
Generating Recombinant Antibodies to Membrane Proteins through Phage Display

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
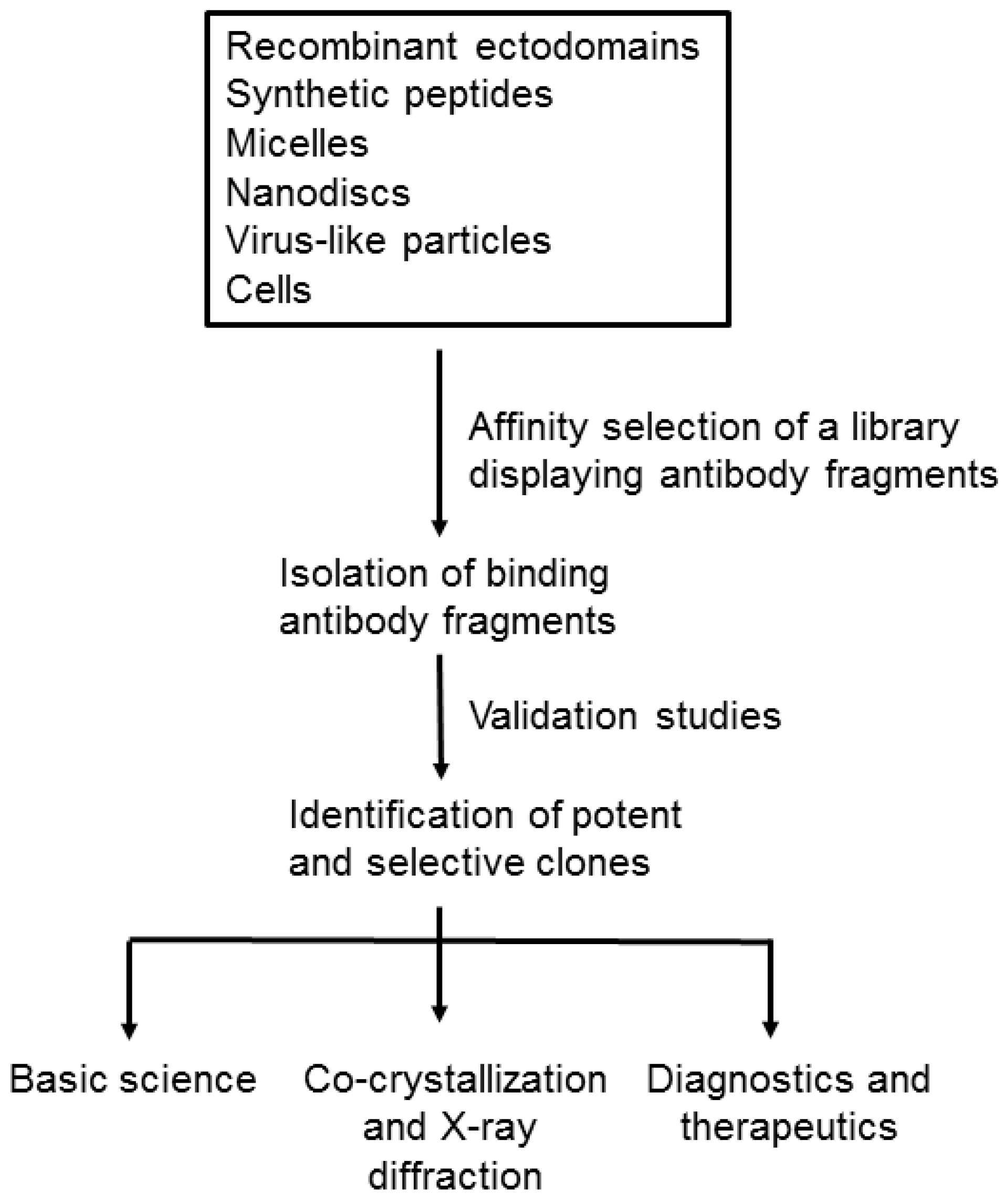
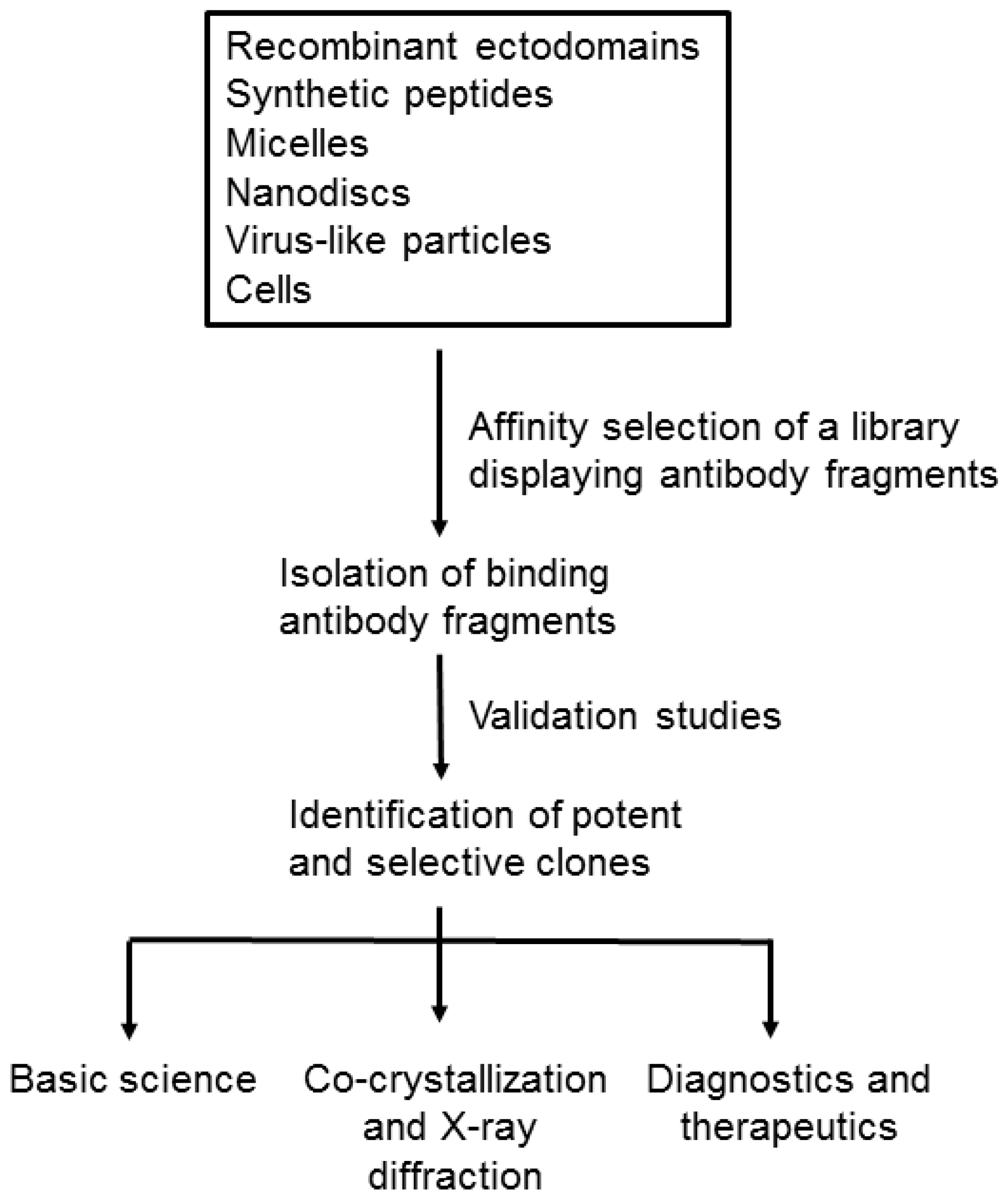
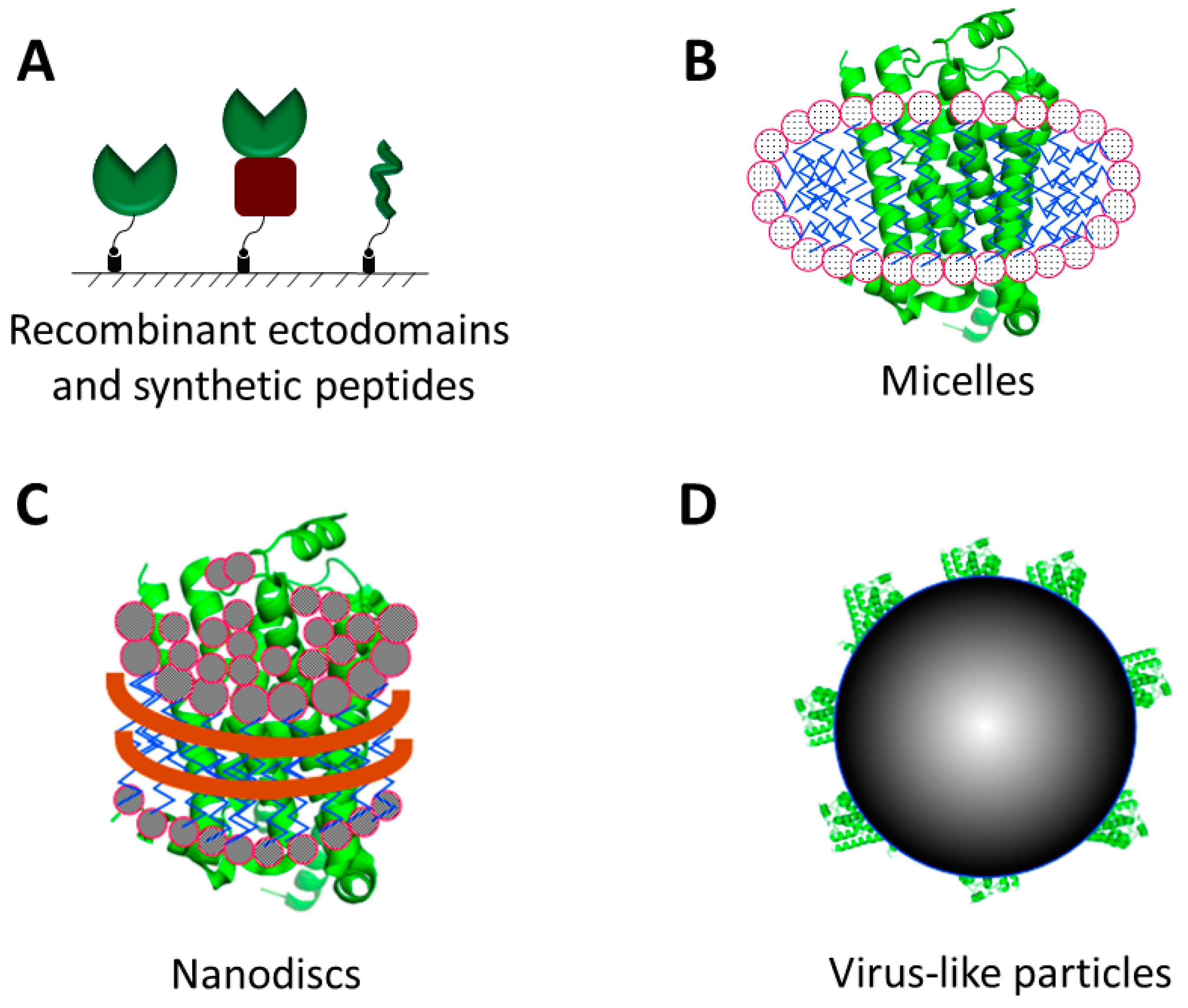
2. Formats of Membrane Proteins for Affinity Selection
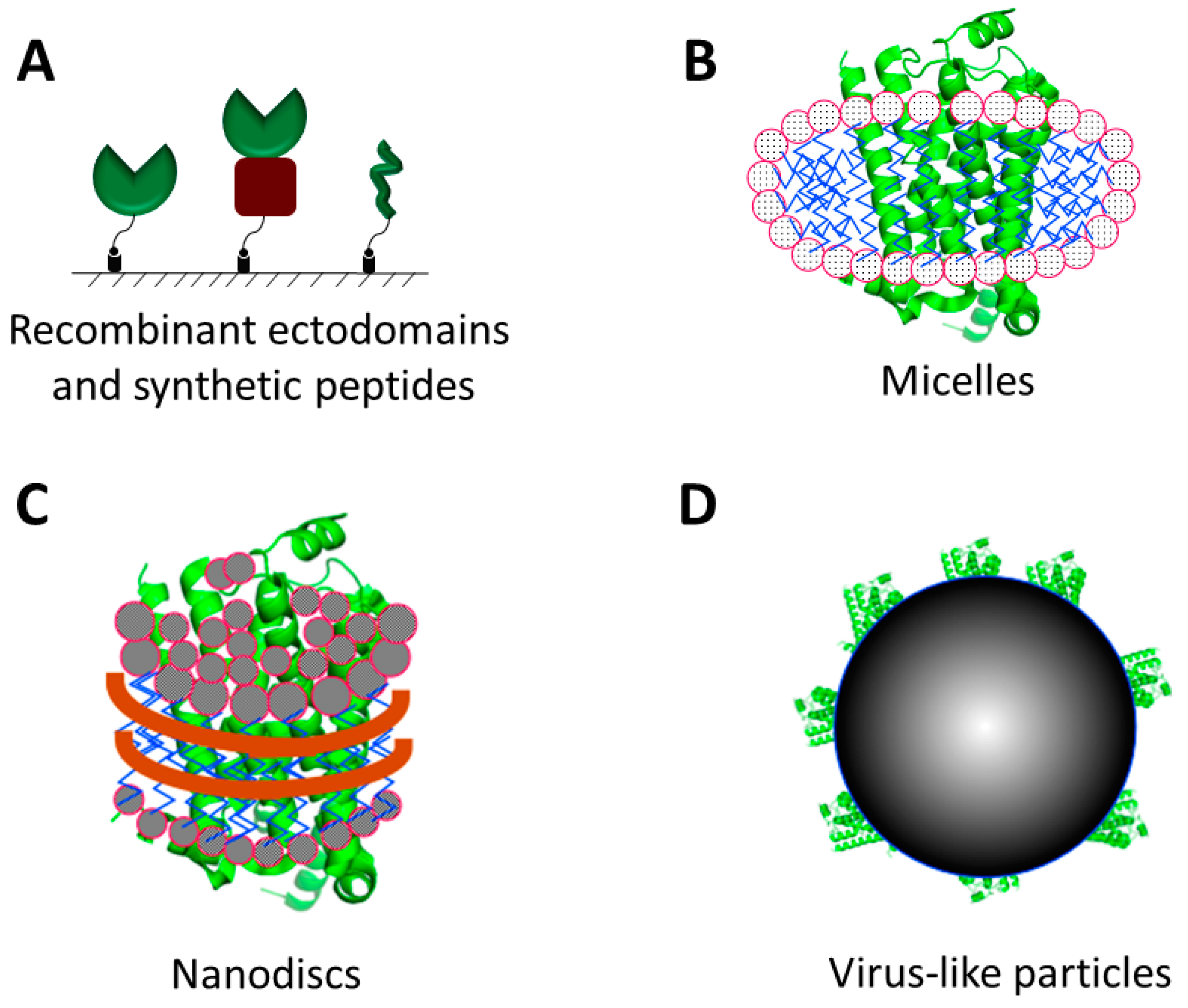
2.1. Recombinant Proteins and Synthetic Peptides
2.2. Detergent Micelles, Liposomes and Nanodiscs
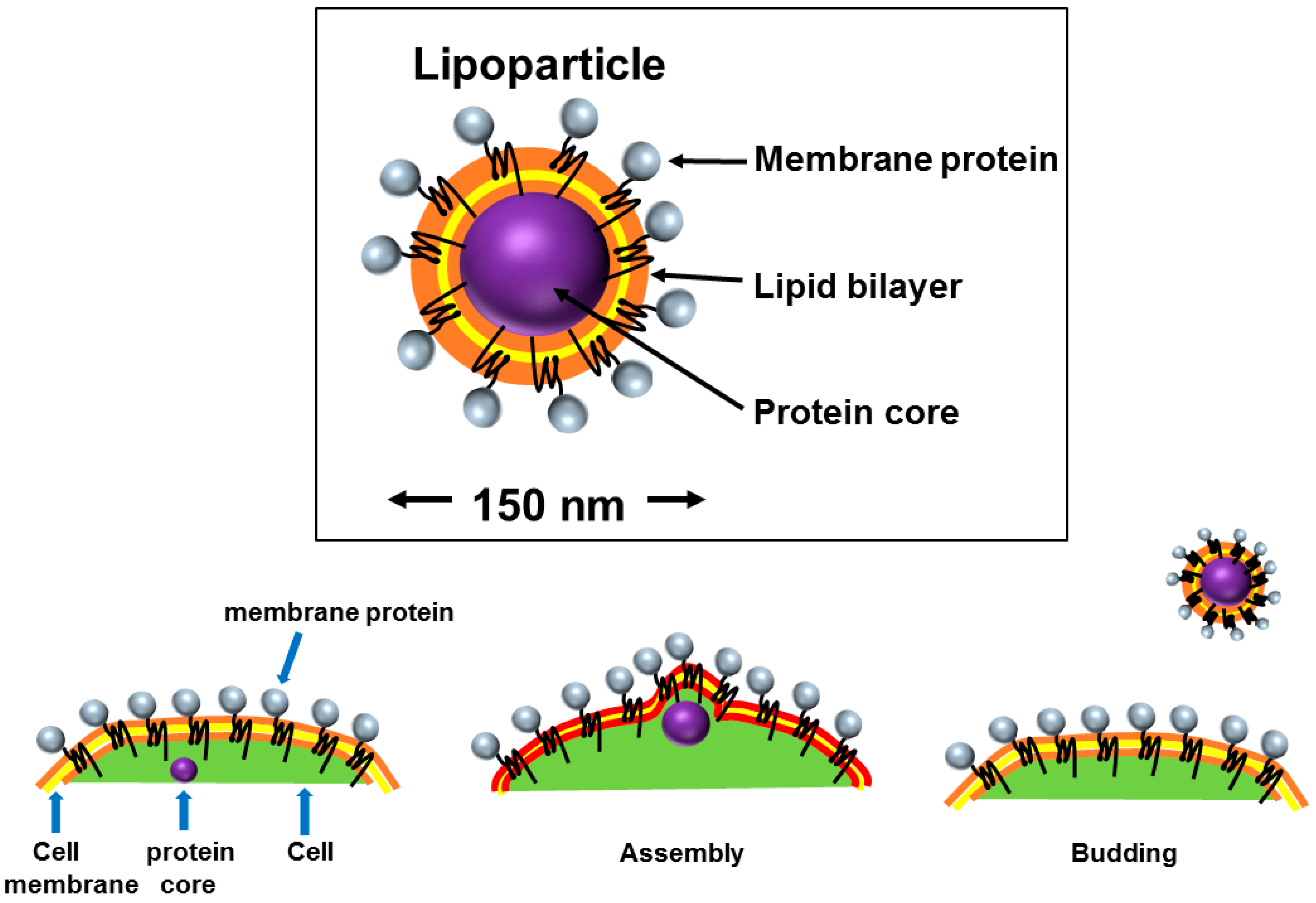
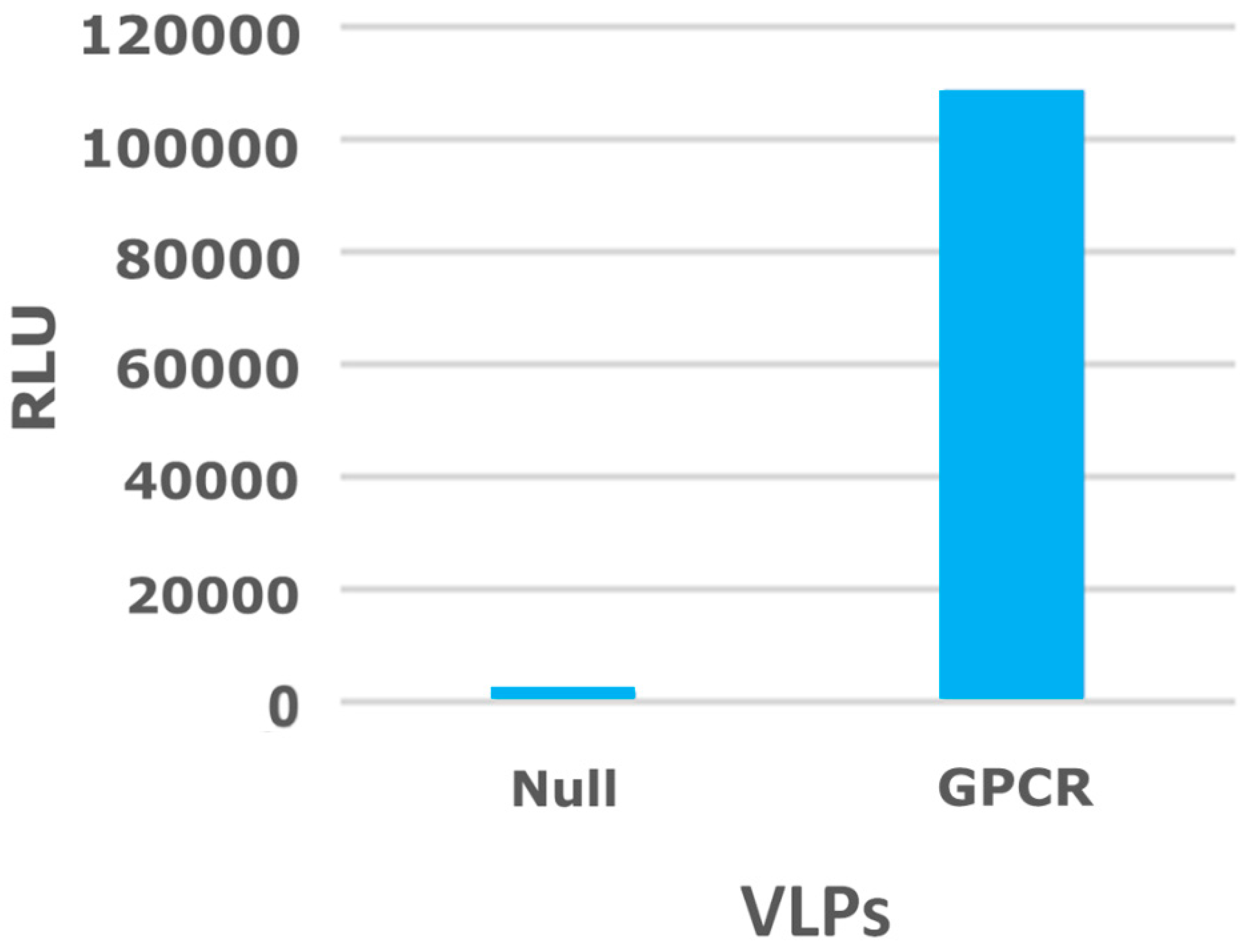
2.3. Virus-Like Particles
2.4. Native Cells
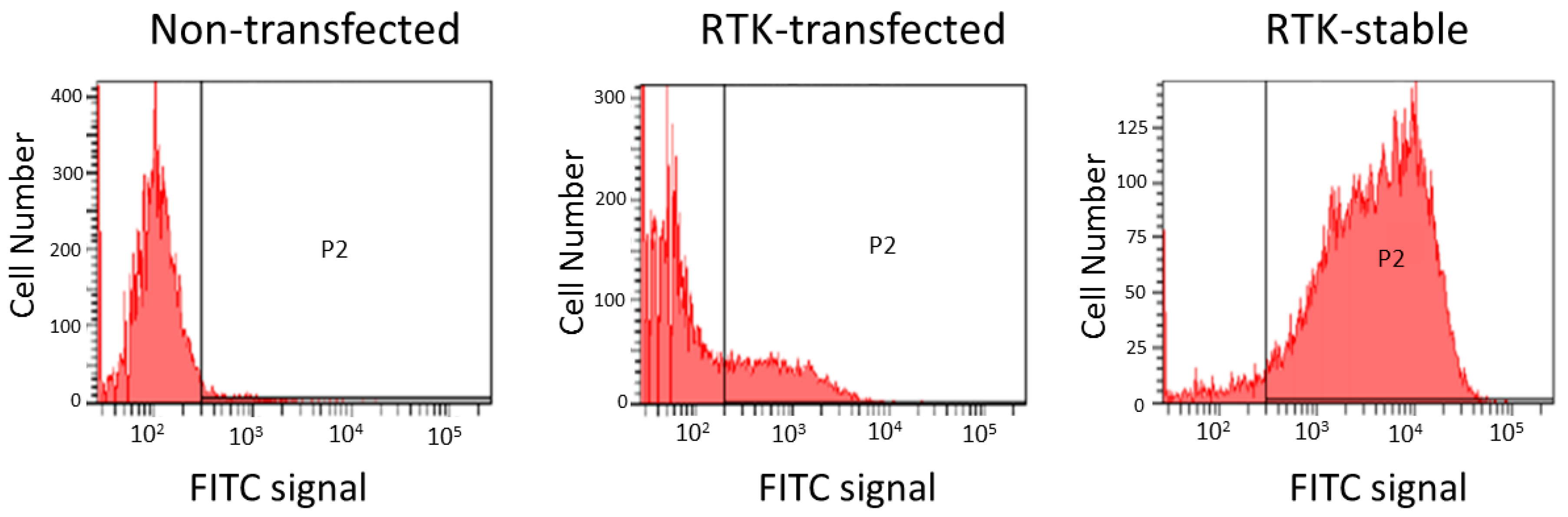
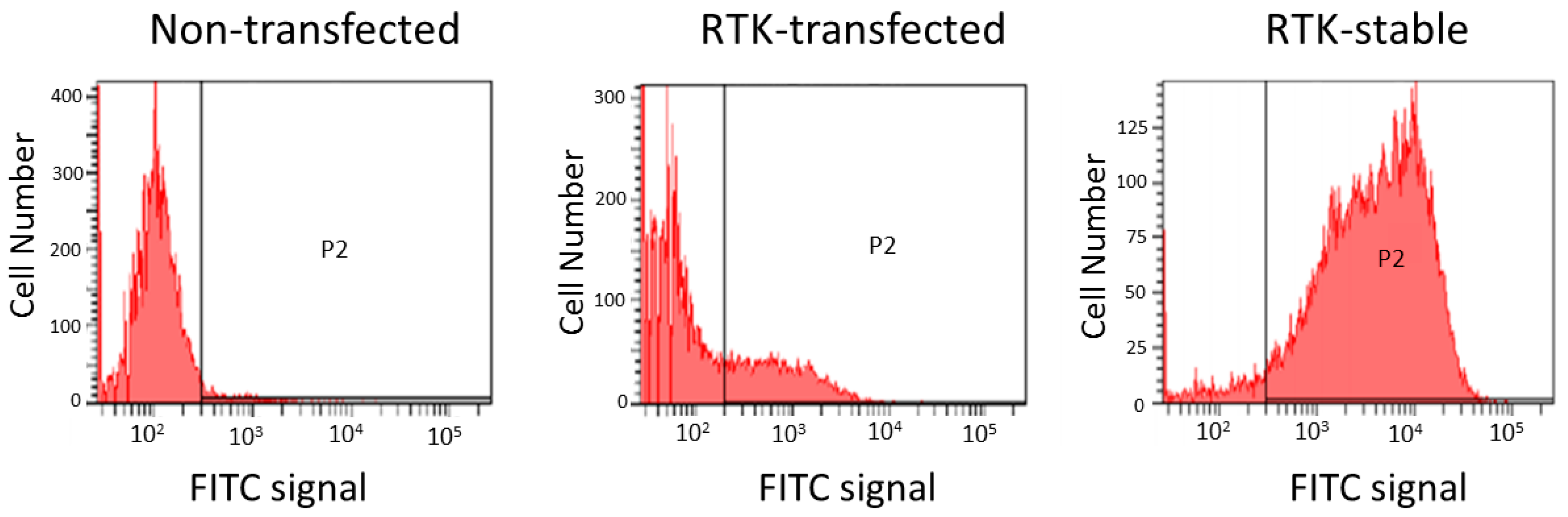
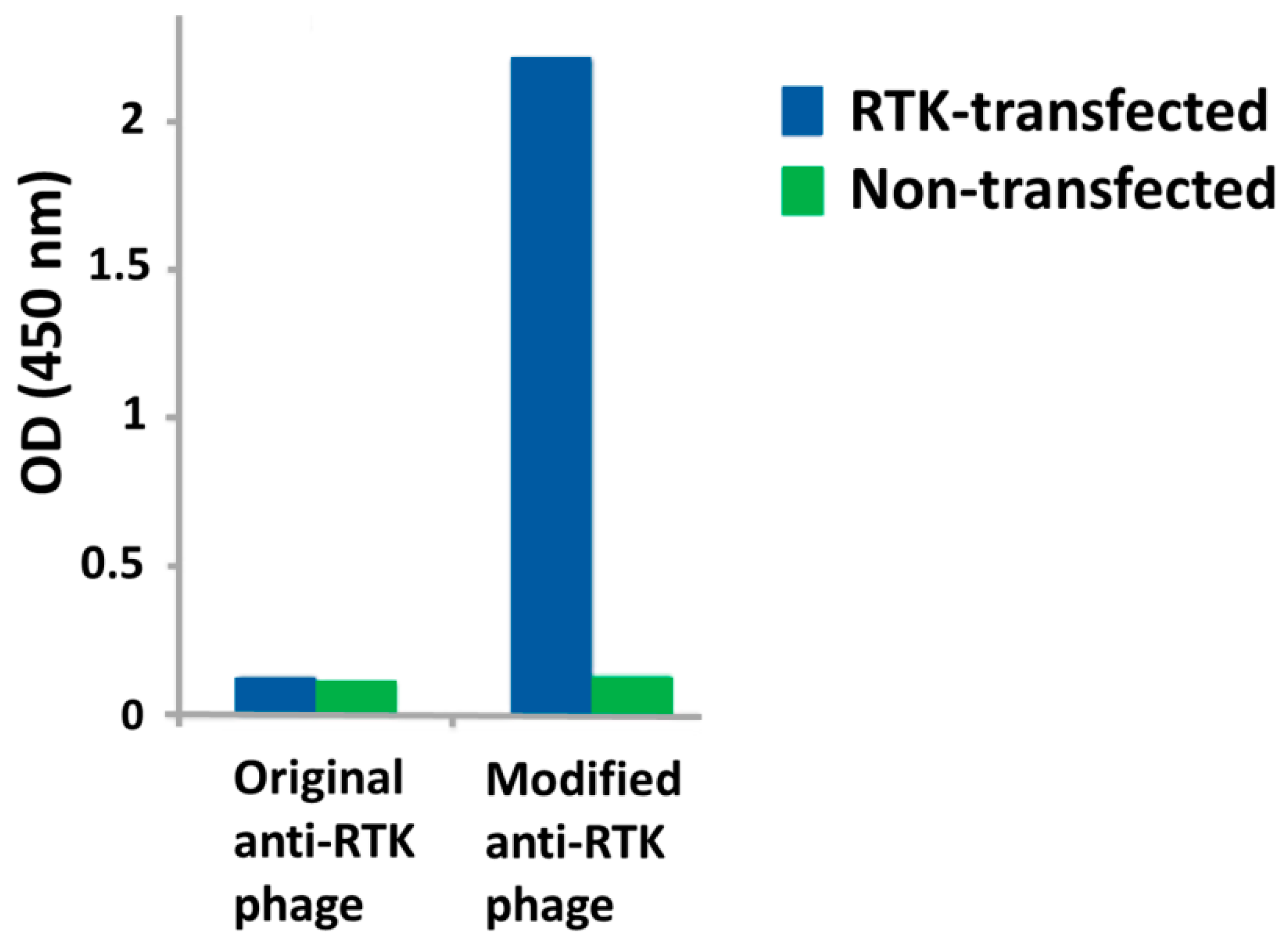
2.5. Engineered Cell Lines
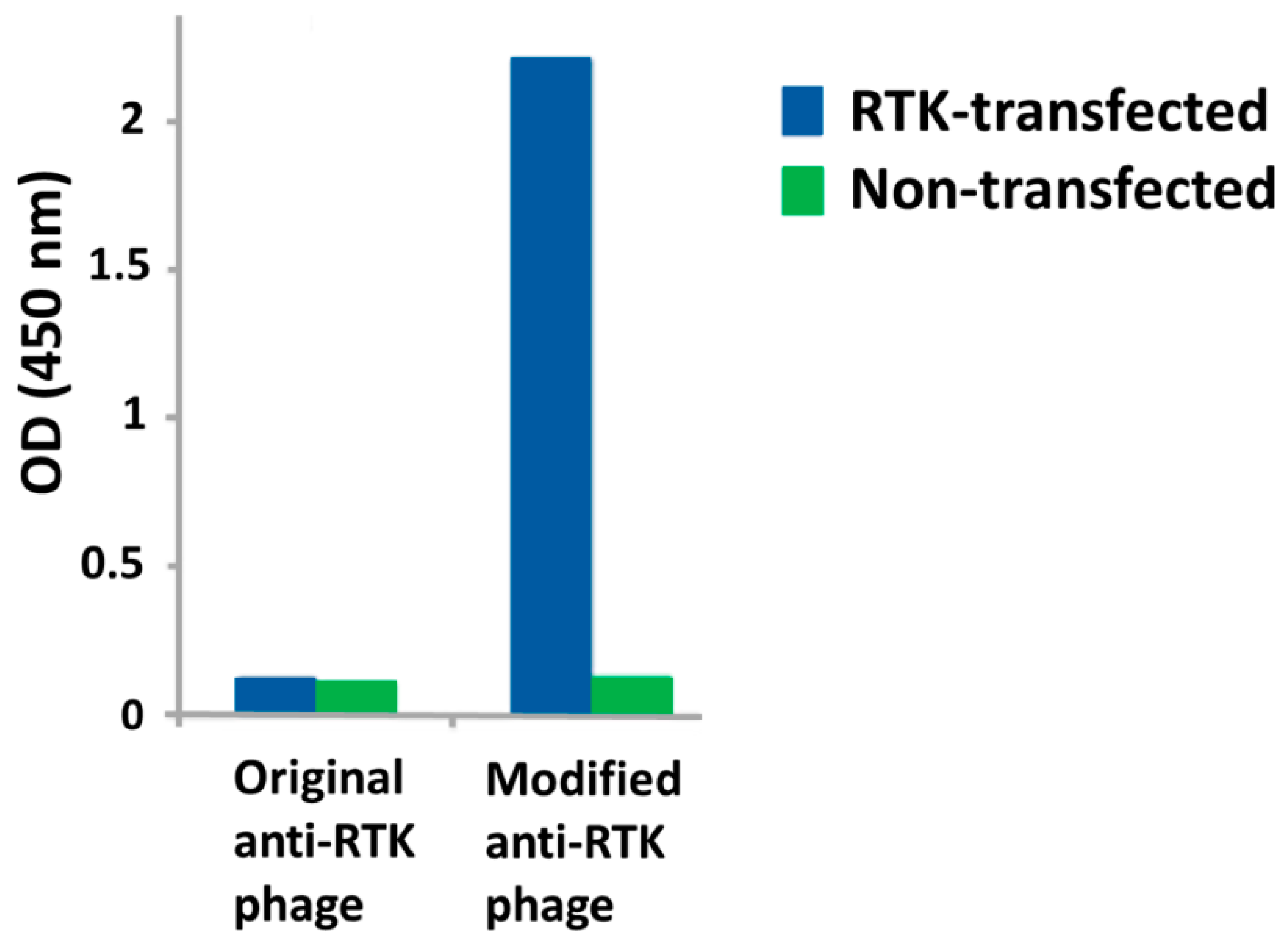
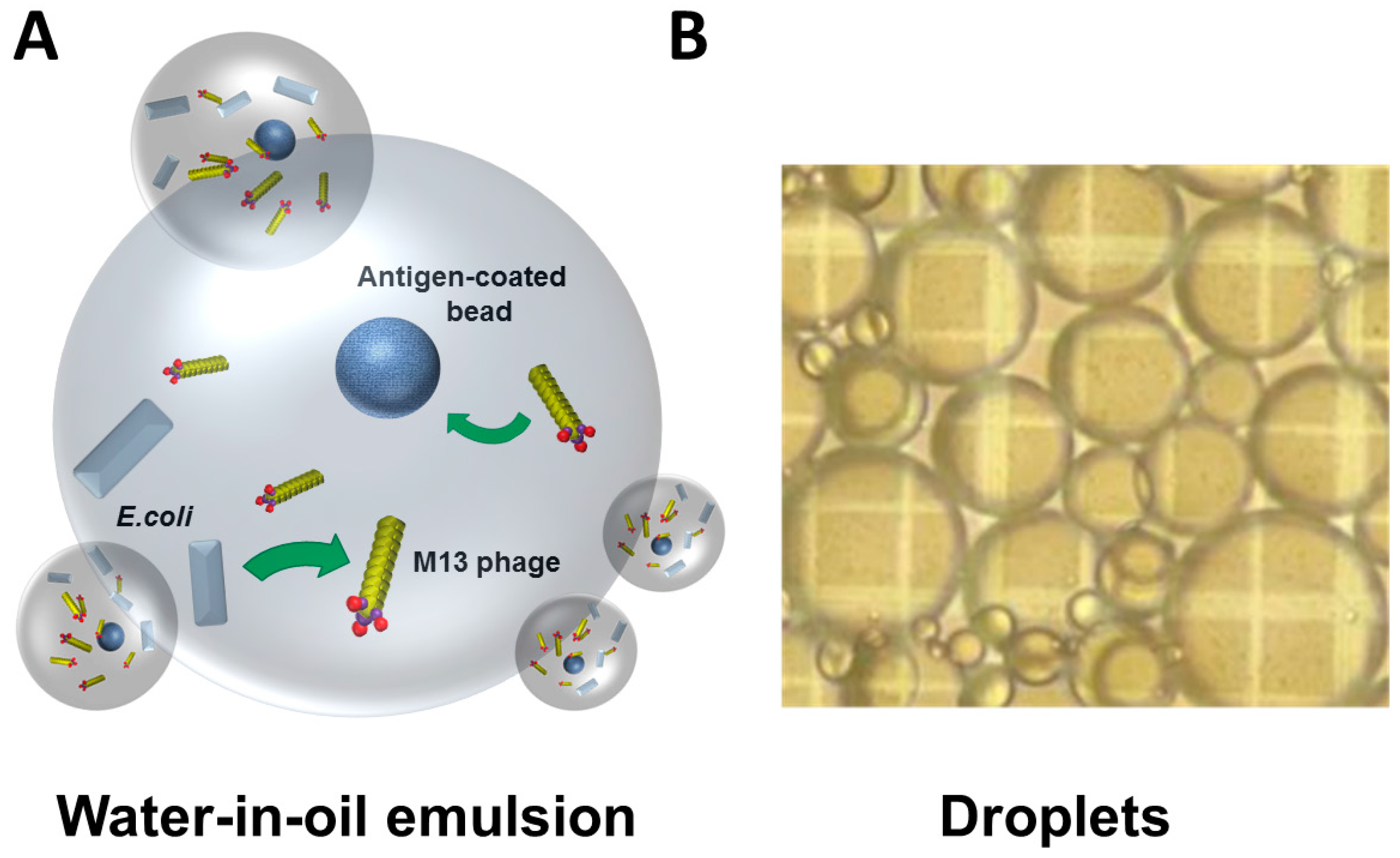
3. Challenges and Novel Methods for Affinity Selecting Recombinant Antibodies against Membrane Proteins
4. Various Applications of Recombinant Antibodies
Acknowledgments
Conflicts of Interest
References
- Bradbury, A.; Pluckthun, A. Reproducibility: Standardize antibodies used in research. Nature 2015, 518, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Gram, H.; Marconi, L.A.; Barbas, C.F., 3rd; Collet, T.A.; Lerner, R.A.; Kang, A.S. In vitro selection and affinity maturation of antibodies from a naive combinatorial immunoglobulin library. Proc. Natl. Acad. Sci. USA 1992, 89, 3576–3580. [Google Scholar] [PubMed]
- Schofield, D.J.; Pope, A.R.; Clementel, V.; Buckell, J.; Chapple, S.; Clarke, K.F.; Conquer, J.S.; Crofts, A.M.; Crowther, S.R.; Dyson, M.R.; et al. Application of phage display to high throughput antibody generation and characterization. Genome Biol. 2007, 8. [Google Scholar] [CrossRef] [PubMed]
- Colwill, K.; Graslund, S. A roadmap to generate renewable protein binders to the human proteome. Nat. Methods 2011, 8, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Hust, M.; Meyer, T.; Voedisch, B.; Rulker, T.; Thie, H.; El-Ghezal, A.; Kirsch, M.I.; Schutte, M.; Helmsing, S.; Meier, D.; et al. A human scFv antibody generation pipeline for proteome research. J. Biotechnol. 2011, 152, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Taussig, M.J.; Stoevesandt, O.; Borrebaeck, C.A.; Bradbury, A.R.; Cahill, D.; Cambillau, C.; de Daruvar, A.; Dubel, S.; Eichler, J.; Frank, R.; et al. Proteomebinders: Planning a European resource of affinity reagents for analysis of the human proteome. Nat. Methods 2007, 4, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Stoevesandt, O.; Taussig, M.J. European and international collaboration in affinity proteomics. New Biotechnol. 2012, 29, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Geyer, C.R.; McCafferty, J.; Dubel, S.; Bradbury, A.R.; Sidhu, S.S. Recombinant antibodies and in vitro selection technologies. Methods Mol. Biol. 2012, 901, 11–32. [Google Scholar] [PubMed]
- Huang, R.; Gorman, K.T.; Vinci, C.R.; Dobrovetsky, E.; Graslund, S.; Kay, B.K. Streamlining the pipeline for generation of recombinant affinity reagents by integrating the affinity maturation step. Int. J. Mol. Sci. 2015, 16, 23587–23603. [Google Scholar] [CrossRef] [PubMed]
- Zhong, N.; Loppnau, P.; Seitova, A.; Ravichandran, M.; Fenner, M.; Jain, H.; Bhattacharya, A.; Hutchinson, A.; Paduch, M.; Lu, V.; et al. Optimizing production of antigens and Fabs in the context of generating recombinant antibodies to human proteins. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Nelson, A.L. Antibody fragments: Hope and hype. MAbs 2010, 2, 77–83. [Google Scholar] [PubMed]
- Ahmad, Z.A.; Yeap, S.K.; Ali, A.M.; Ho, W.Y.; Alitheen, N.B.; Hamid, M. scFv antibody: Principles and clinical application. Clin. Dev. Immunol. 2012, 2012. [Google Scholar] [CrossRef]
- Fellouse, F.A.; Wiesmann, C.; Sidhu, S.S. Synthetic antibodies from a four-amino-acid code: A dominant role for tyrosine in antigen recognition. Proc. Natl. Acad. Sci. USA 2004, 101, 12467–12472. [Google Scholar] [CrossRef] [PubMed]
- Birtalan, S.; Zhang, Y.; Fellouse, F.A.; Shao, L.; Schaefer, G.; Sidhu, S.S. The intrinsic contributions of tyrosine, serine, glycine and arginine to the affinity and specificity of antibodies. J. Mol. Biol. 2008, 377, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Miersch, S.; Sidhu, S.S. Synthetic antibodies: Concepts, potential and practical considerations. Methods 2012, 57, 486–498. [Google Scholar] [CrossRef] [PubMed]
- De Marco, A. Biotechnological applications of recombinant single-domain antibody fragments. Microb. Cell Fact. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, T.; Muyldermans, S.; Depicker, A. Nanobody-based products as research and diagnostic tools. Trends Biotechnol. 2014, 32, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Helma, J.; Cardoso, M.C.; Muyldermans, S.; Leonhardt, H. Nanobodies and recombinant binders in cell biology. J. Cell Biol. 2015, 209, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Skerra, A. Alternative binding proteins: Anticalins-harnessing the structural plasticity of the lipocalin ligand pocket to engineer novel binding activities. FEBS J. 2008, 275, 2677–2683. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, F.Y.; Tolmachev, V. Affibody molecules: New protein domains for molecular imaging and targeted tumor therapy. Curr. Opin. Drug Discov. Dev. 2007, 10, 167–175. [Google Scholar]
- Boersma, Y.L.; Pluckthun, A. DARPins and other repeat protein scaffolds: Advances in engineering and applications. Curr. Opin. Biotechnol. 2011, 22, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Koide, S. Monobodies: Antibody mimics based on the scaffold of the fibronectin type iii domain. Methods Mol. Biol. 2007, 352, 95–109. [Google Scholar] [PubMed]
- Carpenter, E.P.; Beis, K.; Cameron, A.D.; Iwata, S. Overcoming the challenges of membrane protein crystallography. Curr. Opin. Struct. Biol. 2008, 18, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Yasui, N.; Mihara, E.; Nampo, M.; Tamura-Kawakami, K.; Unno, H.; Matsumoto, K.; Takagi, J. Detection of endogenous LRP6 expressed on human cells by monoclonal antibodies specific for the native conformation. J. Immunol. Methods 2010, 352, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Verstraete, K.; Remmerie, B.; Elegheert, J.; Lintermans, B.; Haegeman, G.; Vanhoenacker, P.; Van Craenenbroeck, K.; Savvides, S.N. Inducible production of recombinant human Flt3 ectodomain variants in mammalian cells and preliminary crystallographic analysis of Flt3 ligand-receptor complexes. Acta Crystallogr. Sect. F 2011, 67, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.C.; Tsai, W.Y.; Nerurkar, V.R.; Wang, W.K. Characterization of the ectodomain of the envelope protein of dengue virus type 4: Expression, membrane association, secretion and particle formation in the absence of precursor membrane protein. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Goehring, A.; Lee, C.H.; Wang, K.H.; Michel, J.C.; Claxton, D.P.; Baconguis, I.; Althoff, T.; Fischer, S.; Garcia, K.C.; Gouaux, E. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat. Protoc. 2014, 9, 2574–2585. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, X.X.; Zhang, S.Y.; Lu, M.; Kong, Y.Y.; Wang, Y.; Li, G.D. Expression of hepatitis C virus E2 ectodomain in E. Coli and its application in the detection of anti-E2 antibodies in human sera. Acta Biochim. Biophys. Sin. 2004, 36, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Lay, C.S.; Wilson, K.A.; Kobe, B.; Kemp, B.E.; Drummer, H.E.; Poumbourios, P. Expression and biochemical analysis of the entire HIV-2 gp41 ectodomain: Determinants of stability map to N- and C-terminal sequences outside the 6-helix bundle core. FEBS Lett. 2004, 567, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Laible, P.D.; Scott, H.N.; Henry, L.; Hanson, D.K. Towards higher-throughput membrane protein production for structural genomics initiatives. J. Struct. Funct. Genomics 2004, 5, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Jacquez, P.; Lei, N.; Weigt, D.; Xiao, C.; Sun, J. Expression and purification of the functional ectodomain of human anthrax toxin receptor 2 in Escherichia coli Origami B cells with assistance of bacterial Trigger Factor. Protein Expr. Purif. 2014, 95, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Hays, F.A.; Roe-Zurz, Z.; Stroud, R.M. Overexpression and purification of integral membrane proteins in yeast. Methods Enzymol. 2010, 470, 695–707. [Google Scholar] [PubMed]
- Mizutani, K.; Yoshioka, S.; Mizutani, Y.; Iwata, S.; Mikami, B. High-throughput construction of expression system using yeast pichia pastoris, and its application to membrane proteins. Protein Expr. Purif. 2011, 77, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Trometer, C.; Falson, P. Mammalian membrane protein expression in baculovirus-infected insect cells. Methods Mol. Biol. 2010, 601, 105–117. [Google Scholar] [PubMed]
- Hengen, P. Purification of His-Tag fusion proteins from Escherichia coli. Trends Biochem. Sci. 1995, 20, 285–286. [Google Scholar] [CrossRef]
- Einhauer, A.; Jungbauer, A. The FLAG™ peptide, a versatile fusion tag for the purification of recombinant proteins. J. Biochem. Biophys. Methods 2001, 49, 455–465. [Google Scholar] [CrossRef]
- Engel, C.K.; Chen, L.; Prive, G.G. Insertion of carrier proteins into hydrophilic loops of the Escherichia coli lactose permease. Biochim. Biophys. Acta 2002, 1564, 38–46. [Google Scholar] [CrossRef]
- Serrano-Vega, M.J.; Magnani, F.; Shibata, Y.; Tate, C.G. Conformational thermostabilization of the β1-adrenergic receptor in a detergent-resistant form. Proc. Natl. Acad. Sci. USA 2008, 105, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, C.A.; Dodevski, I.; Kenig, M.; Dudli, S.; Mohr, A.; Hermans, E.; Pluckthun, A. Directed evolution of a G protein-coupled receptor for expression, stability, and binding selectivity. Proc. Natl. Acad. Sci. USA 2008, 105, 14808–14813. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.M.; Cherezov, V.; Hanson, M.A.; Rasmussen, S.G.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Yao, X.J.; Weis, W.I.; Stevens, R.C.; et al. GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science 2007, 318, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Chun, E.; Thompson, A.A.; Liu, W.; Roth, C.B.; Griffith, M.T.; Katritch, V.; Kunken, J.; Xu, F.; Cherezov, V.; Hanson, M.A.; et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 2012, 20, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.A.; Liu, W.; Chun, E.; Katritch, V.; Wu, H.; Vardy, E.; Huang, X.P.; Trapella, C.; Guerrini, R.; Calo, G.; et al. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 2012, 485, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Magnani, F.; Shibata, Y.; Serrano-Vega, M.J.; Tate, C.G. Co-evolving stability and conformational homogeneity of the human adenosine A2a receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 10744–10749. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Tate, C.G. Engineering an ultra-thermostable β1-adrenoceptor. J. Mol. Biol. 2011, 413, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Herzenberg, L.A.; Parks, D.; Sahaf, B.; Perez, O.; Roederer, M. The history and future of the fluorescence activated cell sorter and flow cytometry: A view from stanford. Clin. Chem. 2002, 48, 1819–1827. [Google Scholar] [PubMed]
- Dodevski, I.; Pluckthun, A. Evolution of three human GPCRs for higher expression and stability. J. Mol. Biol. 2011, 408, 599–615. [Google Scholar] [CrossRef] [PubMed]
- Schlinkmann, K.M.; Hillenbrand, M.; Rittner, A.; Kunz, M.; Strohner, R.; Pluckthun, A. Maximizing detergent stability and functional expression of a GPCR by exhaustive recombination and evolution. J. Mol. Biol. 2012, 422, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Egloff, P.; Hillenbrand, M.; Klenk, C.; Batyuk, A.; Heine, P.; Balada, S.; Schlinkmann, K.M.; Scott, D.J.; Schutz, M.; Pluckthun, A. Structure of signaling-competent neurotensin receptor 1 obtained by directed evolution in Escherichia coli. Proc. Natl. Acad. Sci. USA 2014, 111, E655–E662. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, E.; Kumari, P.; Jaiman, D.; Shukla, A.K. Methodological advances: The unsung heroes of the GPCR structural revolution. Nat. Rev. Mol. Cell Biol. 2015, 16, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.J.; Kummer, L.; Tremmel, D.; Pluckthun, A. Stabilizing membrane proteins through protein engineering. Curr. Opin. Chem. Biol. 2013, 17, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Memic, A.; Volgina, V.V.; Gussin, H.A.; Pepperberg, D.R.; Kay, B.K. Generation of recombinant guinea pig antibody fragments to the human GABAC receptor. J. Immunol. Methods 2011, 368, 36–44. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Koduvayur, S.P.; Gussin, H.A.; Parthasarathy, R.; Hao, Z.; Kay, B.K.; Pepperberg, D.R. Generation of recombinant antibodies to rat GABAA receptor subunits by affinity selection on synthetic peptides. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Kierny, M.R.; Cunningham, T.D.; Bouhenni, R.A.; Edward, D.P.; Kay, B.K. Generating recombinant antibodies against putative biomarkers of retinal injury. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Korepanova, A.; Moore, J.D.; Nguyen, H.B.; Hua, Y.; Cross, T.A.; Gao, F. Expression of membrane proteins from Mycobacterium tuberculosis in Escherichia coli as fusions with maltose binding protein. Protein Expr. Purif. 2007, 53, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Jin, J.; Liu, X.; Zhang, Y.; Li, M.; Shao, M.; Qian, Y.; Zhang, D.; Zhu, H.; Ruan, Y.; et al. Functional expression of the Fc-fused extracellular domains of group II membrane proteins. Glycoconj. J. 2015, 32, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Kierstan, M.P.; Coughlan, M.P. Immobilization of proteins by noncovalent procedures: Principles and applications. Bioprocess Technol. 1991, 14, 13–71. [Google Scholar] [PubMed]
- Seddon, A.M.; Curnow, P.; Booth, P.J. Membrane proteins, lipids and detergents: Not just a soap opera. Biochim. Biophys. Acta 2004, 1666, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.E. Determination of critical micelle concentration of surfactants by capillary electrophoresis. J. Chromatogr. A 2004, 1037, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Rothlisberger, D.; Pos, K.M.; Pluckthun, A. An antibody library for stabilizing and crystallizing membrane proteins—Selecting binders to the citrate carrier CitS. FEBS Lett. 2004, 564, 340–348. [Google Scholar] [CrossRef]
- Huber, T.; Steiner, D.; Rothlisberger, D.; Pluckthun, A. In vitro selection and characterization of DARPins and Fab fragments for the co-crystallization of membrane proteins: The Na+-citrate symporter CitS as an example. J. Struct. Biol. 2007, 159, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Stockbridge, R.B.; Kolmakova-Partensky, L.; Shane, T.; Koide, A.; Koide, S.; Miller, C.; Newstead, S. Crystal structures of a double-barrelled fluoride ion channel. Nature 2015, 525, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Raschle, T.; Hiller, S.; Etzkorn, M.; Wagner, G. Nonmicellar systems for solution NMR spectroscopy of membrane proteins. Curr. Opin. Struct. Biol. 2010, 20, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Jesorka, A.; Orwar, O. Liposomes: Technologies and analytical applications. Annu. Rev. Anal. Chem. 2008, 1, 801–832. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, L.K.; Kuusinen, A.; Orellana, A.; Keinanen, K.; Engberg, J. Use of proteoliposomes to generate phage antibodies against native AMPA receptor. Eur. J. Biochem. 2000, 267, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Civjan, N.R.; Bayburt, T.H.; Schuler, M.A.; Sligar, S.G. Direct solubilization of heterologously expressed membrane proteins by incorporation into nanoscale lipid bilayers. Biotechniques 2003, 35, 556–563. [Google Scholar] [PubMed]
- Breslow, J.L.; Ross, D.; McPherson, J.; Williams, H.; Kurnit, D.; Nussbaum, A.L.; Karathanasis, S.K.; Zannis, V.I. Isolation and characterization of cDNA clones for human apolipoprotein A–I. Proc. Natl. Acad. Sci. USA 1982, 79, 6861–6865. [Google Scholar] [CrossRef] [PubMed]
- Knott, T.J.; Pease, R.J.; Powell, L.M.; Wallis, S.C.; Rall, S.C., Jr.; Innerarity, T.L.; Blackhart, B.; Taylor, W.H.; Marcel, Y.; Milne, R.; et al. Complete protein sequence and identification of structural domains of human apolipoprotein B. Nature 1986, 323, 734–738. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Scharadin, T.M.; Saldana, M.; Gellner, C.; Hoang-Phou, S.; Takanishi, C.; Hura, G.L.; Tainer, J.A.; Carraway, K.L., 3rd; Henderson, P.T.; et al. Cell-free expression of functional receptor tyrosine kinases. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Grinkova, Y.V.; Lazarides, A.A.; Sligar, S.G. Directed self-assembly of monodisperse phospholipid bilayer nanodiscs with controlled size. J. Am. Chem. Soc. 2004, 126, 3477–3487. [Google Scholar] [CrossRef] [PubMed]
- Bayburt, T.H.; Sligar, S.G. Membrane protein assembly into nanodiscs. FEBS Lett. 2010, 584, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.A.; Denisov, I.G.; Sligar, S.G. Nanodiscs as a new tool to examine lipid-protein interactions. Methods Mol. Biol. 2013, 974, 415–433. [Google Scholar] [PubMed]
- Chromy, B.A.; Arroyo, E.; Blanchette, C.D.; Bench, G.; Benner, H.; Cappuccio, J.A.; Coleman, M.A.; Henderson, P.T.; Hinz, A.K.; Kuhn, E.A.; et al. Different apolipoproteins impact nanolipoprotein particle formation. J. Am. Chem. Soc. 2007, 129, 14348–14354. [Google Scholar] [CrossRef] [PubMed]
- Cappuccio, J.A.; Blanchette, C.D.; Sulchek, T.A.; Arroyo, E.S.; Kralj, J.M.; Hinz, A.K.; Kuhn, E.A.; Chromy, B.A.; Segelke, B.W.; Rothschild, K.J.; et al. Cell-free co-expression of functional membrane proteins and apolipoprotein, forming soluble nanolipoprotein particles. Mol. Cell Proteomics 2008, 7, 2246–2253. [Google Scholar] [CrossRef] [PubMed]
- Cappuccio, J.A.; Hinz, A.K.; Kuhn, E.A.; Fletcher, J.E.; Arroyo, E.S.; Henderson, P.T.; Blanchette, C.D.; Walsworth, V.L.; Corzett, M.H.; Law, R.J.; et al. Cell-free expression for nanolipoprotein particles: Building a high-throughput membrane protein solubility platform. Methods Mol. Biol. 2009, 498, 273–296. [Google Scholar] [PubMed]
- He, W.; Luo, J.; Bourguet, F.; Xing, L.; Yi, S.K.; Gao, T.; Blanchette, C.; Henderson, P.T.; Kuhn, E.; Malfatti, M.; et al. Controlling the diameter, monodispersity, and solubility of apoa1 nanolipoprotein particles using telodendrimer chemistry. Protein Sci. 2013, 22, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Pavlidou, M.; Hanel, K.; Mockel, L.; Willbold, D. Nanodiscs allow phage display selection for ligands to non-linear epitopes on membrane proteins. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Dominik, P.K.; Kossiakoff, A.A. Phage display selections for affinity reagents to membrane proteins in nanodiscs. Methods Enzymol. 2015, 557, 219–245. [Google Scholar] [PubMed]
- Dominik, P.K.; Borowska, M.T.; Dalmas, O.; Kim, S.S.; Perozo, E.; Keenan, R.J.; Kossiakoff, A.A. Conformational chaperones for structural studies of membrane proteins using antibody phage display with nanodiscs. Structure 2016, 24, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Kay, B.K.; Thai, S.; Volgina, V.V. High-throughput biotinylation of proteins. Methods Mol. Biol. 2009, 498, 185–196. [Google Scholar] [PubMed]
- Chackerian, B.; Rangel, M.; Hunter, Z.; Peabody, D.S. Virus and virus-like particle-based immunogens for Alzheimer's disease induce antibody responses against amyloid-β without concomitant T cell responses. Vaccine 2006, 24, 6321–6331. [Google Scholar] [CrossRef] [PubMed]
- Thrane, S.; Janitzek, C.M.; Agerbaek, M.O.; Ditlev, S.B.; Resende, M.; Nielsen, M.A.; Theander, T.G.; Salanti, A.; Sander, A.F. A novel virus-like particle based vaccine platform displaying the placental malaria antigen VAR2CSA. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Kee, G.S.; Pujar, N.S.; Titchener-Hooker, N.J. Study of detergent-mediated liberation of hepatitis B virus-like particles from S. cerevisiae homogenate: Identifying a framework for the design of future-generation lipoprotein vaccine processes. Biotechnol. Prog. 2008, 24, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Burden, C.S.; Jin, J.; Podgornik, A.; Bracewell, D.G. A monolith purification process for virus-like particles from yeast homogenate. J. Chromatogr. B 2012, 880, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Brune, K.D.; Leneghan, D.B.; Brian, I.J.; Ishizuka, A.S.; Bachmann, M.F.; Draper, S.J.; Biswas, S.; Howarth, M. Plug-and-display: Decoration of virus-like particles via isopeptide bonds for modular immunization. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Gurramkonda, C.; Zahid, M.; Nemani, S.K.; Adnan, A.; Gudi, S.K.; Khanna, N.; Ebensen, T.; Lunsdorf, H.; Guzman, C.A.; Rinas, U. Purification of hepatitis B surface antigen virus-like particles from recombinant pichia pastoris and in vivo analysis of their immunogenic properties. J. Chromatogr. B 2013, 940, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, J.C.; Peabody, D.S. Stability and assembly in vitro of bacteriophage PP7 virus-like particles. J. Nanobiotechnol. 2007, 5. [Google Scholar] [CrossRef] [PubMed]
- Lynch, A.; Meyers, A.E.; Williamson, A.L.; Rybicki, E.P. Stability studies of HIV-1 Pr55gag virus-like particles made in insect cells after storage in various formulation media. Virol. J. 2012, 9. [Google Scholar] [CrossRef] [PubMed]
- Peabody, D.S. A viral platform for chemical modification and multivalent display. J. Nanobiotechnol. 2003, 1. [Google Scholar] [CrossRef] [PubMed]
- Even-Desrumeaux, K.; Chames, P. Phage display and selections on cells. Methods Mol. Biol. 2012, 907, 225–235. [Google Scholar] [PubMed]
- Dias-Neto, E.; Nunes, D.N.; Giordano, R.J.; Sun, J.; Botz, G.H.; Yang, K.; Setubal, J.C.; Pasqualini, R.; Arap, W. Next-generation phage display: Integrating and comparing available molecular tools to enable cost-effective high-throughput analysis. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Song, J.M.; Ryu, C.J.; Kim, Y.G.; Lee, E.K.; Kang, S.; Kim, S.J. An efficient strategy for cell-based antibody library selection using an integrated vector system. BMC Biotechnol. 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, A.; Verma, I.M. Gene therapy: Promises and problems. Annu. Rev. Genomics Hum. Genet. 2001, 2, 177–211. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Eberwine, J.H. Mammalian cell transfection: The present and the future. Anal. Bioanal. Chem. 2010, 397, 3173–3178. [Google Scholar] [CrossRef] [PubMed]
- Recillas-Targa, F. Multiple strategies for gene transfer, expression, knockdown, and chromatin influence in mammalian cell lines and transgenic animals. Mol. Biotechnol. 2006, 34, 337–354. [Google Scholar] [CrossRef]
- Rondot, S.; Koch, J.; Breitling, F.; Dubel, S. A helper phage to improve single-chain antibody presentation in phage display. Nat. Biotechnol. 2001, 19, 75–78. [Google Scholar] [PubMed]
- Lama, J.; Carrasco, L. Inducible expression of a toxic poliovirus membrane protein in Escherichia coli: Comparative studies using different expression systems based on T7 promoters. Biochem. Biophys. Res. Commun. 1992, 188, 972–981. [Google Scholar] [CrossRef]
- Meyer-Ficca, M.L.; Meyer, R.G.; Kaiser, H.; Brack, A.R.; Kandolf, R.; Kupper, J.H. Comparative analysis of inducible expression systems in transient transfection studies. Anal. Biochem. 2004, 334, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Bussow, K. Stable mammalian producer cell lines for structural biology. Curr. Opin. Struct. Biol. 2015, 32, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Andrell, J.; Tate, C.G. Overexpression of membrane proteins in mammalian cells for structural studies. Mol. Membr. Biol. 2013, 30, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Schiedner, G.; Hertel, S.; Bialek, C.; Kewes, H.; Waschutza, G.; Volpers, C. Efficient and reproducible generation of high-expressing, stable human cell lines without need for antibiotic selection. BMC Biotechnol. 2008, 8. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.; Pak, J.E.; Pedersen, B.P.; Bang, L.J.; Zhang, L.B.; Ngaw, S.M.; Green, R.G.; Sharma, V.; Stroud, R.M. Efficient expression screening of human membrane proteins in transiently transfected Human Embryonic Kidney 293S cells. Methods 2011, 55, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Huang, Z.; Wen, W.; Wu, A.; Wang, C.; Niu, L. Enhancing protein expression in HEK-293 cells by lowering culture temperature. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Shaffer, P.L.; Huang, X.; Rose, P.E. Rapid screening of membrane protein expression in transiently transfected insect cells. Protein Expr. Purif. 2013, 88, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Buhr, D.L.; Acca, F.E.; Holland, E.G.; Johnson, K.; Maksymiuk, G.M.; Vaill, A.; Kay, B.K.; Weitz, D.A.; Weiner, M.P.; Kiss, M.M. Use of micro-emulsion technology for the directed evolution of antibodies. Methods 2012, 58, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Mayer, G.; Bendayan, M. Biotinyl-tyramide: A novel approach for electron microscopic immunocytochemistry. J. Histochem. Cytochem. 1997, 45, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Osbourn, J.K.; Earnshaw, J.C.; Johnson, K.S.; Parmentier, M.; Timmermans, V.; McCafferty, J. Directed selection of MIP-1α neutralizing CCR5 antibodies from a phage display human antibody library. Nat. Biotechnol. 1998, 16, 778–781. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.; Bai, J.; St. Clair Tallarico, A.; Xu, C.; Marasco, W.A. Identification of CD4 and transferrin receptor antibodies by CXCR4 antibody-guided Pathfinder selection. Eur. J. Biochem. 2003, 270, 4497–4506. [Google Scholar] [CrossRef] [PubMed]
- Giordano, R.J.; Cardo-Vila, M.; Lahdenranta, J.; Pasqualini, R.; Arap, W. Biopanning and rapid analysis of selective interactive ligands. Nat. Med. 2001, 7, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Ye, Z. Auto T cells expressing chimeric antigen receptor derived from auto antibody might be a new treatment for osteosarcoma. Med. Hypotheses 2012, 78, 616–618. [Google Scholar]
- Lipes, B.D.; Chen, Y.H.; Ma, H.; Staats, H.F.; Kenan, D.J.; Gunn, M.D. An entirely cell-based system to generate single-chain antibodies against cell surface receptors. J. Mol. Biol. 2008, 379, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, A.P.; Reis, C.F.; Morari, E.C.; Maia, Y.C.; Nascimento, R.; Bonatto, J.M.; de Souza, M.A.; Goulart, L.R.; Ward, L.S. A putative OTU domain-containing protein 1 deubiquitinating enzyme is differentially expressed in thyroid cancer and identifies less-aggressive tumours. Br. J. Cancer 2014, 111, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Adams, J.D.; Turner, K.; Cochran, F.V.; Gambhir, S.S.; Soh, H.T. Controlling the selection stringency of phage display using a microfluidic device. Lab. Chip 2009, 9, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Teesalu, T.; Sugahara, K.N.; Kotamrajua, V.R.; Adams, J.D.; Ferguson, B.S.; Gong, Q.; Oh, S.S.; Csordas, A.T.; et al. Selection of phage-displayed peptides on live adherent cells in microfluidic channels. Proc. Natl. Acad. Sci. USA 2011, 108, 6909–6914. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, T.; Boon-Chieng, S.; Mitaku, S. SOSUI: Classification and secondary structure prediction system for membrane proteins. Bioinformatics 1998, 14, 378–379. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Stroud, R.M.; Craik, C.S. Rapid identification of recombinant Fabs that bind to membrane proteins. Methods 2011, 55, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Morais-Cabral, J.H.; Kaufman, A.; MacKinnon, R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature 2001, 414, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Uysal, S.; Vasquez, V.; Tereshko, V.; Esaki, K.; Fellouse, F.A.; Sidhu, S.S.; Koide, S.; Perozo, E.; Kossiakoff, A. Crystal structure of full-length KcsA in its closed conformation. Proc. Natl. Acad. Sci. USA 2009, 106, 6644–6649. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.; Choi, H.J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.B.; Szewczyk, P.; Grimard, V.; Lee, C.W.; Martinez, L.; Doshi, R.; Caya, A.; Villaluz, M.; Pardon, E.; Cregger, C.; et al. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. USA 2013, 110, 13386–13391. [Google Scholar] [CrossRef] [PubMed]
- Sennhauser, G.; Amstutz, P.; Briand, C.; Storchenegger, O.; Grutter, M.G. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol. 2007, 5. [Google Scholar] [CrossRef] [PubMed]
- Pitaksajjakul, P.; Lekcharoensuk, P.; Upragarin, N.; Barbas, C.F., 3rd; Ibrahim, M.S.; Ikuta, K.; Ramasoota, P. Fab MAbs specific to HA of influenza virus with H5N1 neutralizing activity selected from immunized chicken phage library. Biochem. Biophys. Res. Commun. 2010, 395, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Sakurai, A.; Nomura, N.; Park, E.Y.; Shibasaki, F.; Ueda, H. Isolation of recombinant phage antibodies targeting the hemagglutinin cleavage site of highly pathogenic avian influenza virus. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zeng, X.Q.; Zhang, H.B.; Ni, H.Z.; Pei, L.; Zou, L.R.; Liang, L.J.; Zhang, X.; Lin, J.Y.; Ke, C.W. Novel phage display-derived H5N1-specific scFvs with potential use in rapid avian flu diagnosis. J. Microbiol. Biotechnol. 2014, 24, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, M.I.; Hulseweh, B.; Nacke, C.; Rulker, T.; Schirrmann, T.; Marschall, H.J.; Hust, M.; Dubel, S. Development of human antibody fragments using antibody phage display for the detection and diagnosis of venezuelan equine encephalitis virus (VEEV). BMC Biotechnol. 2008, 8. [Google Scholar] [CrossRef] [PubMed]
- Rothe, A.; Klimka, A.; Tur, M.K.; Pfitzner, T.; Huhn, M.; Sasse, S.; Mallmann, P.; Engert, A.; Barth, S. Construction of phage display libraries from reactive lymph nodes of breast carcinoma patients and selection for specifically binding human single chain Fv on cell lines. Int. J. Mol. Med. 2004, 14, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, C.G.; Rasmussen, N.; Laenkholm, A.V.; Ditzel, H.J. Phage display-derived human monoclonal antibodies isolated by binding to the surface of live primary breast cancer cells recognize GRP78. Cancer Res. 2007, 67, 9507–9517. [Google Scholar] [CrossRef] [PubMed]
- Araujo, T.G.; Paiva, C.E.; Rocha, R.M.; Maia, Y.C.; Sena, A.A.; Ueira-Vieira, C.; Carneiro, A.P.; Almeida, J.F.; de Faria, P.R.; Santos, D.W.; et al. A novel highly reactive Fab antibody for breast cancer tissue diagnostics and staging also discriminates a subset of good prognostic triple-negative breast cancers. Cancer Lett. 2014, 343, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Gainkam, L.O.; Caveliers, V.; Vanhove, C.; Keyaerts, M.; De Baetselier, P.; Bossuyt, A.; Revets, H.; Lahoutte, T. SPECT imaging with 99mTc-labeled EGFR-specific nanobody for in vivo monitoring of EGFR expression. Mol. Imaging Biol. 2008, 10, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Vaneycken, I.; Devoogdt, N.; Van Gassen, N.; Vincke, C.; Xavier, C.; Wernery, U.; Muyldermans, S.; Lahoutte, T.; Caveliers, V. Preclinical screening of anti-HER2 nanobodies for molecular imaging of breast cancer. FASEB J. 2011, 25, 2433–2446. [Google Scholar] [CrossRef] [PubMed]
- Broisat, A.; Hernot, S.; Toczek, J.; De Vos, J.; Riou, L.M.; Martin, S.; Ahmadi, M.; Thielens, N.; Wernery, U.; Caveliers, V.; et al. Nanobodies targeting mouse/human VCAM1 for the nuclear imaging of atherosclerotic lesions. Circ. Res. 2012, 110, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.; Nanchahal, J.; Taylor, P.; Feldmann, M. Anti-TNF therapy: Past, present and future. Int. Immunol. 2015, 27, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Brenner, T.L.; Adams, V.R. First MAb approved for treatment of metastatic breast cancer. J. Am. Pharm. Assoc. 1999, 39, 236–238. [Google Scholar] [CrossRef]
- Molineux, G.; Newland, A. Development of romiplostim for the treatment of patients with chronic immune thrombocytopenia: From bench to bedside. Br. J. Haematol. 2010, 150, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Nixon, A.E.; Sexton, D.J.; Ladner, R.C. Drugs derived from phage display: From candidate identification to clinical practice. MAbs 2014, 6, 73–85. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, R.; Kiss, M.M.; Batonick, M.; Weiner, M.P.; Kay, B.K. Generating Recombinant Antibodies to Membrane Proteins through Phage Display. Antibodies 2016, 5, 11. https://doi.org/10.3390/antib5020011
Huang R, Kiss MM, Batonick M, Weiner MP, Kay BK. Generating Recombinant Antibodies to Membrane Proteins through Phage Display. Antibodies. 2016; 5(2):11. https://doi.org/10.3390/antib5020011
Chicago/Turabian StyleHuang, Renhua, Margaret M. Kiss, Melissa Batonick, Michael P. Weiner, and Brian K. Kay. 2016. "Generating Recombinant Antibodies to Membrane Proteins through Phage Display" Antibodies 5, no. 2: 11. https://doi.org/10.3390/antib5020011
APA StyleHuang, R., Kiss, M. M., Batonick, M., Weiner, M. P., & Kay, B. K. (2016). Generating Recombinant Antibodies to Membrane Proteins through Phage Display. Antibodies, 5(2), 11. https://doi.org/10.3390/antib5020011