgG solution structure analyzed by small-angle X-ray scattering. J. Phys. Chem. B 2012, 116, 9611–9618. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.J.; Zhang, H.; Krueger, S.; Lee, H.J.; Ketchem, R.R.; Kerwin, B.; Kanapuram, S.R.; Treuheit, M.J.; McAuley, A.; Curtis, J.E. Small-Angle Neutron Scattering Study of a Monoclonal Antibody Using Free-Energy Constraints. J. Phys. Chem. B 2013, 117, 14029–14038. [Google Scholar] [CrossRef] [PubMed]
- Castellanos, M.M.; Pathak, J.A.; Leach, W.; Bishop, S.M.; Colby, R.H. Explaining the non-Newtonian Character of Aggregating Monoclonal Antibody Solutions Using Small-Angle Neutron Scattering. Biophys. J. 2014, 107, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Langkilde, A.E.; Thorolfsson, M.; Rasmussen, H.B.; Vestergaard, B. Small-Angle X-ray Scattering Screening Complements Conventional Biophysical Analysis: Comparative Structural and Biophysical Analysis of Monoclonal Antibodies IgG1, IgG2, and IgG4. J. Pharm. Sci. 2017, 103, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Vestergaard, B.; Thorolfsson, M.; Yang, Z.; Rasmussen, H.B.; Langkilde, A.E. In-depth analysis of subclass-specific conformational preferences of IgG antibodies. IUCrJ 2015, 2, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Rayner, L.E.; Hui, G.K.; Gor, J.; Heenan, R.K.; Dalby, P.A.; Perkins, S.J. The Solution Structures of Two Human IgG1 Antibodies Show Conformational Stability and Accommodate Their C1q and FcγR Ligands. J. Biol. Chem. 2015, 290, 8420–8438. [Google Scholar] [CrossRef] [PubMed]
- Yearley, E.; Zarraga, I.; Shire, S.; Scherer, T.; Gokarn, Y.; Wagner, N.; Liu, Y. Small-Angle Neutron Scattering Characterization of Monoclonal Antibody Conformations and Interactions at High Concentrations. Biophys. J. 2013, 105, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Yearley, E.J.; Godfrin, P.D.; Perevozchikova, T.; Zhang, H.; Falus, P.; Porcar, L.; Nagao, M.; Curtis, J.E.; Gawande, P.; Taing, R.; et al. Observation of Small Cluster Formation in Concentrated Monoclonal Antibody Solutions and Its Implications to Solution Viscosity. Biophys. J. 2014, 106, 1763–1770. [Google Scholar] [CrossRef] [PubMed]
- Hui, G.K.; Wright, D.W.; Vennard, O.L.; Rayner, L.E.; Pang, M.; Yeo, S.C.; Gor, J.; Molyneux, K.; Barratt, J.; Perkins, S.J. The solution structures of native and patient monomeric human IgA1 reveal asymmetric extended structures: Implications for function and IgAN disease. Biochem. J. 2015, 471, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Castellanos, M.M.; Clark, N.J.; Watson, M.C.; Krueger, S.; McAuley, A.; Curtis, J.E. Role of Molecular Flexibility and Colloidal Descriptions of Proteins in Crowded Environments from Small-Angle Scattering. J. Phys. Chem. B 2016, 120, 12511–12518. [Google Scholar] [CrossRef] [PubMed]
- Yguerabide, J.; Epstein, H.F.; Stryer, L. Segmental flexibility in an antibody molecule. J. Mol. Biol. 1970, 51, 573–590. [Google Scholar] [CrossRef]
- McCammon, J.A.; Karplus, M. Internal motions of antibody molecules. Nature 1977, 268, 765–766. [Google Scholar] [CrossRef] [PubMed]
- Hanson, D.C.; Yguerabide, J.; Schumaker, V.N. Segmental flexibility of immunoglobulin G antibody molecules in solution: A new interpretation. Biochemistry 1981, 20, 6842–6852. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.J.; Skaletsky, E.; McPherson, A. Crystallographic structure of an intact IgG1 monoclonal antibody. J. Mol. Biol. 1998, 275, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Saphire, E.O.; Parren, P.W.H.I.; Barbas, C.F., III; Burton, D.R.; Wilson, I.A. Crystallization and preliminary structure determination of an intact human immunoglobulin, b12: An antibody that broadly neutralizes primary isolates of HIV-1. Acta Crystallogr. Sect. D 2001, 57, 168–171. [Google Scholar] [CrossRef]
- Harris, L.J.; Larson, S.B.; Hasel, K.W.; Day, J.; Greenwood, A.; McPherson, A. The three-dimensional structure of an intact monoclonal antibody for canine lymphoma. Nature 1992, 360, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Chaves, R.C.; Teulon, J.M.; Odorico, M.; Parot, P.; Chen, S.W.W.; Pellequer, J.L. Conformational dynamics of individual antibodies using computational docking and AFM. J. Mol. Recognit. 2013, 26, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, L.; Tong, H.; Peng, B.; Rames, M.J.; Zhang, S.; Ren, G. 3D Structural Fluctuation of IgG1 Antibody Revealed by Individual Particle Electron Tomography. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, P.; Friguet, B.; Djavadi-Ohaniance, L.; Pilz, I.; Goldberg, M.E. Epitope localization in antigen-monoclonal-antibody complexes by small-angle X-ray scattering. Eur. J. Biochem. 1987, 164, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Tuncbag, N.; Gursoy, A. Predicting Protein–Protein Interactions from the Molecular to the Proteome Level. Chem. Rev. 2016, 116, 4884–4909. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.E.; Grossman, J.P.; Bank, J.A.; Batson, B.; Butts, J.A.; Chao, J.C.; Deneroff, M.M.; Dror, R.O.; Even, A.; Fenton, C.H.; et al. Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer. In Proceedings of the SC14: International Conference for High Performance Computing, Networking, Storage and Analysis, New Orleans, LA, USA, 16–21 November 2014; pp. 41–53. [Google Scholar]
- Friedrichs, M.S.; Eastman, P.; Vaidyanathan, V.; Houston, M.; Legrand, S.; Beberg, A.L.; Ensign, D.L.; Bruns, C.M.; Pande, V.S. Accelerating Molecular Dynamic Simulation on Graphics Processing Units. J. Comput. Chem. 2009, 30, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.E.; Phillips, J.C.; Freddolino, P.L.; Hardy, D.J.; Trabuco, L.G.; Schulten, K. Accelerating molecular modeling applications with graphics processors. J. Comput. Chem. 2007, 28, 2618–2640. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.P.; Patapoff, T.W.; Aragon, S.R. Construction, {MD} Simulation, and Hydrodynamic Validation of an All-Atom Model of a Monoclonal IgG Antibody. Biophys. J. 2010, 99, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, M.E.; Colina, C.M. Effects of Galactosylation in Immunoglobulin G from All-Atom Molecular Dynamics Simulations. J. Phys. Chem. B 2014, 118, 9844–9851. [Google Scholar] [CrossRef] [PubMed]
- Lapelosa, M.; Patapoff, T.W.; Zarraga, I.E. Molecular Simulations of the Pairwise Interaction of Monoclonal Antibodies. J. Phys. Chem. B 2014, 118, 13132–13141. [Google Scholar] [CrossRef] [PubMed]
- Castellanos, M.M.; Howell, S.; Gallagher, D.T.; Curtis, J.E. Characterization of the NISTmAb Reference Material using Small-Angle Scattering and Molecular Simulation Part I: Dilute Solution Structures. Anal. Bioanal. Chem. 2017. [Google Scholar] [CrossRef]
- Chaudhri, A.; Zarraga, I.E.; Kamerzell, T.J.; Brandt, J.P.; Patapoff, T.W.; Shire, S.J.; Voth, G.A. Coarse-Grained Modeling of the Self-Association of Therapeutic Monoclonal Antibodies. J. Phys. Chem. B 2012, 116, 8045–8057. [Google Scholar] [CrossRef] [PubMed]
- Franco-Gonzalez, J.F.; Ramos, J.; Cruz, V.L.; Martinez-Salazar, J. Exploring the dynamics and interaction of a full ErbB2 receptor and Trastuzumab-Fab antibody in a lipid bilayer model using Martini coarse-grained force field. J. Comput.-Aided Mol. Des. 2014, 28, 1093–1107. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tsao, H.K.; Sheng, Y.J.; Jiang, S. Monte Carlo simulations of antibody adsorption and orientation on charged surfaces. J. Chem. Phys. 2004, 121, 1050–1057. [Google Scholar] [CrossRef] [PubMed]
- Calero-Rubio, C.; Saluja, A.; Roberts, C.J. Coarse-Grained Antibody Models for “Weak” Protein–Protein Interactions from Low to High Concentrations. J. Phys. Chem. B 2016, 120, 6592–6605. [Google Scholar] [CrossRef] [PubMed]
- De Michele, C.; De Los Rios, P.; Foffi, G.; Piazza, F. Simulation and Theory of Antibody Binding to Crowded Antigen-Covered Surfaces. PLoS Comput. Biol. 2016, 12, e1004752. [Google Scholar] [CrossRef] [PubMed]
- Corbett, D.; Hebditch, M.; Keeling, R.; Ke, P.; Ekizoglou, S.; Sarangapani, P.; Pathak, J.; Van Der Walle, C.F.; Uddin, S.; Baldock, C.; et al. Coarse-Grained Modeling of Antibodies from Small-Angle Scattering Profiles. J. Phys. Chem. B 2017, 121, 8276–8290. [Google Scholar] [CrossRef] [PubMed]
- Arzenšek, D.; Kuzman, D.; Podgornik, R. Hofmeister Effects in Monoclonal Antibody Solution Interactions. J. Phys. Chem. B 2015, 119, 10375–10389. [Google Scholar] [CrossRef] [PubMed]
- Sivasubramanian, A.; Sircar, A.; Chaudhury, S.; Gray, J.J. Toward high-resolution homology modeling of antibody F(v) regions and application to antibody-antigen docking. Proteins 2009, 74, 497–514. [Google Scholar] [CrossRef] [PubMed]
- Weitzner, B.D.; Jeliazkov, J.R.; Lyskov, S.; Marze, N.; Kuroda, D.; Frick, R.; Adolf-Bryfogle, J.; Biswas, N.; Dunbrack, R.L., Jr.; Gray, J.J. Modeling and docking of antibody structures with Rosetta. Nat. Protoc. 2017, 12, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A Protein-Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, G.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.; Karaca, E.; Melquiond, A.; van Dijk, M.; de Vries, S.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Almagro, J.C.; Teplyakov, A.; Luo, J.; Sweet, R.W.; Kodangattil, S.; Hernandez-Guzman, F.; Gilliland, G.L. Second antibody modeling assessment (AMA-II). Proteins Struct. Funct. Bioinform. 2014, 82, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.S.; Toft, K.N.; Snakenborg, D.; Jeppesen, M.G.; Jacobsen, J.K.; Vestergaard, B.; Kutter, J.P.; Arleth, L. BioXTAS RAW, a software program for high-throughput automated small-angle X-ray scattering data reduction and preliminary analysis. J. Appl. Crystallogr. 2009, 42, 959–964. [Google Scholar] [CrossRef]
- Petoukhov, M.V.; Franke, D.; Shkumatov, A.V.; Tria, G.; Kikhney, A.G.; Gajda, M.; Gorba, C.; Mertens, H.D.T.; Konarev, P.V.; Svergun, D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012, 45, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Wypych, J.; Li, M.; Guo, A.; Zhang, Z.; Martinez, T.; Allen, M.J.; Fodor, S.; Kelner, D.N.; Flynn, G.C.; Liu, Y.D.; et al. Human IgG2 Antibodies Display Disulfide-mediated Structural Isoforms. J. Biol. Chem. 2008, 283, 16194–16205. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.E.; Raghunandan, S.; Nanda, H.; Krueger, S. SASSIE: A program to study intrinsically disordered biological molecules and macromolecular ensembles using experimental scattering restraints. Comput. Phys. Commun. 2012, 183, 382–389. [Google Scholar] [CrossRef]
- Freitag, S.; Le Trong, I.; Klumb, L.; Stayton, P.S.; Stenkamp, R.E. Structural studies of the streptavidin binding loop. Protein Sci. 1997, 6, 1157–1166. [Google Scholar] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Gray, J.J.; Moughon, S.; Wang, C.; Schueler-Furman, O.; Kuhlman, B.; Rohl, C.A.; Baker, D. Protein–Protein Docking with Simultaneous Optimization of Rigid-body Displacement and Side-chain Conformations. J. Mol. Biol. 2003, 331, 281–299. [Google Scholar] [CrossRef]
- Li, S.C.; Ng, Y.K. Calibur: A tool for clustering large numbers of protein decoys. BMC Bioinform. 2010, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.C.; Curtis, J.E. Rapid and accurate calculation of small-angle scattering profiles using the golden ratio. J. Appl. Crystallogr. 2013, 46, 1171–1177. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Molecular Weight (kDa) | Number of Measurements |
|---|---|---|
| tSA | 63.3 ± 0.8 | 3 |
| ASA-IgG2 | 160 ± 3 | 5 |
| main complex | 447 ± 12 | 8 |
| Sample | Method | Radius of Gyration (Å) | |||
|---|---|---|---|---|---|
| tSA | Bulk | 27.4 | 0.52 | 1.3 | 0.98 |
| ASA-IgG2 | SEC | 49.2 | 0.44 | 1.0 | 0.99 |
| ASA-IgG2–tSA complex | SEC | 84.8 | 0.56 | 1.3 | 0.95 |
| ASA-IgG2–tSA complex | Fractionation | 88.9 | 0.69 | 1.3 | 0.91 |
| ASA-IgG2–tSA complex | Bulk | 123 | 1.0 | 1.2 | 0.62 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castellanos, M.M.; Snyder, J.A.; Lee, M.; Chakravarthy, S.; Clark, N.J.; McAuley, A.; Curtis, J.E. Characterization of Monoclonal Antibody–Protein Antigen Complexes Using Small-Angle Scattering and Molecular Modeling. Antibodies 2017, 6, 25. https://doi.org/10.3390/antib6040025
Castellanos MM, Snyder JA, Lee M, Chakravarthy S, Clark NJ, McAuley A, Curtis JE. Characterization of Monoclonal Antibody–Protein Antigen Complexes Using Small-Angle Scattering and Molecular Modeling. Antibodies. 2017; 6(4):25. https://doi.org/10.3390/antib6040025
Chicago/Turabian StyleCastellanos, Maria Monica, James A. Snyder, Melody Lee, Srinivas Chakravarthy, Nicholas J. Clark, Arnold McAuley, and Joseph E. Curtis. 2017. "Characterization of Monoclonal Antibody–Protein Antigen Complexes Using Small-Angle Scattering and Molecular Modeling" Antibodies 6, no. 4: 25. https://doi.org/10.3390/antib6040025
APA StyleCastellanos, M. M., Snyder, J. A., Lee, M., Chakravarthy, S., Clark, N. J., McAuley, A., & Curtis, J. E. (2017). Characterization of Monoclonal Antibody–Protein Antigen Complexes Using Small-Angle Scattering and Molecular Modeling. Antibodies, 6(4), 25. https://doi.org/10.3390/antib6040025