Antagonist Anti-CD28 Therapeutics for the Treatment of Autoimmune Disorders



 ,
, 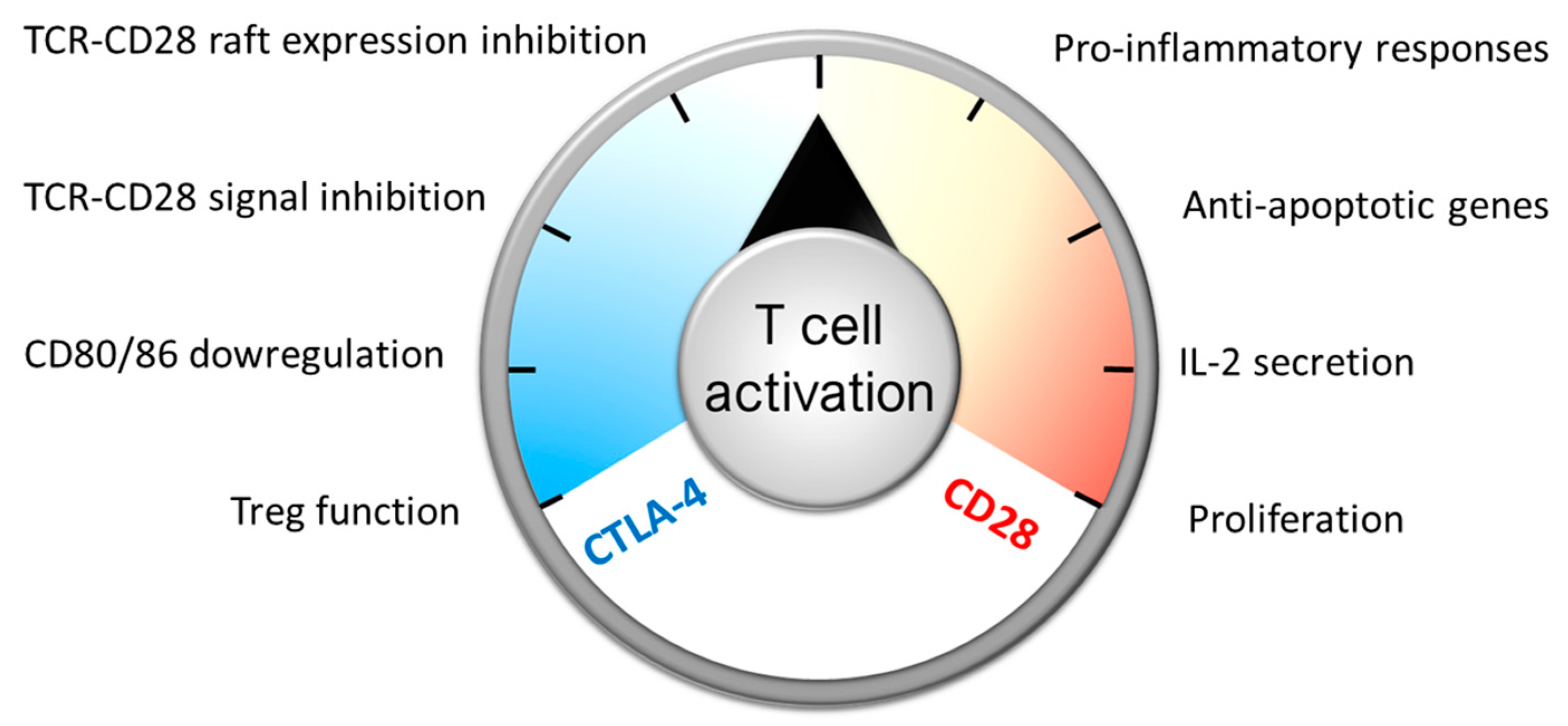{kind=link}
Abstract
1. Introduction
2. Models of Immune-Mediated Diseases of the Skin
3. Models for Neuroinflammatory Diseases
4. Arthritis Models
5. Autoimmune Uveitis Model
6. Lupus Nephritis
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Acuto, O.; Michel, F. CD28-mediated co-stimulation: A quantitative support for TCR signalling. Nat. Rev. Immunol. 2003, 3, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.J.; Heuts, F.; Ovcinnikovs, V.; Wardzinski, L.; Bowers, C.; Schmidt, E.M.; Kogimtzis, A.; Kenefeck, R.; Sansom, D.M.; Walker, L.S. CTLA-4 controls follicular helper T-cell differentiation by regulating the strength of CD28 engagement. Proc. Natl. Acad. Sci. USA 2015, 112, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Marengere, L.E.; Waterhouse, P.; Duncan, G.S.; Mittrucker, H.W.; Feng, G.S.; Mak, T.W. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science 1996, 272, 1170–1173. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Schneider, H.; Azouz, A.; Rudd, C.E. Cytotoxic T lymphocyte antigen 4 and CD28 modulate cell surface raft expression in their regulation of T cell function. J. Exp. Med. 2001, 194, 1675–1681. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef]
- Ueda, H.; Howson, J.M.; Esposito, L.; Heward, J.; Snook, H.; Chamberlain, G.; Rainbow, D.B.; Hunter, K.M.; Smith, A.N.; Di Genova, G.; et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 2003, 423, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Ciszak, L.; Frydecka, I.; Wolowiec, D.; Szteblich, A.; Kosmaczewska, A. Patients with chronic lymphocytic leukaemia (CLL) differ in the pattern of CTLA-4 expression on CLL cells: The possible implications for immunotherapy with CTLA-4 blocking antibody. Tumour Biol. 2016, 37, 4143–4157. [Google Scholar] [CrossRef] [PubMed]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 2007, 27, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Poirier, N.; Azimzadeh, A.M.; Zhang, T.; Dilek, N.; Mary, C.; Nguyen, B.; Tillou, X.; Wu, G.; Reneaudin, K.; Hervouet, J.; et al. Inducing CTLA-4-dependent immune regulation by selective CD28 blockade promotes regulatory T cells in organ transplantation. Sci. Transl. Med. 2010, 2, 17ra10. [Google Scholar] [CrossRef] [PubMed]
- Dilek, N.; Poirier, N.; Hulin, P.; Coulon, F.; Mary, C.; Ville, S.; Vie, H.; Clemenceau, B.; Blancho, G.; Vanhove, B. Targeting CD28, CTLA-4 and PD-L1 costimulation differentially controls immune synapses and function of human regulatory and conventional T-cells. PLoS ONE 2013, 8, e83139. [Google Scholar] [CrossRef] [PubMed]
- Charbonnier, L.M.; Vokaer, B.; Lemaitre, P.H.; Field, K.A.; Leo, O.; Le Moine, A. CTLA4-Ig restores rejection of MHC class-II mismatched allografts by disabling IL-2-expanded regulatory T cells. Am. J. Transplant. 2012, 12, 2313–2321. [Google Scholar] [CrossRef] [PubMed]
- Vogel, I.; Kasran, A.; Cremer, J.; Kim, Y.J.; Boon, L.; van Gool, S.W.; Ceuppens, J.L. CD28/CTLA-4/B7 costimulatory pathway blockade affects regulatory T-cell function in autoimmunity. Eur J. Immunol. 2015, 45, 1832–1841. [Google Scholar] [CrossRef] [PubMed]
- Zaitsu, M.; Issa, F.; Hester, J.; Vanhove, B.; Wood, K.J. Selective blockade of CD28 on human T cells facilitates regulation of alloimmune responses. JCI Insight 2017, 2, 89381. [Google Scholar] [CrossRef] [PubMed]
- Vanhove, B.; Laflamme, G.; Coulon, F.; Mougin, M.; Vusio, P.; Haspot, F.; Tiollier, J.; Soulillou, J.P. Selective blockade of CD28 and not CTLA-4 with a single-chain Fv-alpha1-antitrypsin fusion antibody. Blood 2003, 102, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Suchard, S.J.; Davis, P.M.; Kansal, S.; Stetsko, D.K.; Brosius, R.; Tamura, J.; Schneeweis, L.; Bryson, J.; Salcedo, T.; Wang, H.; et al. A monovalent anti-human CD28 domain antibody antagonist: Preclinical efficacy and safety. J. Immunol. 2013, 191, 4599–4610. [Google Scholar] [CrossRef] [PubMed]
- Krummey, S.M.; Floyd, T.L.; Liu, D.; Wagener, M.E.; Song, M.; Ford, M.L. Candida-elicited murine Th17 cells express high Ctla-4 compared with Th1 cells and are resistant to costimulation blockade. J. Immunol. 2014, 192, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- Poirier, N.; Blancho, G.; Vanhove, B. CD28-specific immunomodulating antibodies: What can be learned from experimental models? Am. J. Transplant. 2012, 12, 1682–1690. [Google Scholar] [CrossRef] [PubMed]
- Mary, C.; Coulon, F.; Poirier, N.; Dilek, N.; Martinet, B.; Blancho, G.; Vanhove, B. Antagonist properties of monoclonal antibodies targeting human CD28: Role of valency and the heavy-chain constant domain. mAbs 2013, 5, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Dengler, T.J.; Szabo, G.; Sido, B.; Nottmeyer, W.; Zimmerman, R.; Vahl, C.F.; Hunig, T.; Meuer, S.C. Prolonged allograft survival but no tolerance induction by modulating CD28 antibody JJ319 after high-responder rat heart transplantation. Transplantation 1999, 67, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Luhder, F.; Huang, Y.; Dennehy, K.M.; Guntermann, C.; Muller, I.; Winkler, E.; Kerkau, T.; Ikemizu, S.; Davis, S.J.; Hanke, T.; et al. Topological requirements and signaling properties of T cell-activating, anti-CD28 antibody superagonists. J. Exp. Med. 2003, 197, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.S.; Pan, F.; Erickson, L.M.; Fisniku, O.; Crews, G.; Wynn, C.; Hong, I.C.; Tamura, K.; Kobayashi, M.; Jiang, H. A blocking anti-CD28-specific antibody induces long-term heart allograft survival by suppression of the PKC theta-JNK signal pathway. Transplantation 2008, 85, 1051–1055. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.P.; Kundu-Raychaudhuri, S.; Tamura, K.; Masunaga, T.; Kubo, K.; Hanaoka, K.; Jiang, W.Y.; Herzenberg, L.A.; Herzenberg, L.A. FR255734, a humanized, Fc-Silent, Anti-CD28 antibody, improves psoriasis in the SCID mouse-psoriasis xenograft model. J. Investig. Dermatol. 2008, 128, 1969–1976. [Google Scholar] [CrossRef] [PubMed]
- Shiao, S.L.; McNiff, J.M.; Masunaga, T.; Tamura, K.; Kubo, K.; Pober, J.S. Immunomodulatory properties of FK734, a humanized anti-CD28 monoclonal antibody with agonistic and antagonistic activities. Transplantation 2007, 83, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Poirier, N.; Mary, C.; Dilek, N.; Hervouet, J.; Minault, D.; Blancho, G.; Vanhove, B. Preclinical efficacy and immunological safety of FR104, an antagonist anti-CD28 monovalent Fab’ antibody. Am. J. Transplant. 2012, 12, 2630–2640. [Google Scholar] [CrossRef] [PubMed]
- Poirier, N.; Blancho, G.; Hiance, M.; Mary, C.; van Assche, T.; Lempoels, J.; Ramael, S.; Wang, W.; Thepenier, V.; Braudeau, C.; et al. First-in-Human Study in Healthy Subjects with FR104, a Pegylated Monoclonal Antibody Fragment Antagonist of CD28. J. Immunol. 2016, 197, 4593–4602. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Haase, I.; Nestle, F.O. Mechanisms regulating skin immunity and inflammation. Nat. Rev. Immunol. 2014, 14, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Kaplan, D.H.; Barker, J. Psoriasis. N. Engl. J. Med. 2009, 361, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Boehncke, W.H.; Brembilla, N.C. Unmet Needs in the Field of Psoriasis: Pathogenesis and Treatment. Clin. Rev. Allergy Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, J.E.; Chan, T.C.; Krueger, J.G. Psoriasis pathogenesis and the development of novel targeted immune therapies. J. Allergy Clin. Immunol. 2017, 140, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Abrams, J.R.; Lebwohl, M.G.; Guzzo, C.A.; Jegasothy, B.V.; Goldfarb, M.T.; Goffe, B.S.; Menter, A.; Lowe, N.J.; Krueger, G.; Brown, M.J.; et al. CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J. Clin. Investig. 1999, 103, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; Gottlieb, A.B.; van der Heijde, D.; FitzGerald, O.; Johnsen, A.; Nys, M.; Banerjee, S.; Gladman, D.D. Efficacy and safety of abatacept, a T-cell modulator, in a randomised, double-blind, placebo-controlled, phase III study in psoriatic arthritis. Ann. Rheum. Dis. 2017, 76, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Krummey, S.M.; Cheeseman, J.A.; Conger, J.A.; Jang, P.S.; Mehta, A.K.; Kirk, A.D.; Larsen, C.P.; Ford, M.L. High CTLA-4 expression on Th17 cells results in increased sensitivity to CTLA-4 coinhibition and resistance to belatacept. Am. J. Transplant. 2014, 14, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Malvezzi, P.; Jouve, T.; Rostaing, L. Costimulation Blockade in Kidney Transplantation: An Update. Transplantation 2016, 100, 2315–2323. [Google Scholar] [CrossRef] [PubMed]
- Vincenti, F.; Charpentier, B.; Vanrenterghem, Y.; Rostaing, L.; Bresnahan, B.; Darji, P.; Massari, P.; Mondragon-Ramirez, G.A.; Agarwal, M.; di Russo, G.; et al. A phase III study of belatacept-based immunosuppression regimens versus cyclosporine in renal transplant recipients (BENEFIT study). Am. J. Transplant. 2010, 10, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Issa, F.; Hester, J.; Goto, R.; Nadig, S.N.; Goodacre, T.E.; Wood, K. Ex vivo-expanded human regulatory T cells prevent the rejection of skin allografts in a humanized mouse model. Transplantation 2010, 90, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Fresnay, S.; Welty, E.; Sangrampurkar, N.; Rybak, E.; Zhou, H.; Cheng, X.F.; Feng, Q.; Avon, C.; Laaris, A.; et al. Selective CD28 blockade attenuates acute and chronic rejection of murine cardiac allografts in a CTLA-4-dependent manner. Am. J. Transplant. 2011, 11, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Issa, F.; Hester, J.; Milward, K.; Wood, K.J. Homing of regulatory T cells to human skin is important for the prevention of alloimmune-mediated pathology in an in vivo cellular therapy model. PLoS ONE 2012, 7, e53331. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Flutter, B.; Rodriguez, R.S.; Sharif-Paghaleh, E.; Barber, L.D.; Lombardi, G.; Nestle, F.O. Xenogeneic graft-versus-host-disease in NOD-scid IL-2Rgammanull mice display a T-effector memory phenotype. PLoS ONE 2012, 7, e44219. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.; Anasetti, C.; Hansen, J.A.; Melrose, J.; Brunvand, M.; Bradshaw, J.; Ledbetter, J.A.; Linsley, P.S. Induction of alloantigen-specific hyporesponsiveness in human T lymphocytes by blocking interaction of CD28 with its natural ligand B7/BB1. J. Exp. Med. 1993, 177, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Riella, L.V.; Liu, T.; Yang, J.; Chock, S.; Shimizu, T.; Mfarrej, B.; Batal, I.; Xiao, X.; Sayegh, M.H.; Chandraker, A. Deleterious effect of CTLA4-Ig on a Treg-dependent transplant model. Am. J. Transplant. 2012, 12, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Levitsky, J.; Miller, J.; Huang, X.; Chandrasekaran, D.; Chen, L.; Mathew, J.M. Inhibitory effects of belatacept on allospecific regulatory T-cell generation in humans. Transplantation 2013, 96, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Gilliet, M.; Conrad, C.; Geiges, M.; Cozzio, A.; Thurlimann, W.; Burg, G.; Nestle, F.O.; Dummer, R. Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch. Dermatol. 2004, 140, 1490–1495. [Google Scholar] [CrossRef] [PubMed]
- Patel, U.; Mark, N.M.; Machler, B.C.; Levine, V.J. Imiquimod 5% cream induced psoriasis: A case report, summary of the literature and mechanism. Br. J. Dermatol. 2011, 164, 670–672. [Google Scholar] [CrossRef] [PubMed]
- Rajan, N.; Langtry, J.A. Generalized exacerbation of psoriasis associated with imiquimod cream treatment of superficial basal cell carcinomas. Clin. Exp. Dermatol. 2006, 31, 140–141. [Google Scholar] [CrossRef] [PubMed]
- Vinter, H.; Iversen, L.; Steiniche, T.; Kragballe, K.; Johansen, C. Aldara(R)-induced skin inflammation: Studies of patients with psoriasis. Br. J. Dermatol. 2015, 172, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Van der Fits, L.; Mourits, S.; Voerman, J.S.; Kant, M.; Boon, L.; Laman, J.D.; Cornelissen, F.; Mus, A.M.; Florencia, E.; Prens, E.P.; et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J. Immunol. 2009, 182, 5836–5845. [Google Scholar] [CrossRef] [PubMed]
- Pantelyushin, S.; Haak, S.; Ingold, B.; Kulig, P.; Heppner, F.L.; Navarini, A.A.; Becher, B. Rorgammat+ innate lymphocytes and gammadelta T cells initiate psoriasiform plaque formation in mice. J. Clin. Investig. 2012, 122, 2252–2256. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Shen, X.; Ding, C.; Qi, C.; Li, K.; Li, X.; Jala, V.R.; Zhang, H.G.; Wang, T.; Zheng, J.; et al. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity 2011, 35, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Poirier, N.; Chevalier, M.; Mary, C.; Hervouet, J.; Minault, D.; le Bas-Bernardet, S.; Belarif, L.; Daguin, V.; Cassagnau, E.; Vanhove, B.; et al. Selective CD28 antagonist prevents induced skin inflammation in non-human primates. Exp. Dermatol. 2016, 25, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Kaneda, K.; Kasama, T. Immunopathogenesis of delayed-type hypersensitivity. Microsc. Res. Tech. 2001, 53, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Poirier, N.; Haudebourg, T.; Brignone, C.; Dilek, N.; Hervouet, J.; Minault, D.; Coulon, F.; de Silly, R.V.; Triebel, F.; Blancho, G.; et al. Antibody-mediated depletion of lymphocyte-activation gene-3 (LAG-3(+))-activated T lymphocytes prevents delayed-type hypersensitivity in non-human primates. Clin. Exp. Immunol. 2011, 164, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Poirier, N.; Chevalier, M.; Mary, C.; Hervouet, J.; Minault, D.; Baker, P.; Ville, S.; le Bas-Bernardet, S.; Dilek, N.; Belarif, L.; et al. Selective CD28 Antagonist Blunts Memory Immune Responses and Promotes Long-Term Control of Skin Inflammation in Nonhuman Primates. J. Immunol. 2016, 196, 274–283. [Google Scholar] [CrossRef] [PubMed]
- ’t Hart, B.A.; Gran, B.; Weissert, R. EAE: Imperfect but useful models of multiple sclerosis. Trends Mol. Med. 2011, 17, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Haanstra, K.G.; Dijkman, K.; Bashir, N.; Bauer, J.; Mary, C.; Poirier, N.; Baker, P.; Scobie, L.; ’t Hart, B.A.; Vanhove, B. Selective blockade of CD28-mediated T cell costimulation protects rhesus monkeys against acute fatal experimental autoimmune encephalomyelitis. J. Immunol. 2015, 194, 1454–1466, Correction in 2015, 195, 749. [Google Scholar] [CrossRef] [PubMed]
- Haanstra, K.G.; Hofman, S.O.; Estevao, D.M.L.; Blezer, E.L.; Bauer, J.; Yang, L.L.; Wyant, T.; Csizmadia, V.; ’t Hart, B.A.; Fedyk, E.R. Antagonizing the alpha4beta1 integrin, but not alpha4beta7, inhibits leukocytic infiltration of the central nervous system in rhesus monkey experimental autoimmune encephalomyelitis. J. Immunol. 2013, 190, 1961–1973. [Google Scholar] [CrossRef] [PubMed]
- Vierboom, M.P.; Breedveld, E.; Kap, Y.S.; Mary, C.; Poirier, N.; ’t Hart, B.A.; Vanhove, B. Clinical efficacy of a new CD28-targeting antagonist of T cell co-stimulation in a non-human primate model of collagen-induced arthritis. Clin. Exp. Immunol. 2016, 183, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Zhu, Y.; Zhu, G.; Augustine, M.; Zheng, L.; Goode, D.J.; Broadwater, M.; Ruff, W.; Flies, S.; Xu, H.; et al. B7-h2 is a costimulatory ligand for CD28 in human. Immunity 2011, 34, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Ville, S.; Poirier, N.; Blancho, G.; Vanhove, B. Co-Stimulatory Blockade of the CD28/CD80–86/CTLA-4 Balance in Transplantation: Impact on Memory T Cells? Front. Immunol. 2015, 6, 411. [Google Scholar] [CrossRef] [PubMed]
- Whitcup, S.M.; Nussenblatt, R.B. Immunologic mechanisms of uveitis. New targets for immunomodulation. Arch. Ophthalmol. 1997, 115, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Gritz, D.C.; Wong, I.G. Incidence and prevalence of uveitis in Northern California; the Northern California Epidemiology of Uveitis Study. Ophthalmology 2004, 111, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Comarmond, C.; Wechsler, B.; Bodaghi, B.; Cacoub, P.; Saadoun, D. Biotherapies in Behcet’s disease. Autoimmun. Rev. 2014, 13, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Selmi, C.; Gershwin, M.E. Diagnosis and classification of reactive arthritis. Autoimmun. Rev. 2014, 13, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Jamilloux, Y.; Kodjikian, L.; Broussolle, C.; Seve, P. Sarcoidosis and uveitis. Autoimmun. Rev. 2014, 13, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Sakata, V.M.; da Silva, F.T.; Hirata, C.E.; Takahashi, W.Y.; Costa, R.A.; Yamamoto, J.H. Choroidal bulging in patients with Vogt-Koyanagi-Harada disease in the non-acute uveitic stage. J. Ophthalmic Inflamm. Infect. 2014, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Lee, C.; Phatak, S.; Pavesio, C. Immunopharmacotherapy of non-infectious uveitis: Where do we stand? Expert Opin. Biol. Ther. 2014, 14, 1719–1722. [Google Scholar] [CrossRef] [PubMed]
- Nussenblatt, R.B.; Fortin, E.; Schiffman, R.; Rizzo, L.; Smith, J.; van Veldhuisen, P.; Sran, P.; Yaffe, A.; Goldman, C.K.; Waldmann, T.A.; et al. Treatment of noninfectious intermediate and posterior uveitis with the humanized anti-Tac mAb: A phase I/II clinical trial. Proc. Natl. Acad. Sci. USA 1999, 96, 7462–7466. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.R.; Levinson, R.D.; Holland, G.N.; Jabs, D.A.; Robinson, M.R.; Whitcup, S.M.; Rosenbaum, J.T. Differential efficacy of tumor necrosis factor inhibition in the management of inflammatory eye disease and associated rheumatic disease. Arthritis Rheum. 2001, 45, 252–257. [Google Scholar] [CrossRef]
- Papotto, P.H.; Marengo, E.B.; Sardinha, L.R.; Goldberg, A.C.; Rizzo, L.V. Immunotherapeutic strategies in autoimmune uveitis. Autoimmun. Rev. 2014, 13, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.R.; Roberge, F.G.; Chan, C.C.; Wiggert, B.; Chader, G.J.; Rozenszajn, L.A.; Lando, Z.; Nussenblatt, R.B. A new model of autoimmune disease. Experimental autoimmune uveoretinitis induced in mice with two different retinal antigens. J. Immunol. 1988, 140, 1490–1495. [Google Scholar] [PubMed]
- Rizzo, L.V.; Silver, P.; Wiggert, B.; Hakim, F.; Gazzinelli, R.T.; Chan, C.C.; Caspi, R.R. Establishment and characterization of a murine CD4+ T cell line and clone that induce experimental autoimmune uveoretinitis in B10.A mice. J. Immunol. 1996, 156, 1654–1660. [Google Scholar] [PubMed]
- Tang, J.; Zhu, W.; Silver, P.B.; Su, S.B.; Chan, C.C.; Caspi, R.R. Autoimmune uveitis elicited with antigen-pulsed dendritic cells has a distinct clinical signature and is driven by unique effector mechanisms: Initial encounter with autoantigen defines disease phenotype. J. Immunol. 2007, 178, 5578–5587. [Google Scholar] [CrossRef] [PubMed]
- Luger, D.; Silver, P.B.; Tang, J.; Cua, D.; Chen, Z.; Iwakura, Y.; Bowman, E.P.; Sgambellone, N.M.; Chan, C.C.; Caspi, R.R. Either a Th17 or a Th1 effector response can drive autoimmunity: Conditions of disease induction affect dominant effector category. J. Exp. Med. 2008, 205, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Papotto, P.H.; Marengo, E.B.; Sardinha, L.R.; Carvalho, K.I.; de Carvalho, A.E.; Castillo-Mendez, S.; Jank, C.C.; Vanhove, B.; Goldberg, A.C.; Rizzo, L.V. Novel CD28 antagonist mPEG PV1-Fab’ mitigates experimental autoimmune uveitis by suppressing CD4+ T lymphocyte activation and IFN-gamma production. PLoS ONE 2017, 12, e0171822. [Google Scholar] [CrossRef] [PubMed]
- Silver, P.B.; Hathcock, K.S.; Chan, C.C.; Wiggert, B.; Caspi, R.R. Blockade of costimulation through B7/CD28 inhibits experimental autoimmune uveoretinitis, but does not induce long-term tolerance. J. Immunol. 2000, 165, 5041–5047. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.T. Co-stimulatory molecules as targets for treatment of lupus. Clin. Immunol. 2013, 148, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.K.; Linsley, P.S.; Wofsy, D. Treatment of murine lupus with CTLA4Ig. Science 1994, 265, 1225–1227. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.; Nicholls, K.; Cheng, T.T.; Houssiau, F.; Burgos-Vargas, R.; Chen, S.L.; Hillson, J.L.; Meadows-Shropshire, S.; Kinaszczuk, M.; Merrill, J.T. Efficacy and safety of abatacept in lupus nephritis: A twelve-month, randomized, double-blind study. Arthritis Rheum. 2014, 66, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.T.; Burgos-Vargas, R.; Westhovens, R.; Chalmers, A.; D’Cruz, D.; Wallace, D.J.; Bae, S.C.; Sigal, L.; Becker, J.C.; Kelly, S.; et al. The efficacy and safety of abatacept in patients with non-life-threatening manifestations of systemic lupus erythematosus: Results of a twelve-month, multicenter, exploratory, phase IIb, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2010, 62, 3077–3087. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.; le Fur, A.; Bloas, R.L.; Neel, M.; Mary, C.; Moreau, A.; Poirier, N.; Vanhove, B.; Fakhouri, F. Prevention of lupus nephritis development in NZB/NZW mice by selective blockade of CD28. Eur. J. Immunol. 2017, 47, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Abe, J.; Ueha, S.; Suzuki, J.; Tokano, Y.; Matsushima, K.; Ishikawa, S. Increased Foxp3(+) CD4(+) regulatory T cells with intact suppressive activity but altered cellular localization in murine lupus. Am. J. Pathol. 2008, 173, 1682–1692. [Google Scholar] [CrossRef] [PubMed]
- Haspot, F.; Seveno, C.; Dugast, A.S.; Coulon, F.; Renaudin, K.; Usal, C.; Hill, M.; Anegon, I.; Heslan, M.; Josien, R.; et al. Anti-CD28 antibody-induced kidney allograft tolerance related to tryptophan degradation and TCR class II B7 regulatory cells. Am. J. Transplant. 2005, 5, 2339–2348. [Google Scholar] [CrossRef] [PubMed]
- Ville, S.; Poirier, N.; Branchereau, J.; Charpy, V.; Pengam, S.; Nerriere-Daguin, V.; le Bas-Bernardet, S.; Coulon, F.; Mary, C.; Chenouard, A.; et al. Anti-CD28 Antibody and Belatacept Exert Differential Effects on Mechanisms of Renal Allograft Rejection. J. Am. Soc. Nephrol. 2016, 27, 3577–3588. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Honczarenko, M.; Zhang, S.; Fleener, C.; Mora, J.; Lee, S.K.; Wang, R.; Liu, X.; Shevell, D.E.; Yang, Z.; et al. Pharmacokinetic, Pharmacodynamic, and Safety Profile of a Novel Anti-CD28 Domain Antibody Antagonist in Healthy Subjects. J. Clin. Pharmacol. 2017, 57, 161–172. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanhove, B.; Poirier, N.; Fakhouri, F.; Laurent, L.; ’t Hart, B.; Papotto, P.H.; Rizzo, L.V.; Zaitsu, M.; Issa, F.; Wood, K.; et al. Antagonist Anti-CD28 Therapeutics for the Treatment of Autoimmune Disorders. Antibodies 2017, 6, 19. https://doi.org/10.3390/antib6040019
Vanhove B, Poirier N, Fakhouri F, Laurent L, ’t Hart B, Papotto PH, Rizzo LV, Zaitsu M, Issa F, Wood K, et al. Antagonist Anti-CD28 Therapeutics for the Treatment of Autoimmune Disorders. Antibodies. 2017; 6(4):19. https://doi.org/10.3390/antib6040019
Chicago/Turabian StyleVanhove, Bernard, Nicolas Poirier, Fadi Fakhouri, Laetitia Laurent, Bert ’t Hart, Pedro H. Papotto, Luiz V. Rizzo, Masaaki Zaitsu, Fadi Issa, Kathryn Wood, and et al. 2017. "Antagonist Anti-CD28 Therapeutics for the Treatment of Autoimmune Disorders" Antibodies 6, no. 4: 19. https://doi.org/10.3390/antib6040019
APA StyleVanhove, B., Poirier, N., Fakhouri, F., Laurent, L., ’t Hart, B., Papotto, P. H., Rizzo, L. V., Zaitsu, M., Issa, F., Wood, K., Soulillou, J.-P., & Blancho, G. (2017). Antagonist Anti-CD28 Therapeutics for the Treatment of Autoimmune Disorders. Antibodies, 6(4), 19. https://doi.org/10.3390/antib6040019