Modulation of BCR Signaling by the Induced Dimerization of Receptor-Associated SYK


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids, DNA Constructs and Antibodies
2.2. Cell Culture and DNA Transfection/Transduction
2.3. TMP-Agarose Pull-Down
2.4. Immunofluorescence
2.5. NFAT- and NFκB-Luciferase Reporter Assays
2.6. Intracellular Calcium Assay
2.7. ImageJ Analyses
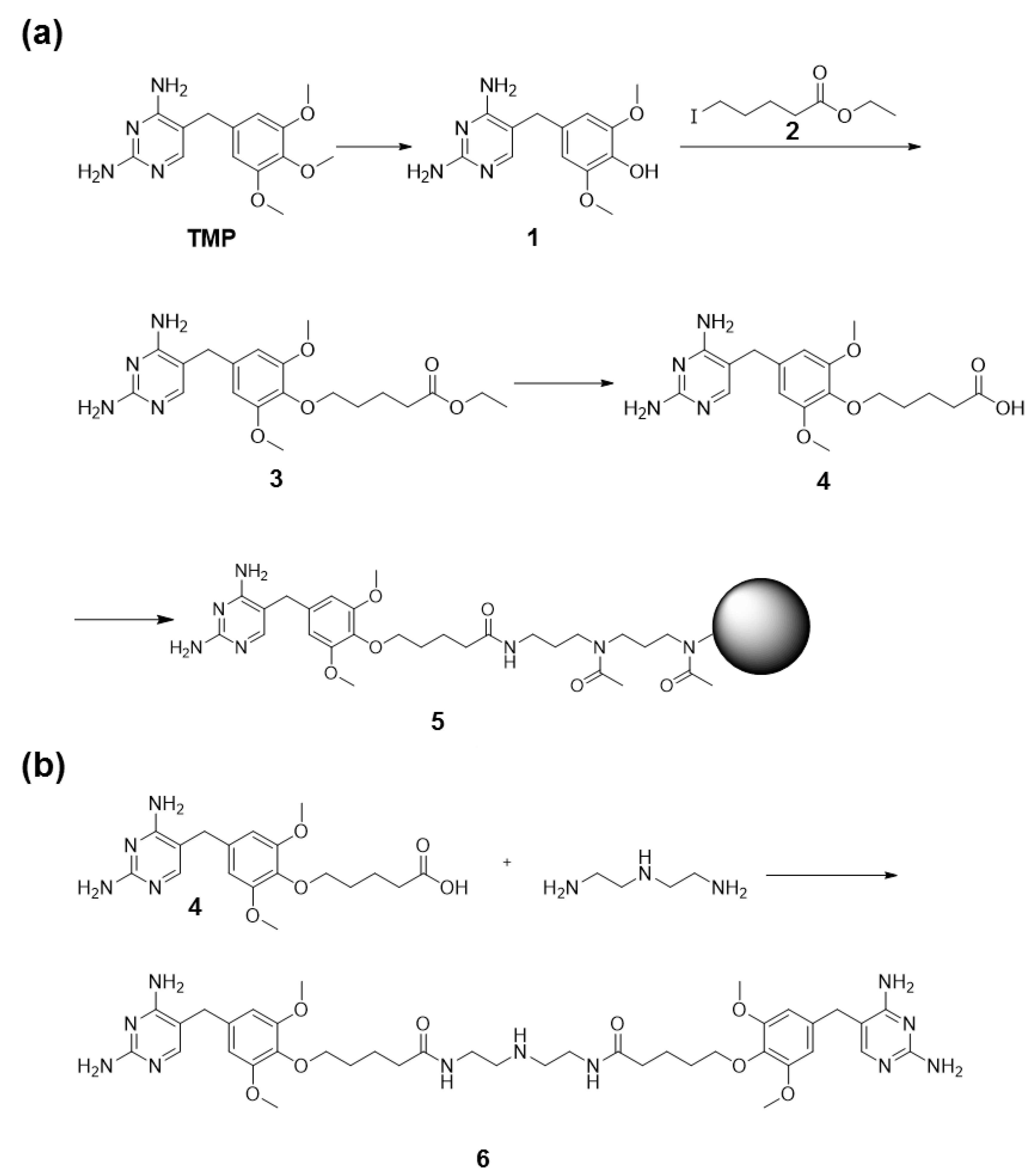
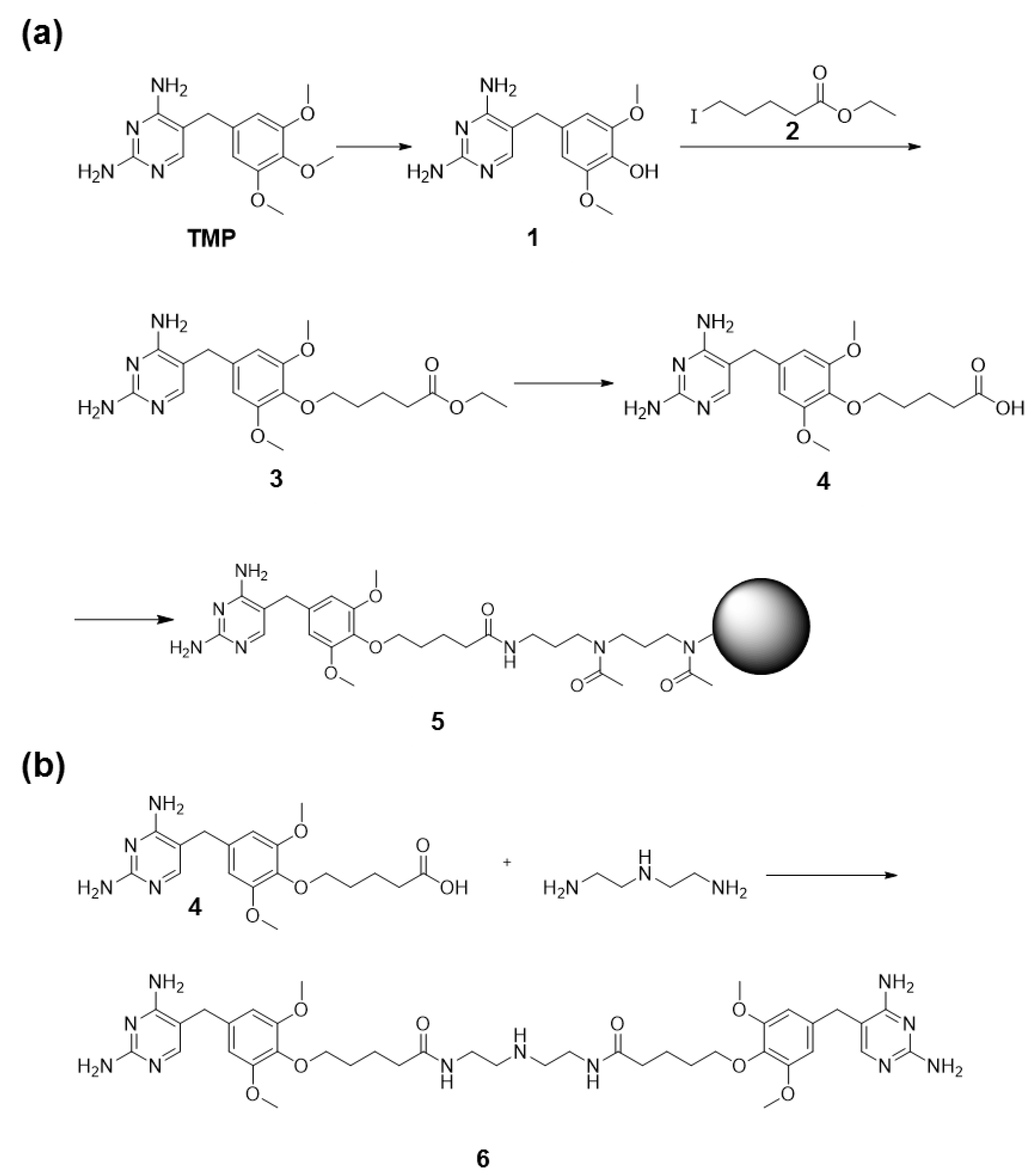
2.8. Synthesis of TMP Coupled to Agarose Beads
2.9. Synthesis of TMP Dimer 6
3. Results
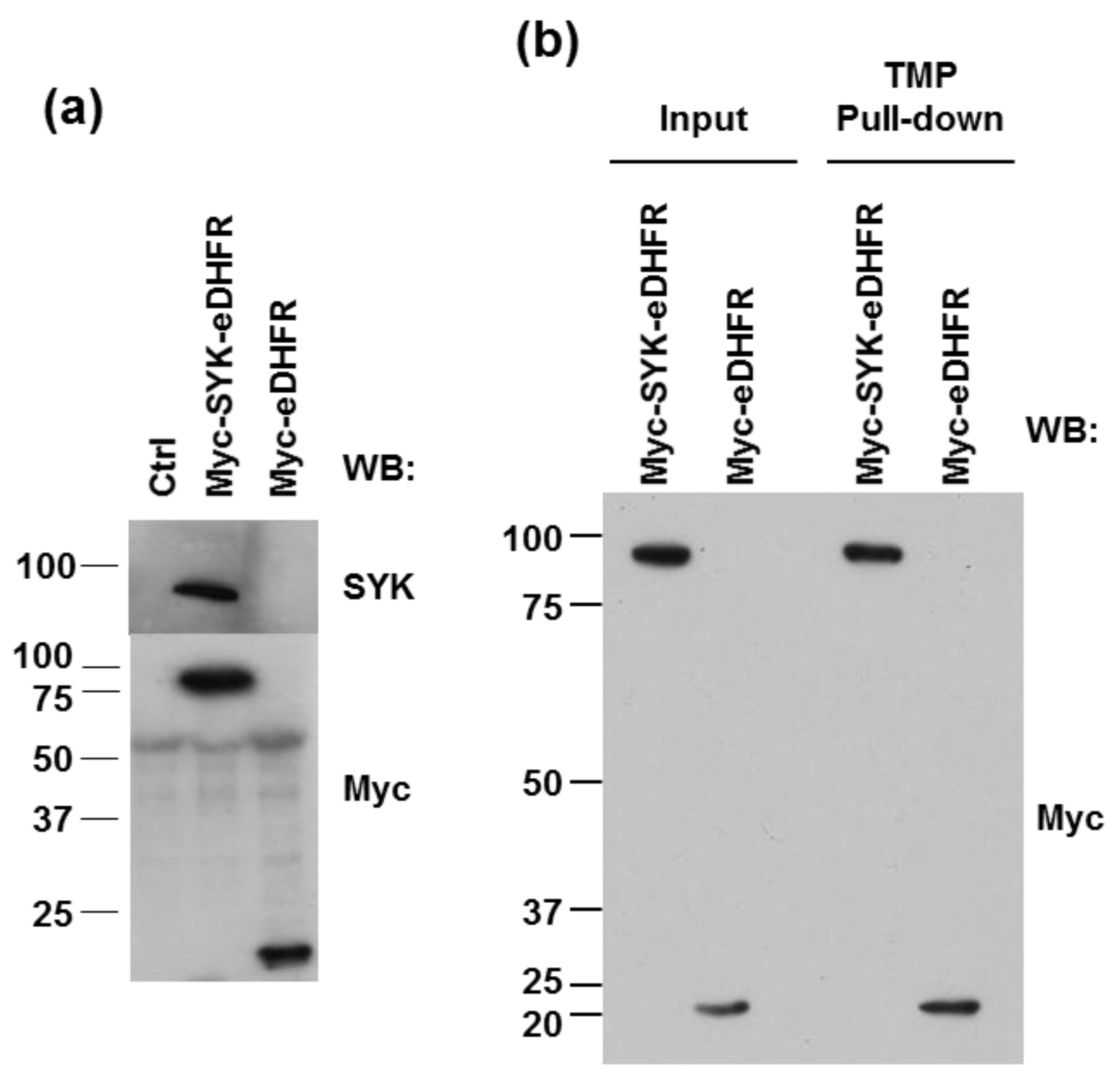
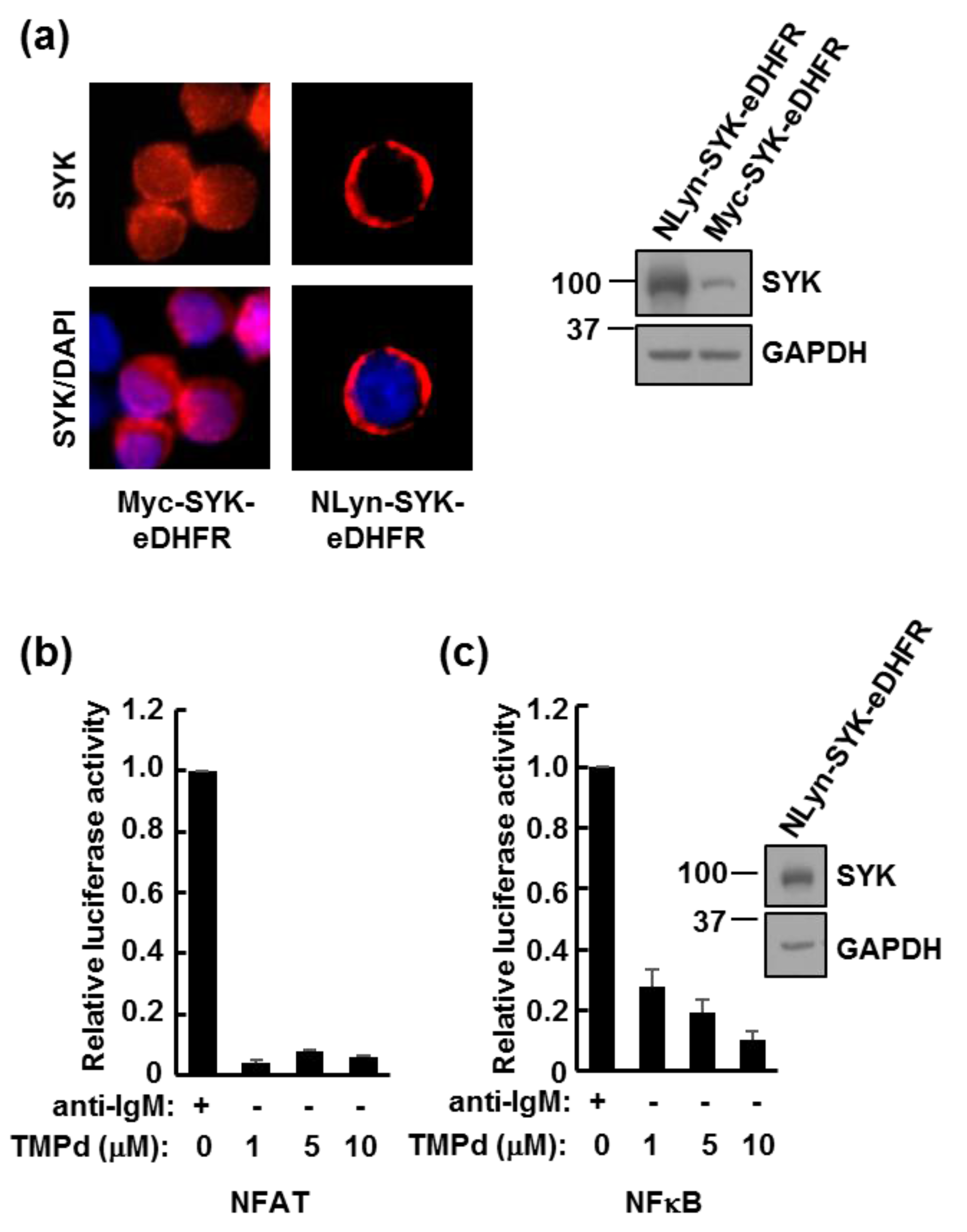
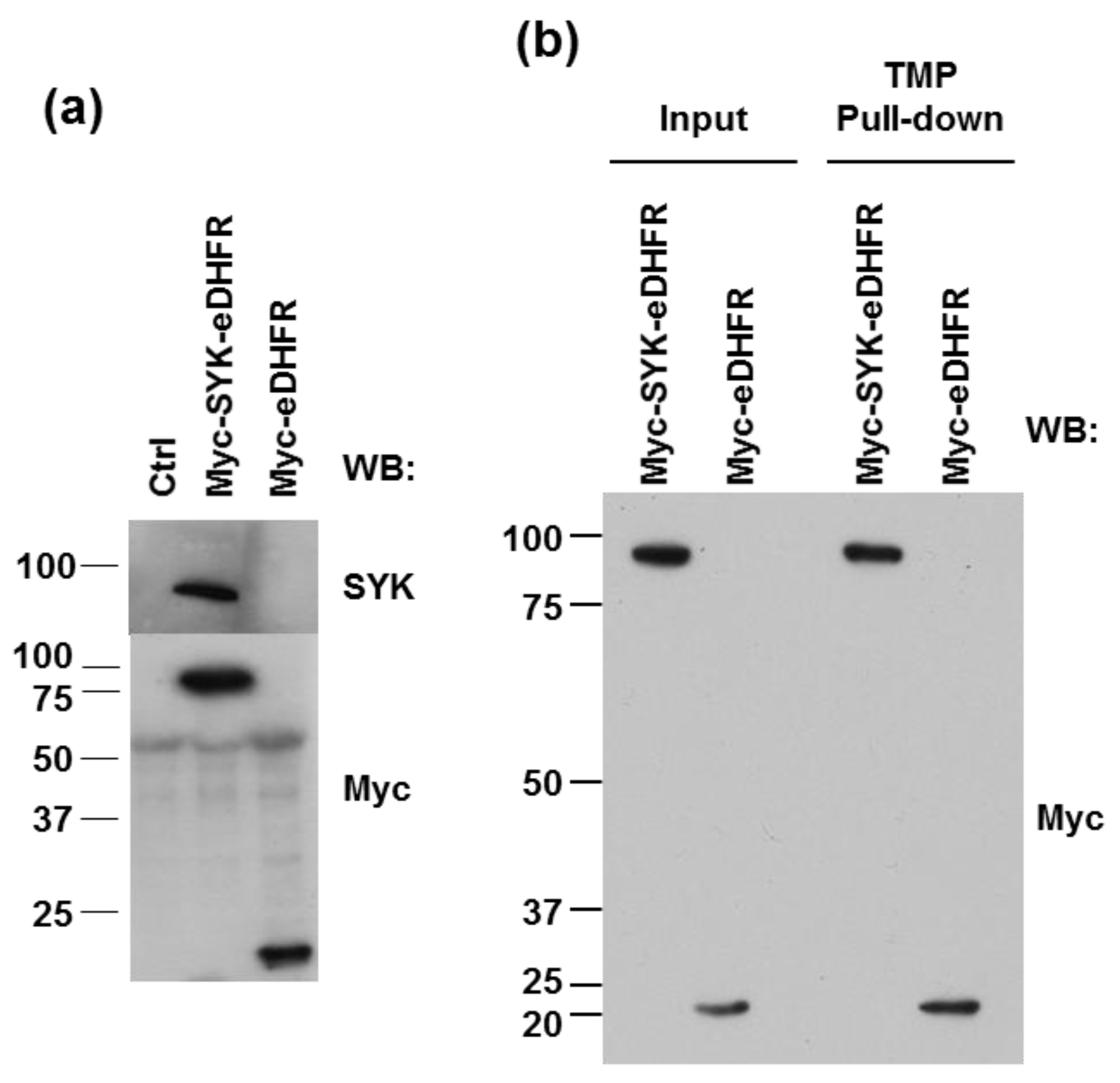
3.1. eDHFR-Tagged SYK Binds TMP
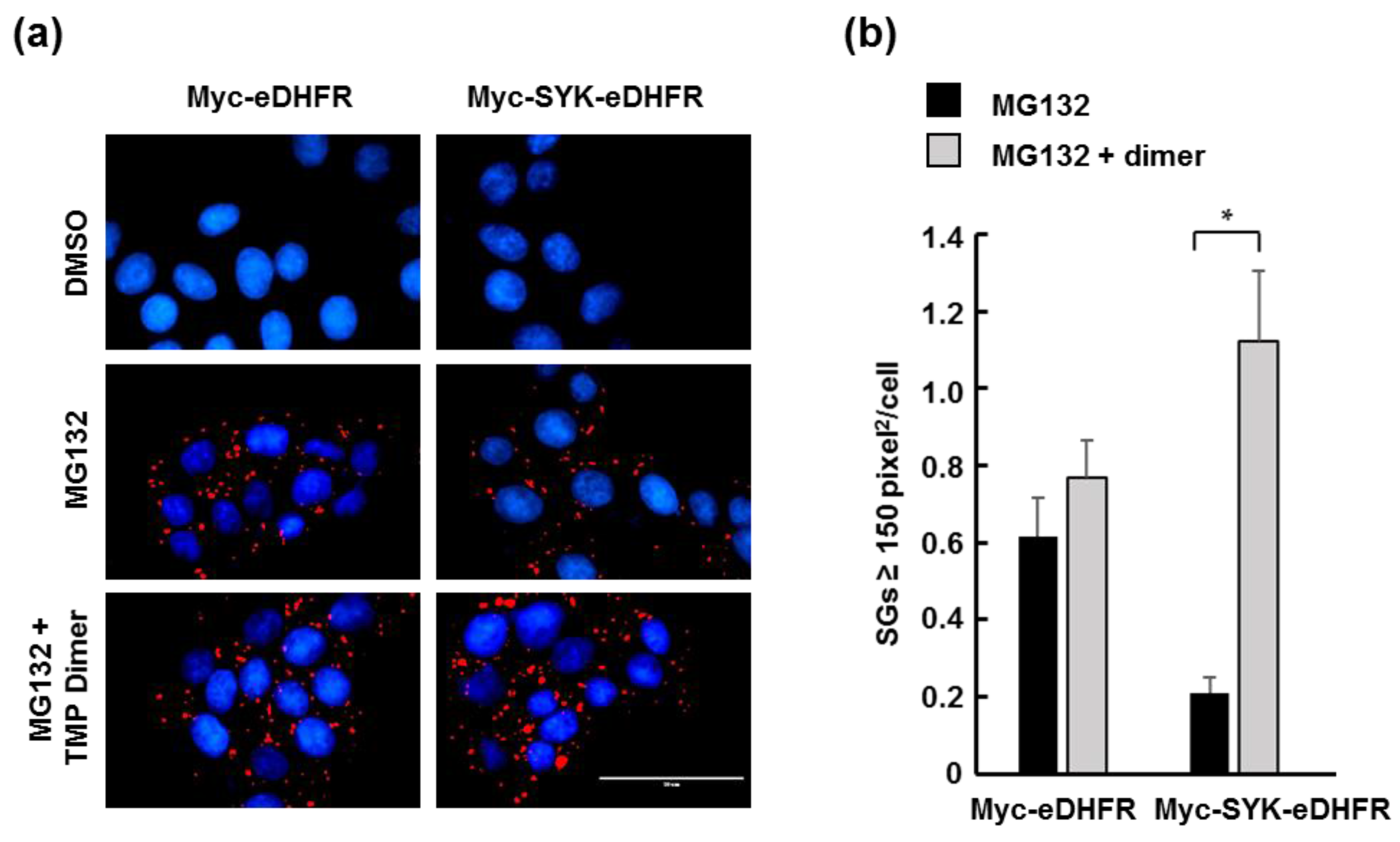
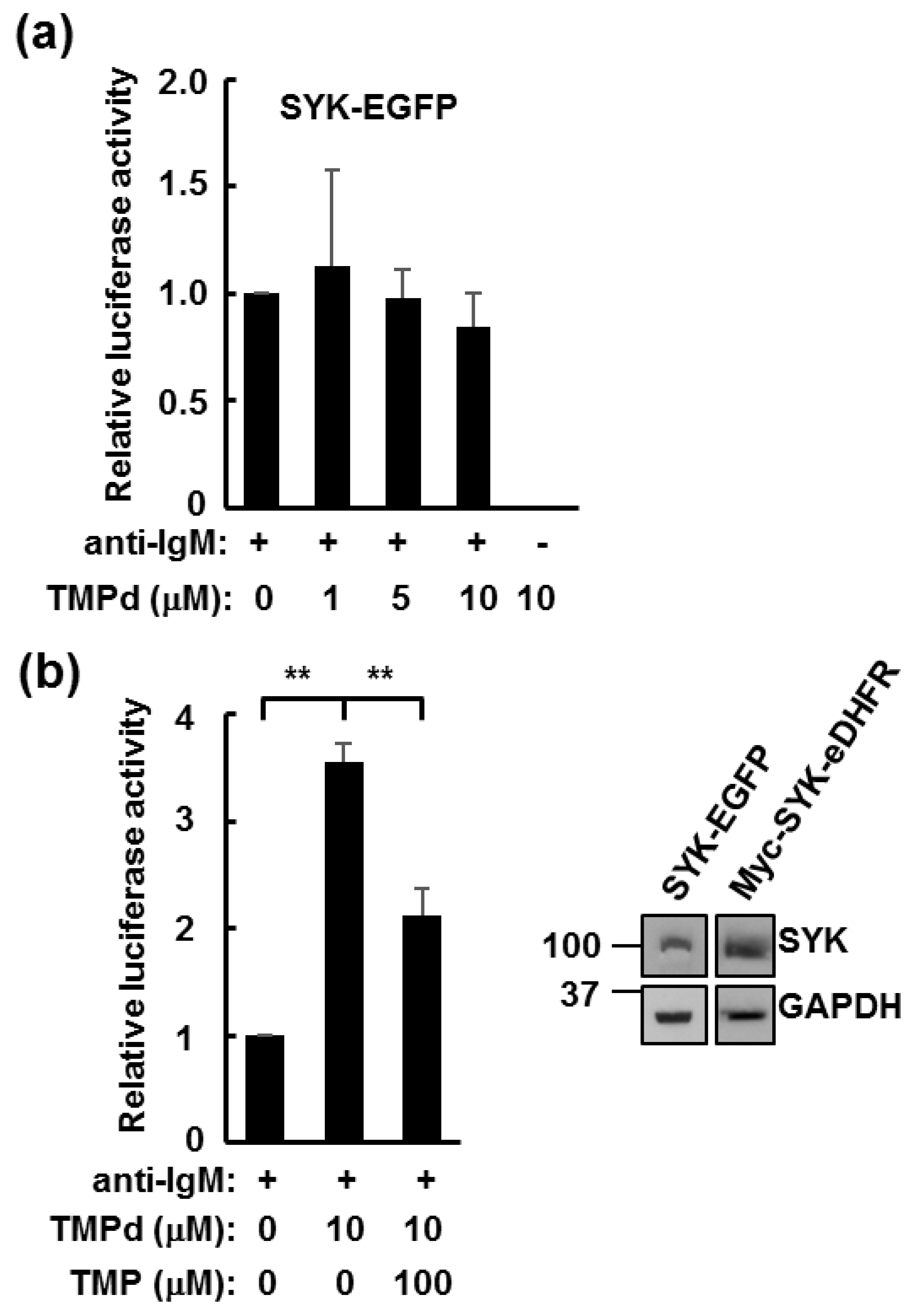
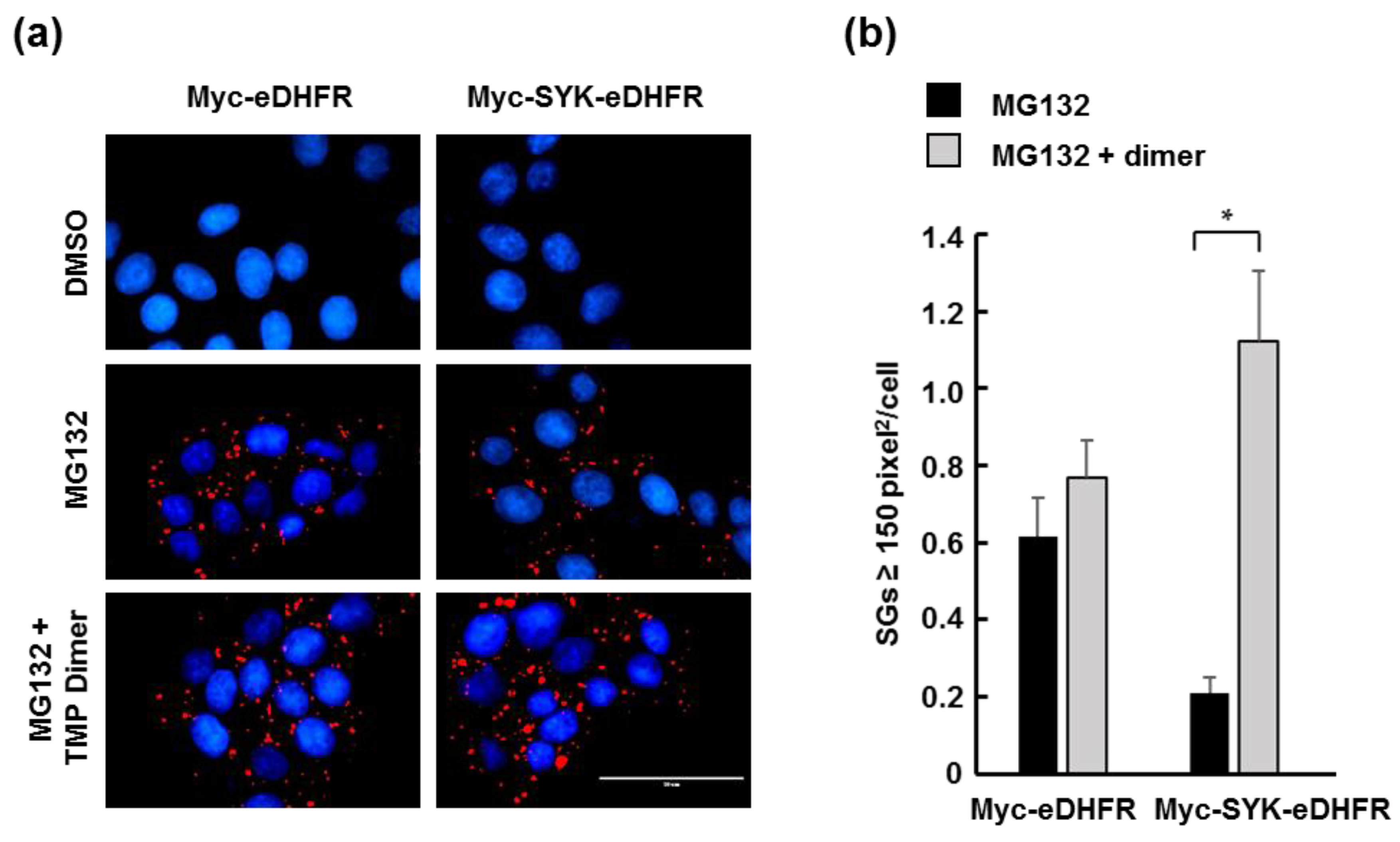
3.2. TMP Dimer Induces SYK Aggregation
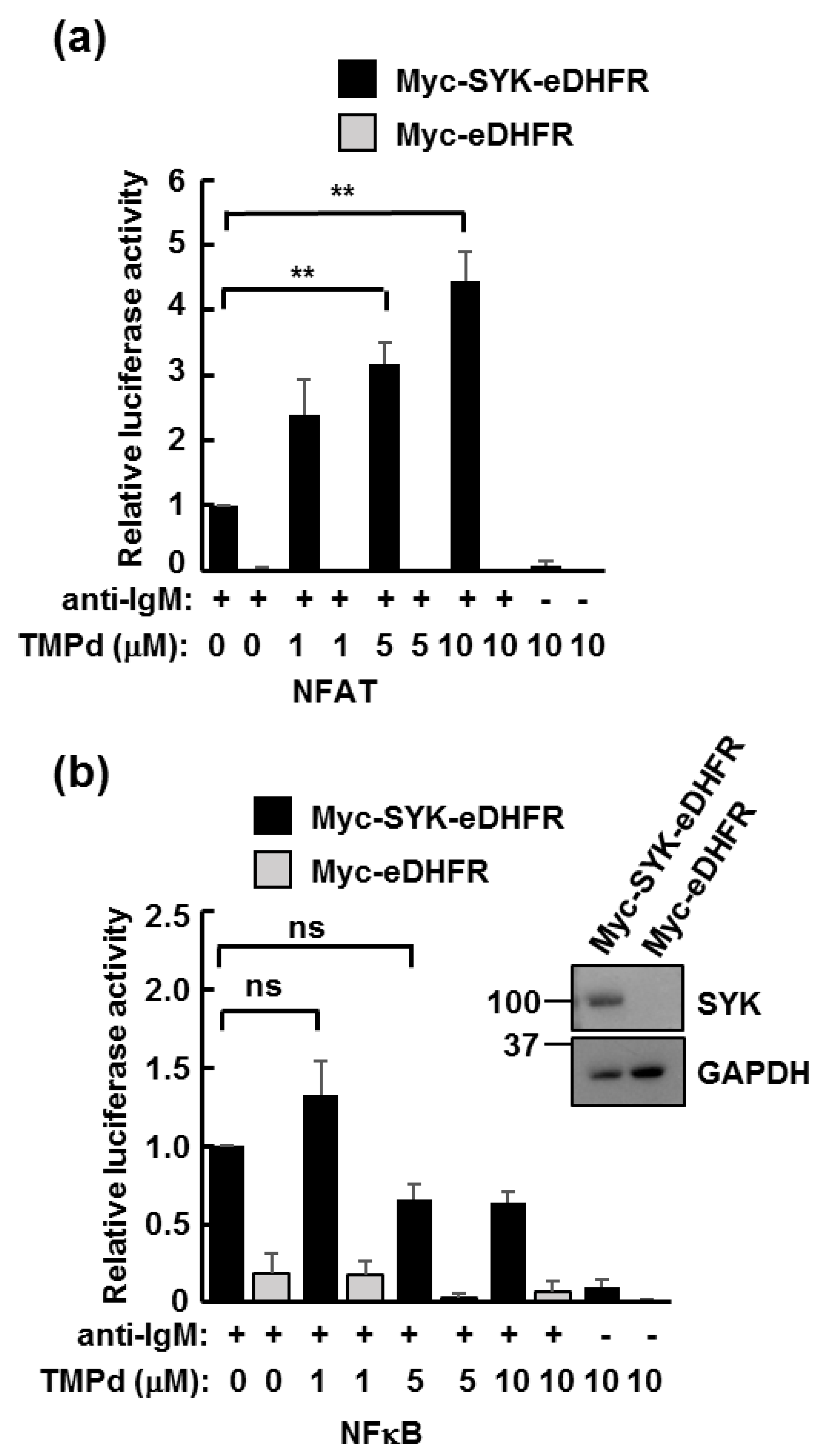
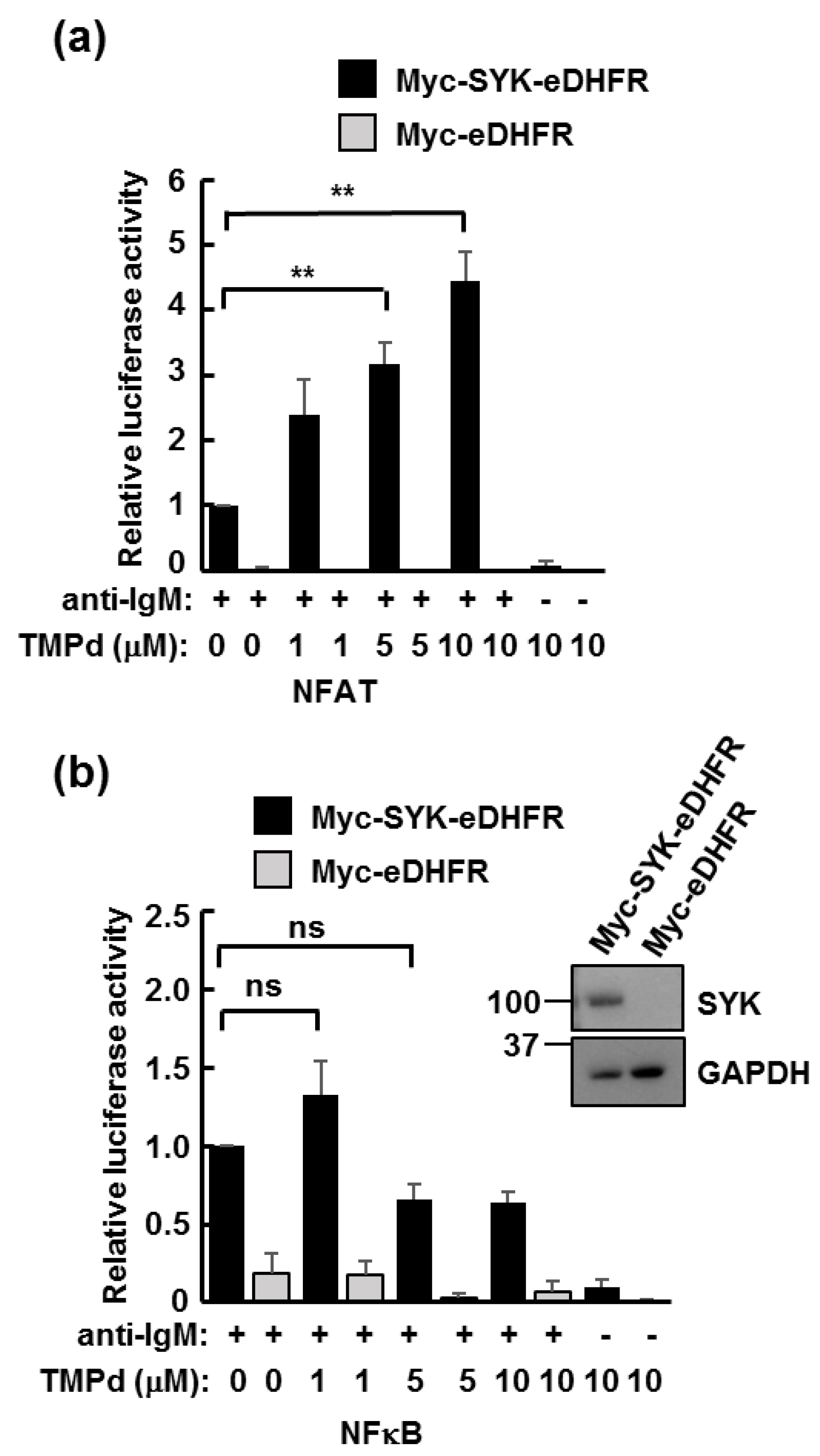
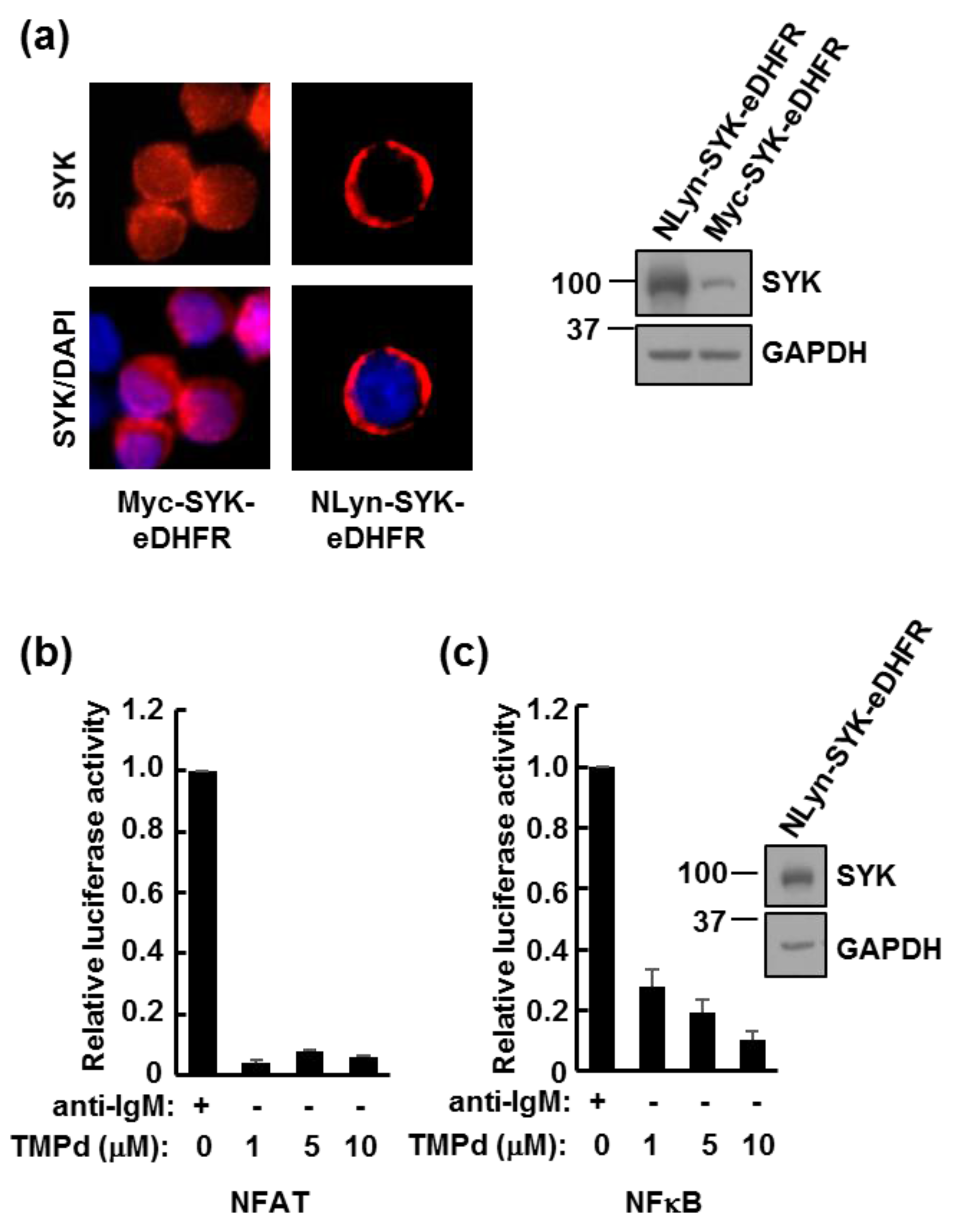
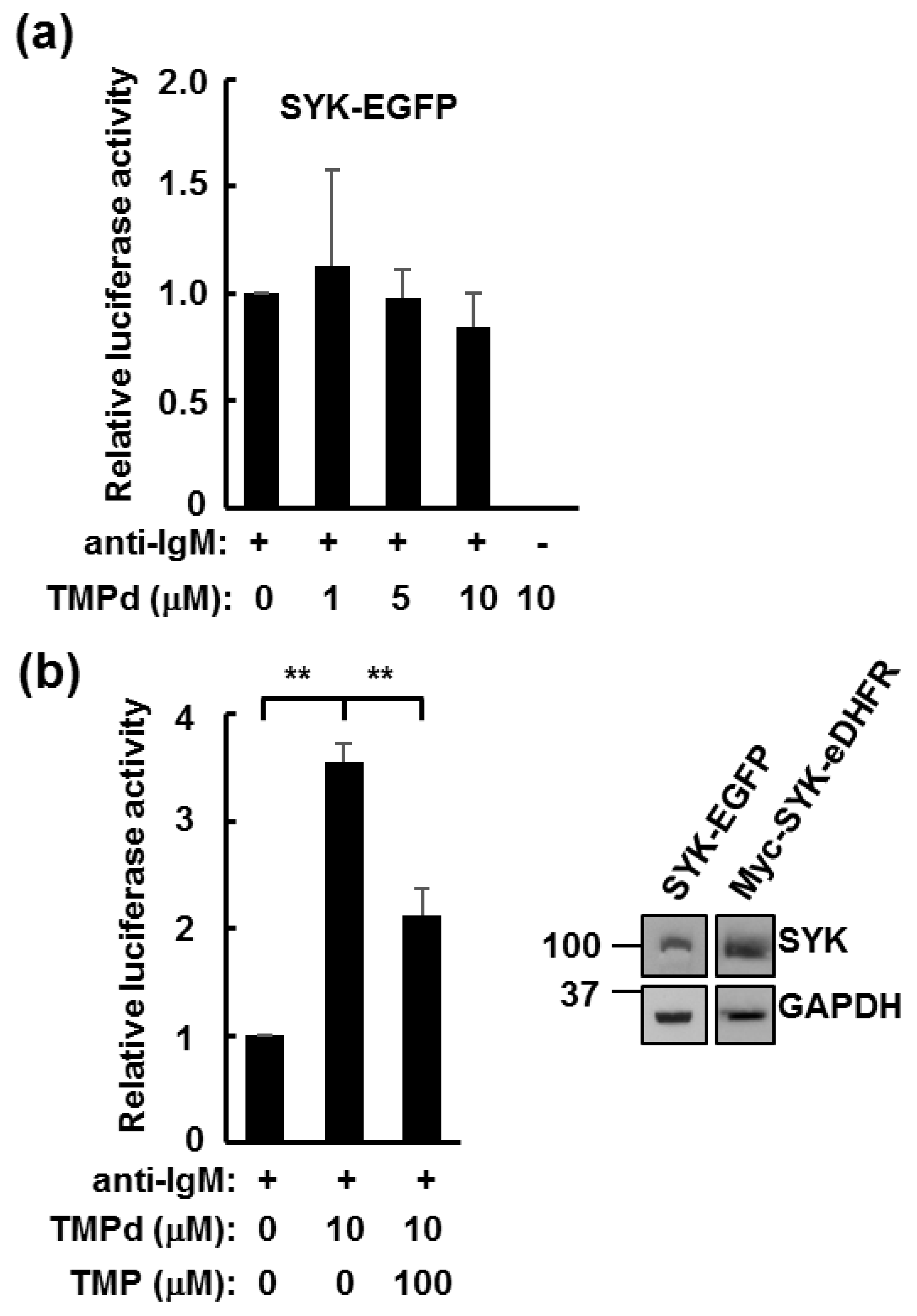
3.3. Dimerized SYK Signals through NFAT or NFκB When Interacting with the BCR
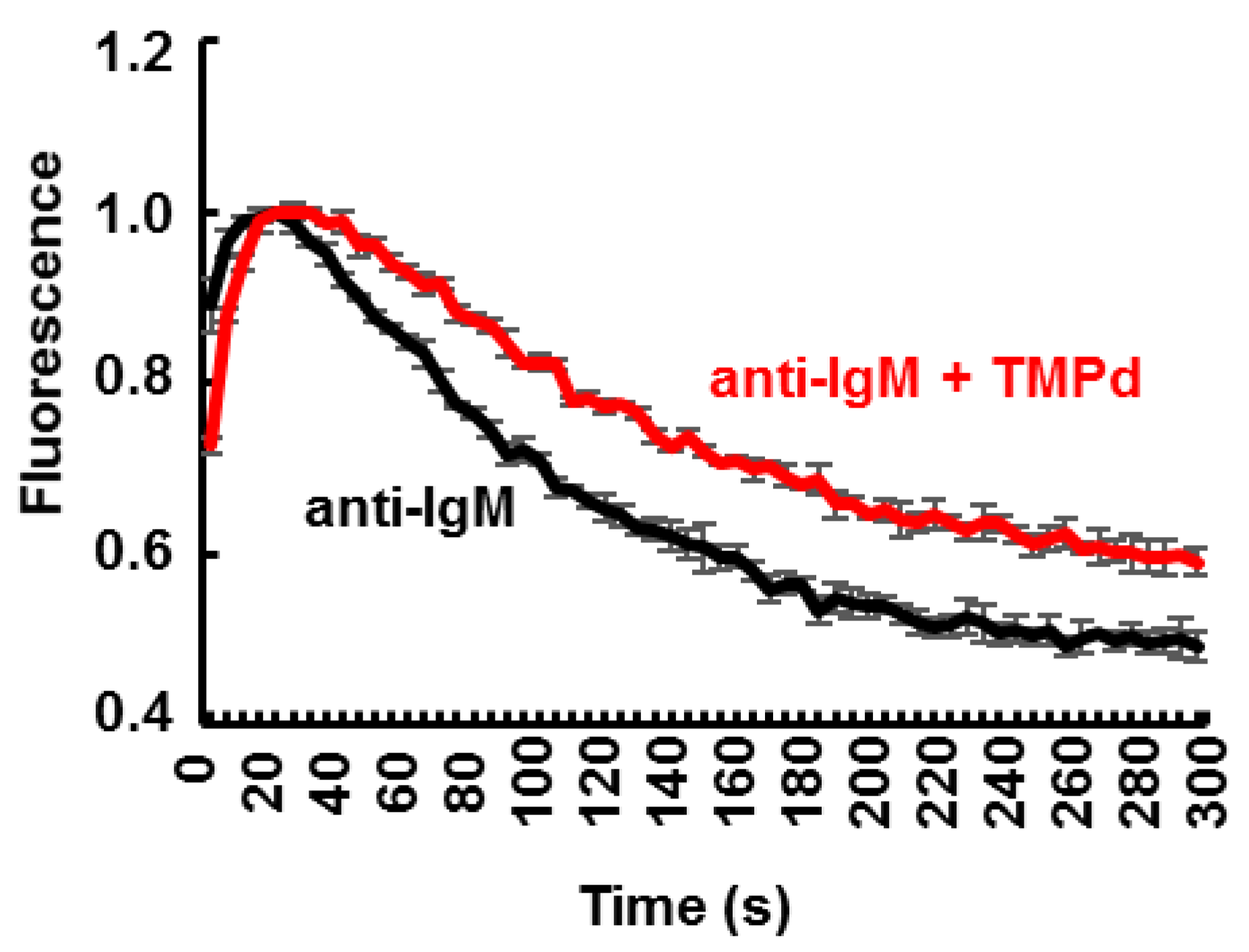
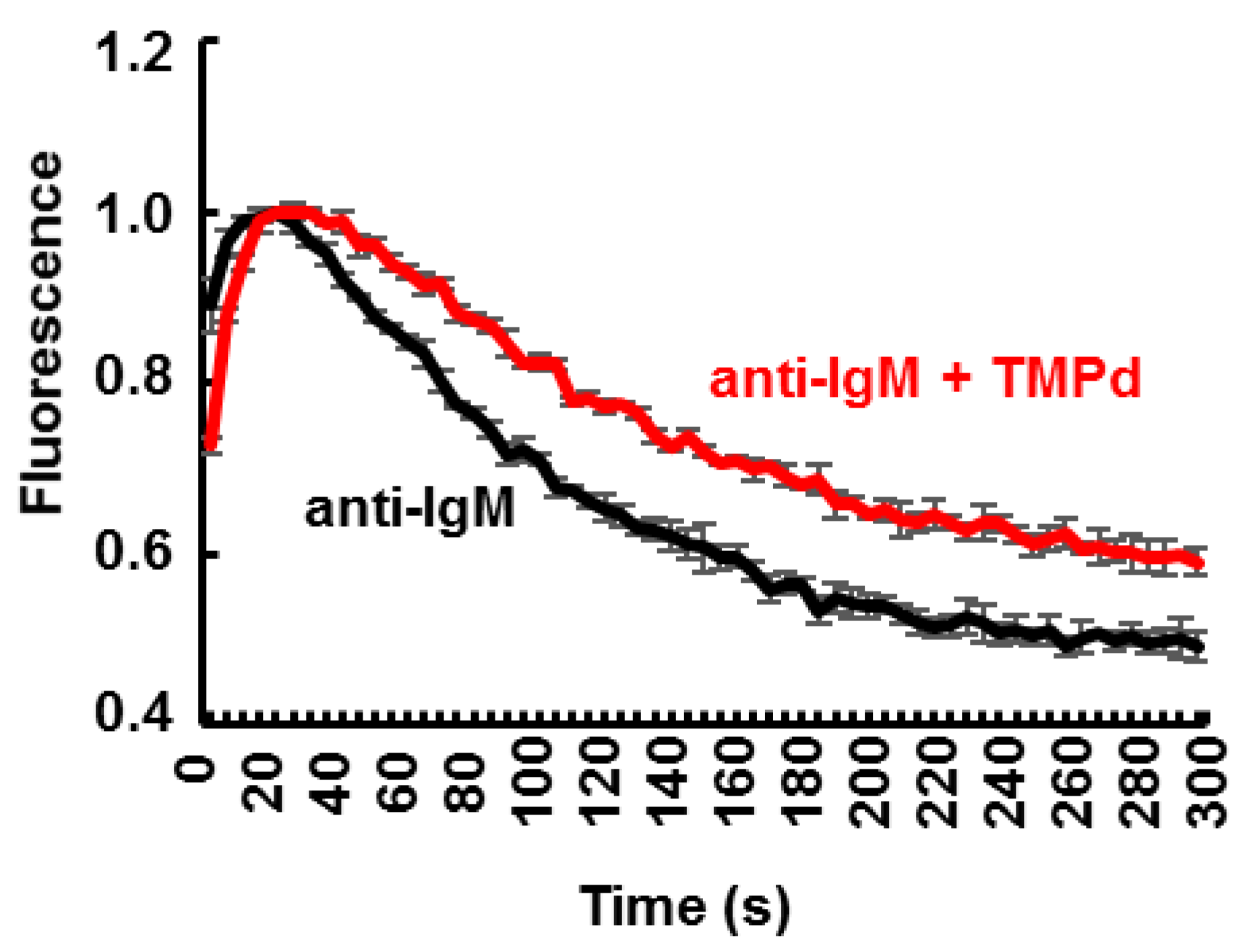
3.4. Dimerization of SYK Promotes Sustained Ca2+ Mobilization
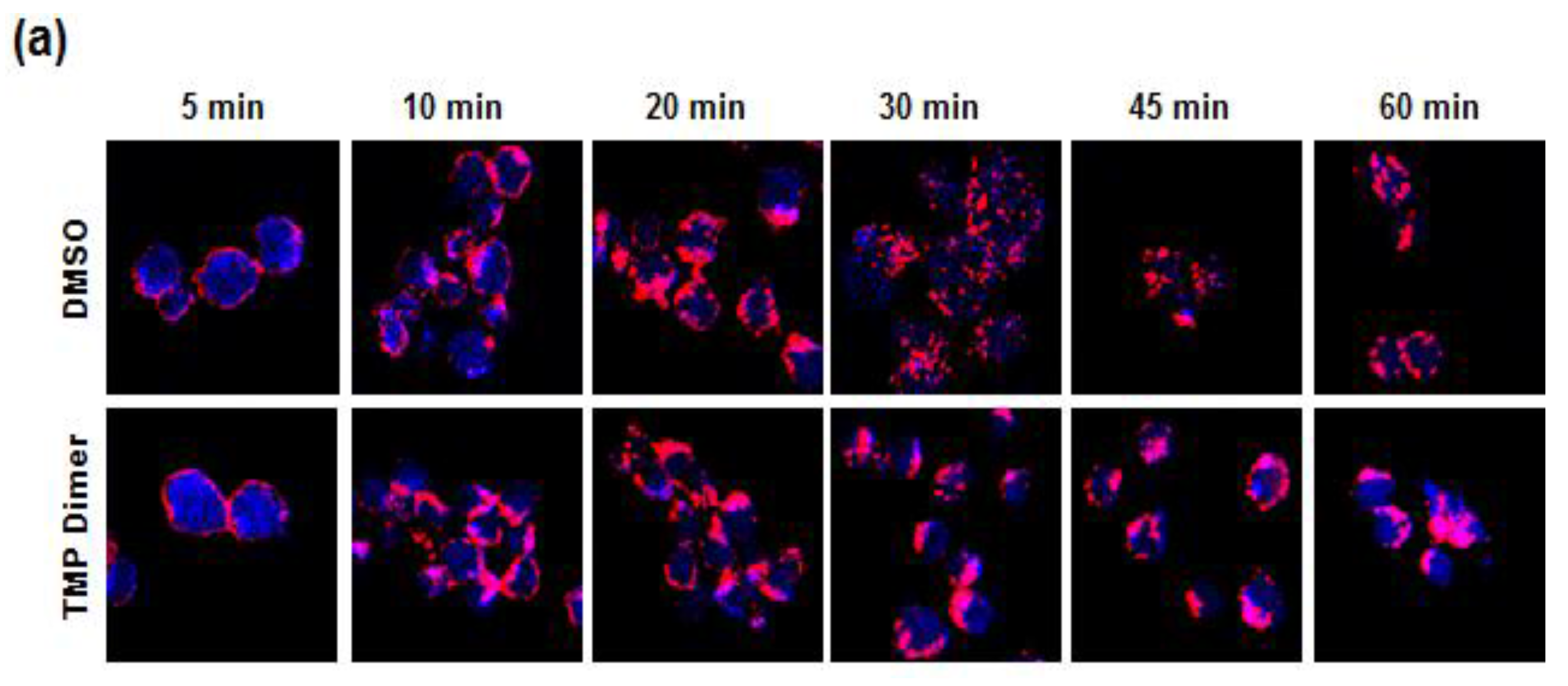
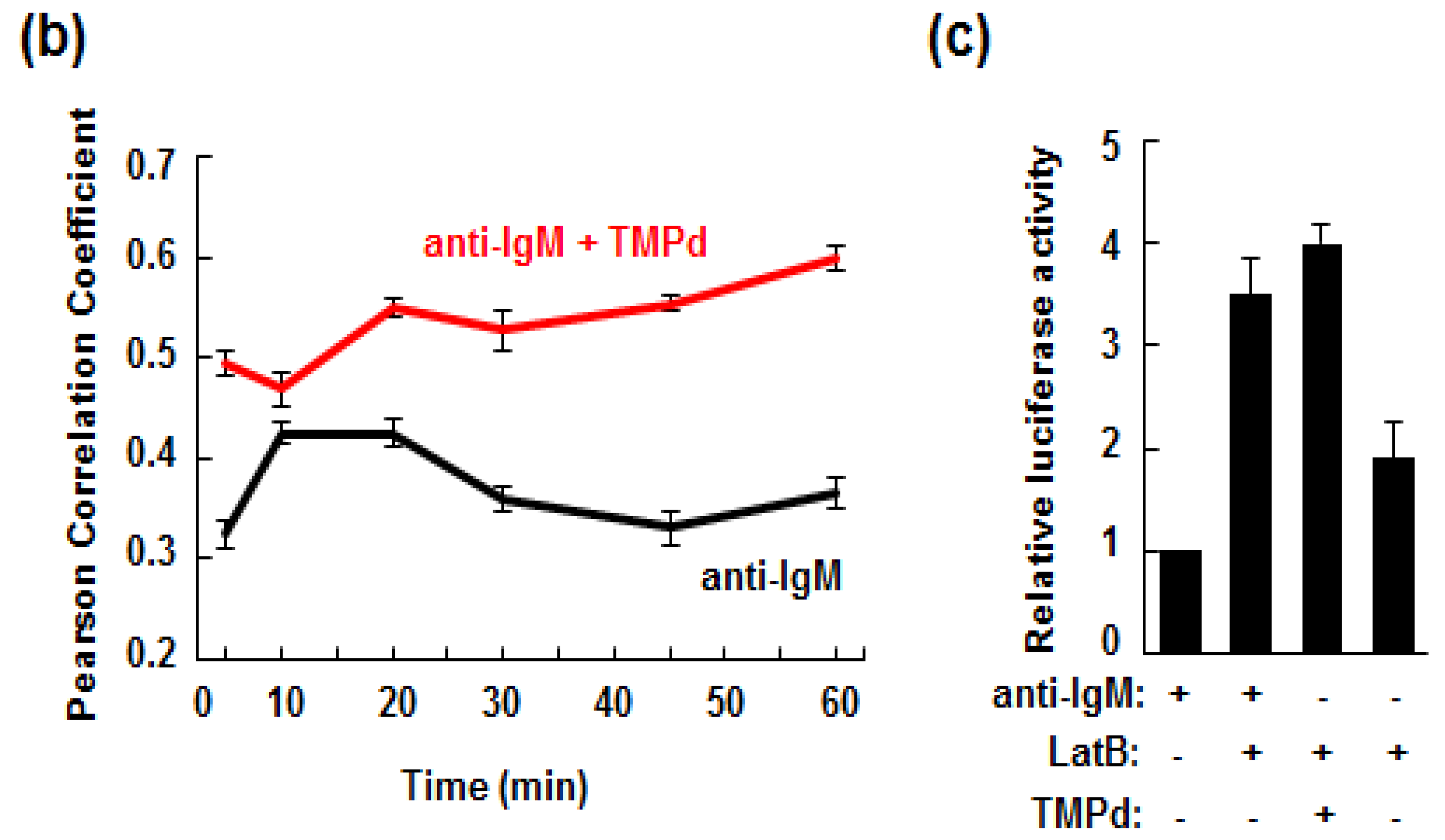
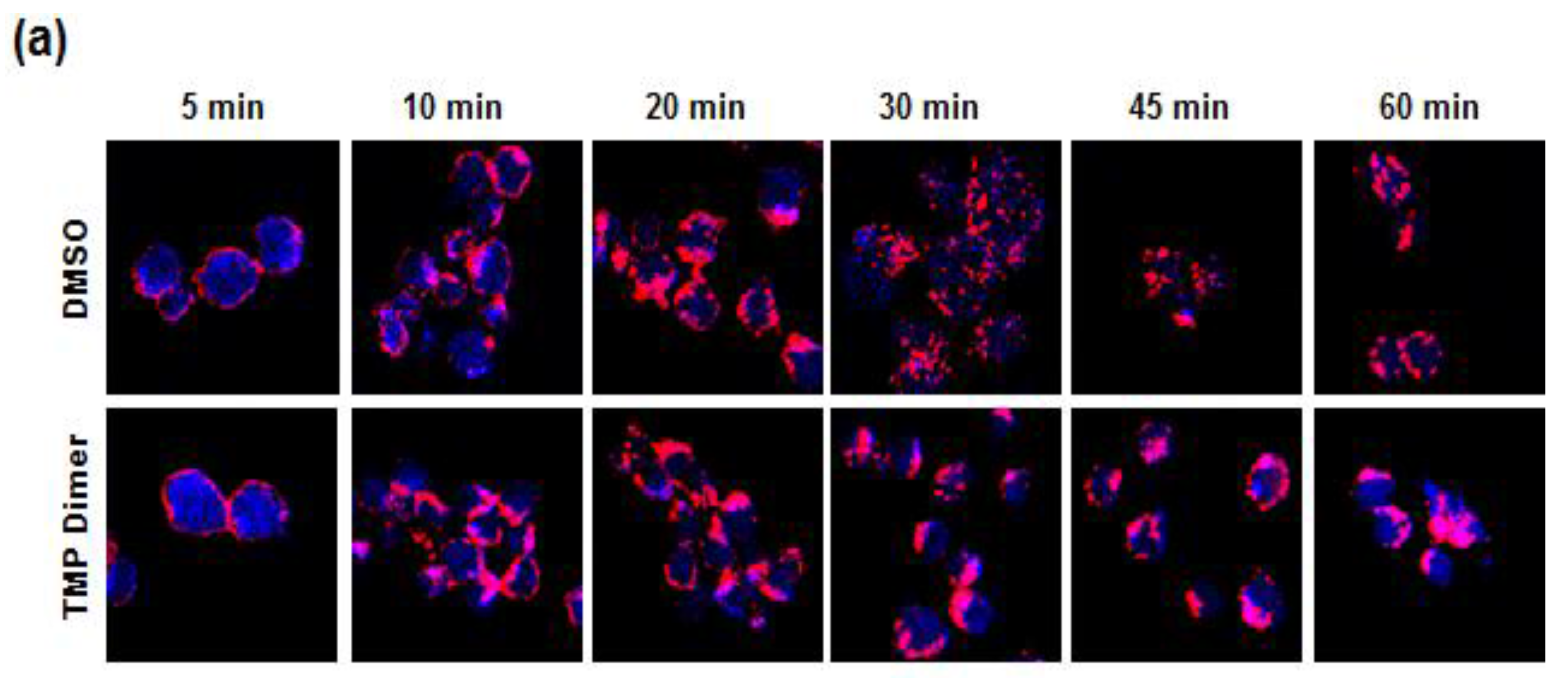
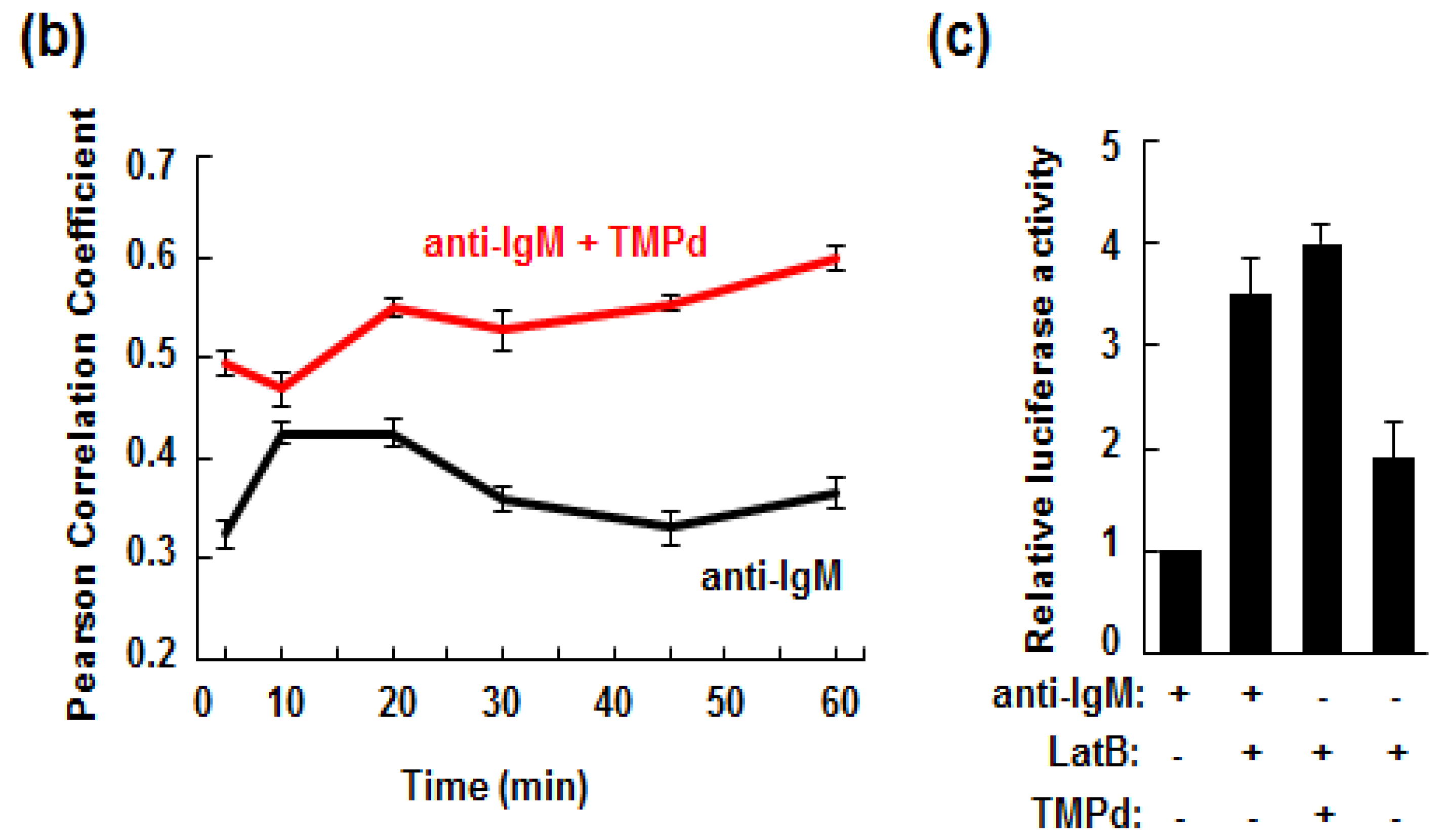
3.5. Retention of the BCR at the Plasma Membrane Enhances Signaling through NFAT
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Geahlen, R. Syk and pTyr’d: Signaling through the B cell antigen receptor. Biochim. Biophys. Acta 2009, 1793, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Dal Porto, J.M.; Gauld, S.B.; Merrell, K.T.; Mills, D.; Pugh-Bernard, A.E.; Cambier, J. B cell antigen receptor signaling 101. Mol. Immunol. 2004, 41, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T. Genetic analysis of B cell antigen receptor signaling. Annu. Rev. Immunol. 1999, 17, 555–592. [Google Scholar] [CrossRef] [PubMed]
- Rowley, R.; Burkhardt, A.; Chao, H.; Matsueda, G.; Bolen, J. Syk protein-tyrosine kinase is regulated by tyrosine-phosphorylated Ig alpha/Ig beta immunoreceptor tyrosine activation motif binding and autophosphorylation. J. Biol. Chem. 1995, 270, 11590–11594. [Google Scholar] [CrossRef] [PubMed]
- Metzger, H. Transmembrane signaling: The joy of aggregation. J. Immunol. 1992, 149, 1477–1487. [Google Scholar] [PubMed]
- Minguet, S.; Dopfer, E.-P.; Schamel, W.W.A. Low-valency, but not monovalent, antigens trigger the B-cell antigen receptor (BCR). Int. Immunol. 2010, 22, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Fanger, M.W.; Hart, D.A.; Wells, J.V.; Nisonoff, A. Requirement for cross-linkage in the stimulation of transformation of rabbit peripheral lymphocytes by antiglobulin reagents. J. Immunol. 1970, 105, 1484–1492. [Google Scholar] [PubMed]
- Maino, V.C.; Hayman, M.J.; Crumpton, M.J. Relationship between enhanced turnover of phosphatidylinositol and lymphocyte activation by mitogens. Biochem. J. 1975, 146, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Riethmüller, G.; Riethmüller, D.; Stein, H.; Hausen, P. In vivo and in vitro properties of intact and pepsin-digested heterologous anti-mouse thymus antibodies. J. Immunol. 1968, 100, 969–973. [Google Scholar] [PubMed]
- Weiner, H.L.; Moorhead, J.W.; Yamaga, K.; Kubo, R.T. Anti-immunoglobulin stimulation of murine lymphocytes. II. Identification of cell surface target molecules and requirements for cross-linkage. J. Immunol. 1976, 117, 1527–1531. [Google Scholar] [CrossRef]
- Woodruff, M.F.A.; Reid, B.; James, K. Effect of antilymphocytic antibody and antibody fragments on human lymphocytes in vitro. Nature 1967, 215, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Tolar, P.; Pierce, S.K. A conformation-induced oligomerization model for B cell receptor microclustering and signaling. In Immunological Synapse; Saito, T., Batista, F.D., Eds.; Springer: Berlin/Heidelberg, 2010; Volume 340, pp. 155–169. [Google Scholar]
- Volkmann, C.; Brings, N.; Becker, M.; Hobeika, E.; Yang, J.; Reth, M. Molecular requirements of the B-cell antigen receptor for sensing monovalent antigens. EMBO 2016, 35, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Reth, M. Oligomeric organization of the B-cell antigen receptor on resting cells. Nature 2010, 467, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; August, A. Actin depolymerization transduces the strength of B-cell receptor stimulation. Mol. Biol. Cell 2005, 16, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Keppler, S.J.; Gasparrini, F.; Burbage, M.; Aggarwal, S.; Frederico, B.; Geha, R.S.; Way, M.; Bruckbauer, A.; Batista, F.D. Wiskott-Aldrich syndrome interacting protein deficiency uncovers the role of the co-receptor CD19 as a generic hub for PI3 kinase signaling in B cells. Immunity 2015, 43, 660–673. [Google Scholar] [CrossRef] [PubMed]
- Mattila, P.K.; Feest, C.; Depoil, D.; Treanor, B.; Montaner, B.; Otipoby, K.L.; Carter, R.; Justement, L.B.; Bruckbauer, A.; Batista, F.D. The actin and tetraspanin networks organize receptor nanoclusters to regulate B cell receptor-mediated signaling. Immunity 2013, 38, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Tolar, P. Cytoskeletal control of B cell responses to antigens. Nat. Rev. Immunol. 2017, 17, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Bennett, N.R.; Zwick, D.B.; Courtney, A.H. Multivalent antigens for promoting B and T cell activation. ACS Chem. Biol. 2015, 10, 1817–1824. [Google Scholar] [CrossRef] [PubMed]
- Puffer, E.B.; Pontrello, J.K.; Hollenbeck, J.J.; Kink, J.A.; Kiessling, L.L. Activating B cell signaling with defined multivalent ligands. ACS Chem. Biol. 2007, 2, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Kläsener, K.; Maity, P.C.; Hobeika, E.; Yang, J.; Reth, M. B cell activation involves nanoscale receptor reorganizations and inside-out signaling by Syk. eLife 2014, 3, e02069. [Google Scholar] [CrossRef] [PubMed]
- Rolli, V.; Gallwitz, M.; Wossning, T.; Flemming, A.; Schamel, W.W.A.; Zürn, C.; Reth, M. Amplification of B cell antigen receptor signaling by a Syk/ITAM positive feedback loop. Mol. Cell 2002, 10, 1057–1069. [Google Scholar] [CrossRef]
- Grädler, U.; Schwarz, D.; Dresing, V.; Musil, D.; Bomke, J.; Frech, M.; Greiner, H.; Jäkel, S.; Rysiok, T.; Müller-Pompalla, D.; et al. Structural and biophysical characterization of the Syk activation switch. J. Mol. Biol. 2013, 425, 309–333. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Pan, J.Y.-J.; Korbel, G.A.; Peperzak, V.; Boes, M.; Ploegh, H.L. Monovalent ligation of the B cell receptor induces receptor activation but fails to promote antigen presentation. PNAS 2006, 103, 3327–3332. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, A.; Jackson, A.P.; Brodsky, F.M. Plasticity of B cell receptor internalization upon conditional depletion of clathrin. Mol. Biol. Cell 2005, 16, 2339–2348. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Yankee, T.M.; Hu, J.; Asai, D.J.; Harrison, M.L.; Geahlen, R.L. Visualization of Syk-antigen receptor interactions using green fluorescent protein: Differential roles for Syk and Lyn in the regulation of receptor capping and internalization. J. Immunol. 2001, 166, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Ozkirimli, E.; Shah, K.; Harrison, M.L.; Geahlen, R.L. Generation of an analog-sensitive Syk tyrosine kinase for the study of signaling dynamics from the B cell antigen receptor. J. Biol. Chem. 2007, 282, 33760–33768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Oh, H.; Burton, R.A.; Burgner, J.W.; Geahlen, R.L.; Post, C.B. Tyr130 phosphorylation triggers Syk release from antigen receptor by long-distance conformational uncoupling. PNAS 2008, 105, 11760–11765. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Geahlen, R.L. The protein-tyrosine kinase Syk interacts with TRAF-interacting protein TRIP in breast epithelial cells. Oncogene 2009, 28, 1348–1356. [Google Scholar] [CrossRef] [PubMed]
- Takata, M.; Sabe, H.; Hata, A.; Inazu, T.; Homma, Y.; Nukada, T.; Yamamura, H.; Kurosaki, T. Tyrosine kinases Lyn and Syk regulate B cell receptor-coupled Ca2+ mobilization through distinct pathways. EMBO 1994, 13, 1341–1349. [Google Scholar] [CrossRef]
- Miller, L.; Cai, Y.; Sheetz, M.; Cornish, V. In vivo protein labeling with trimethoprim conjugates: A flexible chemical tag. Nat. Methods 2005, 2, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.P.; Shea, K.J. Dual function catalysts. Dehydrogenation and asymmetric intramolecular Diels-Alder cycloaddition of N-hydroxy formate esters and hydroxamic acids: Evidence for a ruthenium-acylnitroso intermediate. J. Am. Chem. Soc. 2005, 127, 3678–3679. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.; Aig, E.; Rauckman, B.S.; Strelitz, J.Z.; Phillips, A.P.; Ferone, R.; Bushby, S.R.M.; Sigel, C.W. 2, 4-Diamino-5-benzylpyrimidines and analogs as antibacterial agents. 5. 3′,5′-Dimethoxy-4′-substituted-benzyl analogs of trimethoprim. J. Med. Chem. 1981, 24, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.; Liu, J.-W.; Ollis, D. Directed evolution of trimethoprim resistance in Escherichia coli. FEBS J. 2007, 274, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Geahlen, R.L. Stress granules modulate SYK to cause microglial cell dysfunction in Alzheimer’s disease. EBioMedicine 2015, 2, 1785–1798. [Google Scholar] [CrossRef] [PubMed]
- Krisenko, M.O.; Higgins, R.L.; Ghosh, S.; Zhou, Q.; Trybula, J.S.; Wang, W.-H.; Geahlen, R.L. Syk is recruited to stress granules and promotes their clearance through autophagy. J. Biol. Chem. 2015, 290, 27803–27815. [Google Scholar] [CrossRef] [PubMed]
- Carsetti, L.; Laurenti, L.; Gobessi, S.; Longo, P.G.; Leone, G.; Efremov, D.G. Phosphorylation of the activation loop tyrosines is required for sustained Syk signaling and growth factor-independent B-cell proliferation. Cell. Signal. 2009, 21, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Kuno, Y.; Abe, A.; Emi, N.; Iida, M.; Yokozawa, T.; Towatari, M.; Tanimoto, M.; Saito, H. Constitutive kinase activation of the TEL-Syk fusion gene in myelodysplastic syndrome with t(9;12)(q22;p12). Blood 2001, 97, 1050–1055. [Google Scholar] [CrossRef] [PubMed]
- Dolmetsch, R.E.; Lewis, R.S.; Goodnow, C.C.; Healy, J.I. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 1997, 386, 855–858. [Google Scholar] [CrossRef] [PubMed]
- DeRose, R.; Miyamoto, T.; Inoue, T. Manipulating signaling at will: Chemically-inducible dimerization (CID) techniques resolve problems in cell biology. Pflug. Arch. Eur. J. Physiol. 2013, 465, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.; Wandless, T.; Schreiber, S.; Crabtree, G. Controlling signal transduction with synthetic ligands. Science 1993, 262, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Kopytek, S.J.; Standaert, R.F.; Dyer, J.C.D.; Hu, J.C. Chemically induced dimerization of dihydrofolate reductase by a homobifunctional dimer of methotrexate. Chem. Biol. 2000, 7, 313–321. [Google Scholar] [CrossRef]
- Sasso, S.P.; Gilli, R.M.; Sari, J.C.; Rimet, O.S.; Briand, C.M. Thermodynamic study of dihydrofolate reductase inhibitor selectivity. Biochim. Biophys. Acta 1994, 1207, 74–79. [Google Scholar] [CrossRef]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Araujo, E.; Zhao, T.; Zhang, M.; Massenburg, D.; Veselits, M.; Doyle, C.; Dinner, A.R.; Clark, M.R. B cell antigen receptor signaling and internalization are mutually exclusive events. PLoS Biol. 2006, 4, e200. [Google Scholar] [CrossRef] [PubMed]
- Busman-Sahay, K.; Drake, L.; Sitaram, A.; Marks, M.; Drake, J.R. Cis and Trans Regulatory Mechanisms Control AP2-Mediated B Cell Receptor Endocytosis via Select Tyrosine-Based Motifs. PLoS ONE 2013, 8, e54938. [Google Scholar] [CrossRef] [PubMed]
- Abudula, A.; Grabbe, A.; Brechmann, M.; Polaschegg, C.; Herrmann, N.; Goldbeck, I.; Dittmann, K.; Wienands, J. SLP-65 signal transduction requires Src homology 2 domain-mediated membrane anchoring and a kinase-independent adaptor function of Syk. J. Biol. Chem. 2007, 282, 29059–29066. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Kurosaki, T. Role of Calcium Signaling in B Cell Activation and Biology. In B Cell Receptor Signaling; Kurosaki, T., Wienands, J., Eds.; Springer International Publishing: Cham, Germany, 2016; Volume 393, pp. 143–174. [Google Scholar]
- Gauld, S.B.; Benschop, R.J.; Merrell, K.T.; Cambier, J.C. Maintenance of B cell anergy requires constant antigen receptor occupancy and signaling. Nat. Immunol. 2005, 6, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Scharenberg, A.M.; Humphries, L.A.; Rawlings, D.J. Calcium signalling and cell-fate choice in B cells. Nat. Rev. Immunol. 2007, 7, 778–789. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Westbroek, M.L.; Geahlen, R.L. Modulation of BCR Signaling by the Induced Dimerization of Receptor-Associated SYK. Antibodies 2017, 6, 23. https://doi.org/10.3390/antib6040023
Westbroek ML, Geahlen RL. Modulation of BCR Signaling by the Induced Dimerization of Receptor-Associated SYK. Antibodies. 2017; 6(4):23. https://doi.org/10.3390/antib6040023
Chicago/Turabian StyleWestbroek, Mark L., and Robert L. Geahlen. 2017. "Modulation of BCR Signaling by the Induced Dimerization of Receptor-Associated SYK" Antibodies 6, no. 4: 23. https://doi.org/10.3390/antib6040023
APA StyleWestbroek, M. L., & Geahlen, R. L. (2017). Modulation of BCR Signaling by the Induced Dimerization of Receptor-Associated SYK. Antibodies, 6(4), 23. https://doi.org/10.3390/antib6040023