
Anticytokine Autoantibodies: Association with Infection and Immune Dysregulation


Abstract
:1. Introduction

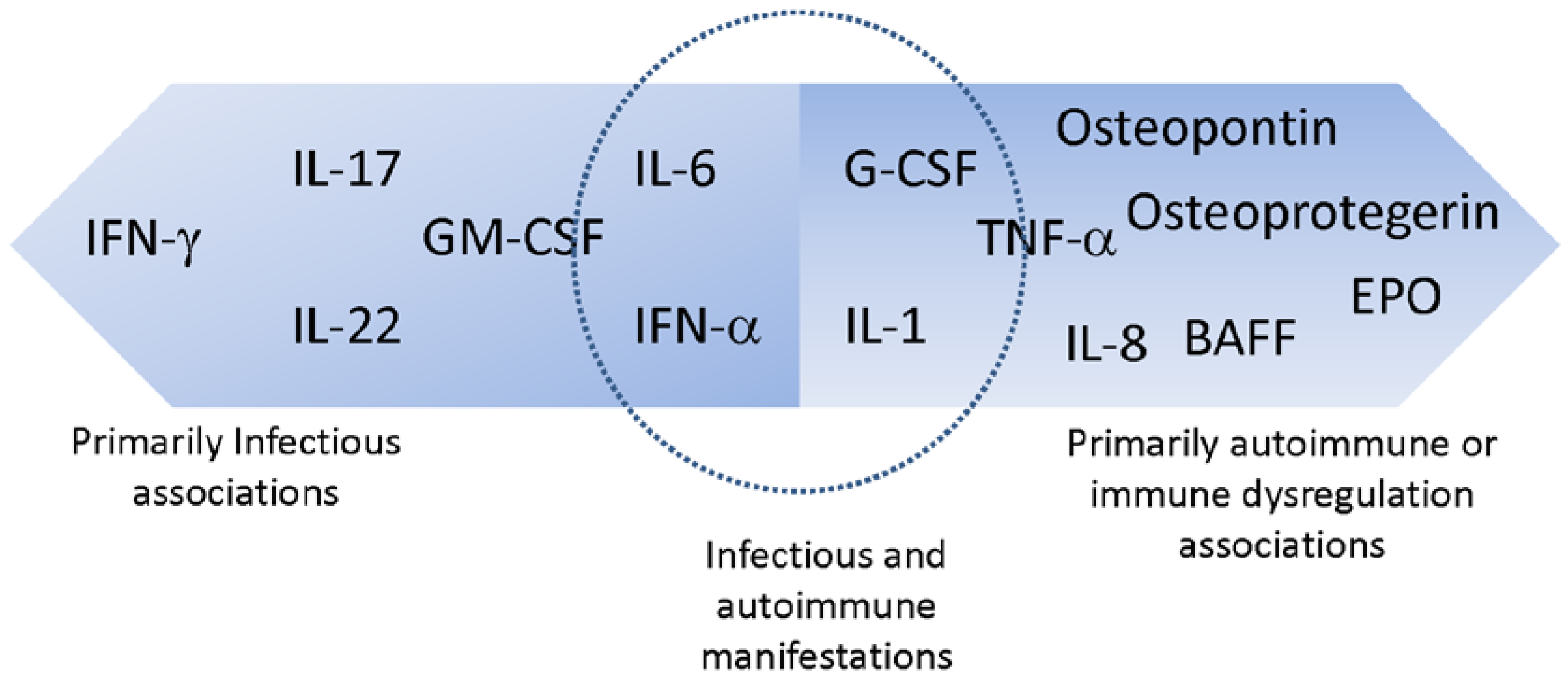{kind=link}
| Non-destructive form of polyarthritis, Sjogren’s syndrome, rheumatoid arthritis, psoriasis, pemphigus. | Neutralizing, negatively correlated with disease severity, may modulate disease. | [21,22] |
| Recurrent staphylococcal infections, associated with low CRP levels. Observed in systemic sclerosis | Neutralizing, leads to decreased CRP levels, increased susceptibility to infection. May form stable complexes with IL-6 and contribute to disease progression in systemic sclerosis | [23,24,25,26] |
| Acute Respiratory Distress Syndrome | Forms immune complex with IL-8, extending proinflammatory activity and neutrophil recruitment | [27] |
| Autoimmune Polyendocrionopathy Syndrome type-1, thymoma associated autoimmune disease. One case of Burkholdaria lymphadenitis | Biological role not well established. Neutralizing activity may contribute to susceptibility to intracellular organisms | [28,29] |
| Autoimmune Polyendocrinopathy Syndrome type-1, Chronic Mucocutaneous Candidiasis | Neutralizing, may contribute to impaired immune responses mediated by IL-17 | [30,31] |
| Felty’s syndrome, neutropenia | Not well established, may contribute to neutropenia through neutralization of G-CSF | [7] |
| Pulmonary Alveolar Proteinosis. Intracellular infections with Cryptococcus, Norcardia, Aspergillus and Mycobacterium avium | Neutralizing, impaired alveolar macrophage development, impaired macrophage function leading to compromised cellular immune responses. | [3,32,33,34,35] |
| Disseminated mycobacterial infections, Infections with Salmonella typhi, CMV and Toxoplasma, reactivation of VZV | Neutralizing, abrogates IFNγ mediated cellular immune responses essential for clearance of intracellular infections | [16,36,37,38] |
| Systemic Lupus Erythematosus, Autoimmune Polyendocrionopathy Syndrome type-1, Thymoma Immune deficiency associated with hypomorphic RAG mutations | Neutralizing, associated with reduction in disease severity in SLE. Neutralizing activity associated with viral infections. | [19,20,39,40] |
| Systemic Lupus Erythematosus | Unclear, associated with elevated levels of IFNγ and increased disease activity. | [41] |
| Rheumatoid arthritis, prostate cancer, hepatocellular carcinoma | Unclear, may have a role in modulating disease activity in RA Potential early serum biomarker for prostate cancer. Diagnostic and prognostic biomarker for hepatocellular carcinoma | [42,43] |
| Systemic Lupus Erythematosus, Multiple Sclerosis | May play a role in disease modulation in SLE. Unclear role in MS. | [44,45] |
| Osteoporosis, Celiac Disease, Increased bone resorption in rheumatoid arthritis | Biological role unclear. | [46,47,48] |
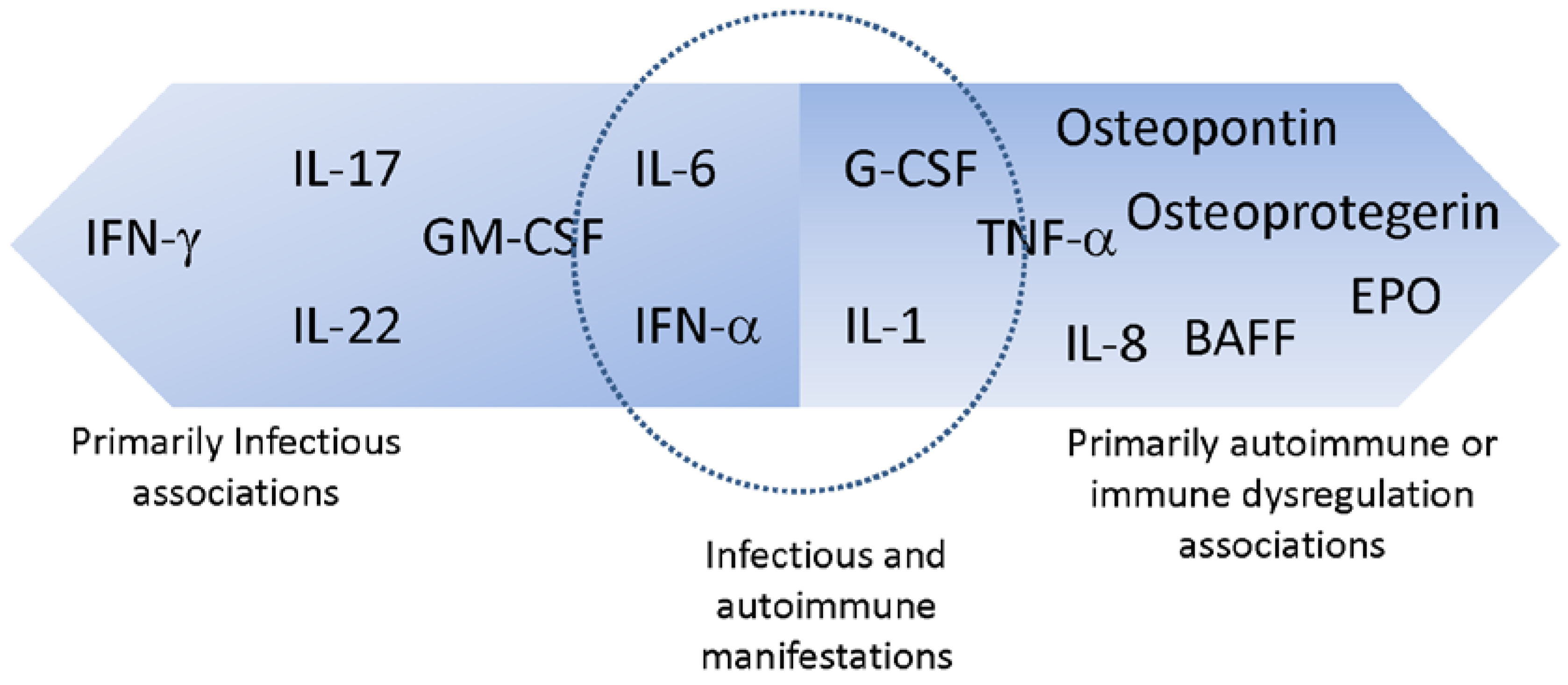
2. Anti-Cytokine AAbs Associated Primarily with Infectious Manifestations
2.1. Interferon Gamma (IFNγ)
2.2. Interleukin-17 (IL-17) and Interleukin-22 (IL-22)
2.3. Granulocyte-Macrophage Colony Stimulating Factor (GM-CSF)
3. Anti-Cytokine AAbs Associated with both Infectious and Autoimmune Manifestations
3.1. Interleukin-6 (IL-6)
3.2. Interferon-Alpha (IFNα)
3.3. Granulocyte Colony-Stimulating Factor (G-CSF)
4. Anti-Cytokine AAbs Associated with Immune Dysregulation or Autoimmune Manifestations
4.1. Interleukin 8 (IL-8)
4.2. Erythropoietin (EPO)
5. Treatment
6. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Browne, S.K. Anticytokine autoantibody-associated immunodeficiency. Ann. Rev. Immunol. 2014, 32, 635–657. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.K.; Holland, S.M. Anticytokine autoantibodies in infectious diseases: Pathogenesis and mechanisms. Lancet Infect. Dis. 2010, 10, 875–885. [Google Scholar] [CrossRef]
- Browne, S.K.; Holland, S.M. Immunodeficiency secondary to anticytokine autoantibodies. Curr. Opin. Allergy Clin. Immunol. 2010, 10, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Boehm, U.; Klamp, T.; Groot, M.; Howard, J.C. Cellular responses to interferon-gamma. Ann. Rev. Immunol. 1997, 15, 749–795. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Berclaz, P.Y.; Chroneos, Z.C.; Yoshida, M.; Whitsett, J.A.; Trapnell, B.C. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity 2001, 15, 557–567. [Google Scholar] [CrossRef]
- Bonfield, T.L.; Raychaudhuri, B.; Malur, A.; Abraham, S.; Trapnell, B.C.; Kavuru, M.S.; Thomassen, M.J. PU.1 regulation of human alveolar macrophage differentiation requires granulocyte-macrophage colony-stimulating factor. Am. J. Physiol. 2003, 285, L1132–L1136. [Google Scholar] [CrossRef] [PubMed]
- Hellmich, B.; Csernok, E.; Schatz, H.; Gross, W.L.; Schnabel, A. Autoantibodies against granulocyte colony-stimulating factor in Felty’s syndrome and neutropenic systemic lupus erythematosus. Arthritis Rheum. 2002, 46, 2384–2391. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Uchida, K.; Nakagaki, K.; Kanazawa, H.; Trapnell, B.C.; Hoshino, Y.; Kagamu, H.; Yoshizawa, H.; Keicho, N.; Goto, H.; et al. Anti-cytokine autoantibodies are ubiquitous in healthy individuals. FEBS Lett. 2007, 581, 2017–2021. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, M.; Meager, A.; Dilger, P.; Bird, C.; Dolman, C.; Das, R.G.; Thorpe, R. Neutralizing antibodies to granulocyte-macrophage colony-stimulating factor, interleukin-1alpha and interferon-alpha but not other cytokines in human immunoglobulin preparations. Immunology 2000, 99, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Courtney, L.P.; Phelps, J.L.; Karavodin, L.M. An anti-IL-2 antibody increases serum half-life and improves anti-tumor efficacy of human recombinant interleukin-2. Immunopharmacology 1994, 28, 223–232. [Google Scholar] [CrossRef]
- Uchida, K.; Nakata, K.; Trapnell, B.C.; Terakawa, T.; Hamano, E.; Mikami, A.; Matsushita, I.; Seymour, J.F.; Oh-Eda, M.; Ishige, I.; et al. High-affinity autoantibodies specifically eliminate granulocyte-macrophage colony-stimulating factor activity in the lungs of patients with idiopathic pulmonary alveolar proteinosis. Blood 2004, 103, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Nagele, E.P.; Han, M.; Acharya, N.K.; DeMarshall, C.; Kosciuk, M.C.; Nagele, R.G. Natural IgG autoantibodies are abundant and ubiquitous in human sera, and their number is influenced by age, gender, and disease. PLoS ONE 2013, 8, e60726. [Google Scholar] [CrossRef] [PubMed]
- Cappellano, G.; Orilieri, E.; Woldetsadik, A.D.; Boggio, E.; Soluri, M.F.; Comi, C.; Sblattero, D.; Chiocchetti, A.; Dianzani, U. Anti-cytokine autoantibodies in autoimmune diseases. Am. J. Clin. Exp. Immunol. 2012, 1, 136–146. [Google Scholar] [PubMed]
- Wolff, A.S.; Sarkadi, A.K.; Marodi, L.; Karner, J.; Orlova, E.; Oftedal, B.E.; Kisand, K.; Olah, E.; Meloni, A.; Myhre, A.G.; et al. Anti-cytokine autoantibodies preceding onset of autoimmune polyendocrine syndrome type I features in early childhood. J. Clin. Immunol. 2013, 33, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Sakagami, T.; Uchida, K.; Suzuki, T.; Carey, B.C.; Wood, R.E.; Wert, S.E.; Whitsett, J.A.; Trapnell, B.C.; Luisetti, M. Human GM-CSF autoantibodies and reproduction of pulmonary alveolar proteinosis. N. Engl. J. Med. 2009, 361, 2679–2681. [Google Scholar] [CrossRef] [PubMed]
- Czaja, C.A.; Merkel, P.A.; Chan, E.D.; Lenz, L.L.; Wolf, M.L.; Alam, R.; Frankel, S.K.; Fischer, A.; Gogate, S.; Perez-Velez, C.M.; et al. Rituximab as successful adjunct treatment in a patient with disseminated nontuberculous mycobacterial infection due to acquired anti-interferon-gamma autoantibody. Clin. Infect. Dis. 2014, 58, e115–e118. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, L.; Campo, I.; Fregni, C.S.; Rodriguez, B.M.; Minola, A.; Sallusto, F.; Luisetti, M.; Corti, D.; Lanzavecchia, A. Neutralization and clearance of GM-CSF by autoantibodies in pulmonary alveolar proteinosis. Nat. Commun. 2015, 6, 375. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.M.; Price, J.V.; Barcenas-Morales, G.; Ceron-Gutierrez, L.; Davies, S.; Kumararatne, D.S.; Doffinger, R.; Utz, P.J. Protein microarrays identify disease-specific anti-cytokine autoantibody profiles in the landscape of immunodeficiency. J. Allergy Clin. Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ching, K.H.; Burbelo, P.D.; Tipton, C.; Wei, C.; Petri, M.; Sanz, I.; Iadarola, M.J. Two major autoantibody clusters in systemic lupus erythematosus. PLoS ONE 2012, 7, e32001. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.E.; Rosen, L.B.; Csomos, K.; Rosenberg, J.M.; Mathew, D.; Keszei, M.; Ujhazi, B.; Chen, K.; Lee, Y.N.; Tirosh, I.; et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J. Clin. Investig. 2015, 125, 4135–4148. [Google Scholar] [CrossRef] [PubMed]
- Jouvenne, P.; Fossiez, F.; Banchereau, J.; Miossec, P. High levels of neutralizing autoantibodies against IL-1 alpha are associated with a better prognosis in chronic polyarthritis: A follow-up study. Scand. J. Immunol. 1997, 46, 413–418. [Google Scholar] [CrossRef] [PubMed]
- 140. Mizutani, H.; Ohmoto, Y.; Kupper, T.S.; Shimizu, M. Endogenous neutralizing anti-IL-1 alpha autoantibodies in inflammatory skin diseases: Possible natural inhibitor for over expressed epidermal IL-1. J. Dermatol. Sci. 1998, 20, 63–71. [Google Scholar] [CrossRef]
- Puel, A.; Picard, C.; Lorrot, M.; Pons, C.; Chrabieh, M.; Lorenzo, L.; Mamani-Matsuda, M.; Jouanguy, E.; Gendrel, D.; Casanova, J.L. Recurrent staphylococcal cellulitis and subcutaneous abscesses in a child with autoantibodies against IL-6. J. Immunol. 2008, 180, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Takemura, H.; Suzuki, H.; Yoshizaki, K.; Ogata, A.; Yuhara, T.; Akama, T.; Yamane, K.; Kashiwagi, H. Anti-interleukin-6 autoantibodies in rheumatic diseases. Increased frequency in the sera of patients with systemic sclerosis. Arthritis Rheum. 1992, 35, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Takemura, H.; Yoshizaki, K.; Koishihara, Y.; Ohsugi, Y.; Okano, A.; Akiyama, Y.; Tojo, T.; Kishimoto, T.; Kashiwagi, H. IL-6-anti-IL-6 autoantibody complexes with IL-6 activity in sera from some patients with systemic sclerosis. J. Immunol. 1994, 152, 935–942. [Google Scholar] [PubMed]
- Nanki, T.; Onoue, I.; Nagasaka, K.; Takayasu, A.; Ebisawa, M.; Hosoya, T.; Shirai, T.; Sugihara, T.; Hirata, S.; Kubota, T.; et al. Suppression of elevations in serum C reactive protein levels by anti-IL-6 autoantibodies in two patients with severe bacterial infections. Ann. Rheum. Dis. 2013, 72, 1100–1102. [Google Scholar] [CrossRef] [PubMed]
- Fudala, R.; Krupa, A.; Stankowska, D.; Allen, T.C.; Kurdowska, A.K. Anti-interleukin-8 autoantibody:interleukin-8 immune complexes in acute lung injury/acute respiratory distress syndrome. Clin. Sci. 2008, 114, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Meager, A.; Vincent, A.; Newsom-Davis, J.; Willcox, N. Spontaneous neutralising antibodies to interferon--alpha and interleukin-12 in thymoma-associated autoimmune disease. Lancet 1997, 350, 1596–1597. [Google Scholar] [CrossRef]
- Sim, B.T.; Browne, S.K.; Vigliani, M.; Zachary, D.; Rosen, L.; Holland, S.M.; Opal, S.M. Recurrent Burkholderia gladioli suppurative lymphadenitis associated with neutralizing anti-IL-12p70 autoantibodies. J. Clin. Immunol. 2013, 33, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Kisand, K.; Boe Wolff, A.S.; Podkrajsek, K.T.; Tserel, L.; Link, M.; Kisand, K.V.; Ersvaer, E.; Perheentupa, J.; Erichsen, M.M.; Bratanic, N.; et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J. Exp. Med. 2010, 207, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Doffinger, R.; Natividad, A.; Chrabieh, M.; Barcenas-Morales, G.; Picard, C.; Cobat, A.; Ouachee-Chardin, M.; Toulon, A.; Bustamante, J.; et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J. Exp. Med. 2010, 207, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.B.; Freeman, A.F.; Yang, L.M.; Jutivorakool, K.; Olivier, K.N.; Angkasekwinai, N.; Suputtamongkol, Y.; Bennett, J.E.; Pyrgos, V.; Williamson, P.R.; et al. Anti-GM-CSF autoantibodies in patients with cryptococcal meningitis. J. Immunol. 2013, 190, 3959–3966. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.B.; Rocha Pereira, N.; Figueiredo, C.; Fiske, L.C.; Ressner, R.A.; Hong, J.C.; Gregg, K.S.; Henry, T.L.; Pak, K.J.; Baumgarten, K.L.; et al. Nocardia-induced granulocyte macrophage colony-stimulating factor is neutralized by autoantibodies in disseminated/extrapulmonary nocardiosis. Clin. Infect. Dis. 2015, 60, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.F.; Doyle, I.R.; Nakata, K.; Presneill, J.J.; Schoch, O.D.; Hamano, E.; Uchida, K.; Fisher, R.; Dunn, A.R. Relationship of anti-GM-CSF antibody concentration, surfactant protein A and B levels, and serum LDH to pulmonary parameters and response to GM-CSF therapy in patients with idiopathic alveolar proteinosis. Thorax 2003, 58, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Beck, D.C.; Yamamoto, T.; Berclaz, P.Y.; Abe, S.; Staudt, M.K.; Carey, B.C.; Filippi, M.D.; Wert, S.E.; Denson, L.A.; et al. GM-CSF autoantibodies and neutrophil dysfunction in pulmonary alveolar proteinosis. N. Engl. J. Med. 2007, 356, 567–579. [Google Scholar] [CrossRef] [PubMed]
- DeLeon, T.T.; Chung, H.H.; Opal, S.M.; Dworkin, J.D. Mycobacterium avium complex empyema in a patient with interferon gamma autoantibodies. Hawai’i J. Med. Public Health 2014, 73, 15–17. [Google Scholar]
- Hanitsch, L.G.; Lobel, M.; Muller-Redetzky, H.; Schurmann, M.; Suttorp, N.; Unterwalder, N.; Monnich, U.; Meisel, C.; Wittke, K.; Volk, H.D.; et al. Salmonella Sepsis in a German Caucasian Patient with Unusual Anti-Interferon-Gamma IgG1 Autoantibodies. J. Clin. Immunol. 2015, 35, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Baerlecken, N.; Jacobs, R.; Stoll, M.; Schmidt, R.E.; Witte, T. Recurrent, multifocal Mycobacterium avium-intercellulare infection in a patient with interferon-gamma autoantibody. Clin. Infect. Dis. 2009, 49, e76–e78. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, A.M.; Flesher, D.T.; Yang, J.; Wolslegel, K.; Wang, X.; Brady, A.; Abbas, A.R.; Quarmby, V.; Wakshull, E.; Richardson, B.; et al. Association of endogenous anti-interferon-alpha autoantibodies with decreased interferon-pathway and disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2011, 63, 2407–2415. [Google Scholar] [CrossRef] [PubMed]
- Shiono, H.; Wong, Y.L.; Matthews, I.; Liu, J.L.; Zhang, W.; Sims, G.; Meager, A.; Beeson, D.; Vincent, A.; Willcox, N. Spontaneous production of anti-IFN-alpha and anti-IL-12 autoantibodies by thymoma cells from myasthenia gravis patients suggests autoimmunization in the tumor. Int. Immunol. 2003, 15, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Price, J.V.; Haddon, D.J.; Kemmer, D.; Delepine, G.; Mandelbaum, G.; Jarrell, J.A.; Gupta, R.; Balboni, I.; Chakravarty, E.F.; Sokolove, J.; et al. Protein microarray analysis reveals BAFF-binding autoantibodies in systemic lupus erythematosus. J. Clin. Investig. 2013, 123, 5135–5145. [Google Scholar] [CrossRef] [PubMed]
- Sakata, M.; Tsuruha, J.I.; Masuko-Hongo, K.; Nakamura, H.; Matsui, T.; Sudo, A.; Nishioka, K.; Kato, T. Autoantibodies to osteopontin in patients with osteoarthritis and rheumatoid arthritis. J. Rheumatol. 2001, 28, 1492–1495. [Google Scholar] [PubMed]
- Ying, X.; Zhao, Y.; Wang, J.L.; Zhou, X.; Zhao, J.; He, C.C.; Guo, X.J.; Jin, G.H.; Wang, L.J.; Zhu, Q.; et al. Serum anti-osteopontin autoantibody as a novel diagnostic and prognostic biomarker in patients with hepatocellular carcinoma. Oncol. Rep. 2014, 32, 1550–1556. [Google Scholar] [CrossRef] [PubMed]
- Sjowall, C.; Ernerudh, J.; Bengtsson, A.A.; Sturfelt, G.; Skogh, T. Reduced anti-TNFalpha autoantibody levels coincide with flare in systemic lupus erythematosus. J. Autoimmun. 2004, 22, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Elkarim, R.A.; Mustafa, M.; Kivisakk, P.; Link, H.; Bakhiet, M. Cytokine autoantibodies in multiple sclerosis, aseptic meningitis and stroke. Eur. J. Clin. Investig. 1998, 28, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Hauser, B.; Riches, P.L.; Gilchrist, T.; Visconti, M.R.; Wilson, J.F.; Ralston, S.H. Autoantibodies to osteoprotegerin are associated with increased bone resorption in rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 1631–1632. [Google Scholar] [CrossRef] [PubMed]
- Real, A.; Gilbert, N.; Hauser, B.; Kennedy, N.; Shand, A.; Gillett, H.; Gillett, P.; Goddard, C.; Cebolla, A.; Sousa, C.; et al. Characterisation of osteoprotegerin autoantibodies in coeliac disease. Calcified Tissue Int. 2015, 97, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Riches, P.L.; McRorie, E.; Fraser, W.D.; Determann, C.; van’t Hof, R.; Ralston, S.H. Osteoporosis associated with neutralizing autoantibodies against osteoprotegerin. N. Engl. J. Med. 2009, 361, 1459–1465. [Google Scholar] [CrossRef] [PubMed]
- Dorman, S.E.; Holland, S.M. Interferon-gamma and interleukin-12 pathway defects and human disease. Cytokine Growth Factor Rev. 2000, 11, 321–333. [Google Scholar] [CrossRef]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leuk. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Carnaud, C.; Lee, D.; Donnars, O.; Park, S.H.; Beavis, A.; Koezuka, Y.; Bendelac, A. Cutting edge: Cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J. Immunol. 1999, 163, 4647–4650. [Google Scholar] [PubMed]
- Gessani, S.; Belardelli, F. IFN-gamma expression in macrophages and its possible biological significance. Cytokine Growth Factor Rev. 1998, 9, 117–123. [Google Scholar] [CrossRef]
- Harris, D.P.; Haynes, L.; Sayles, P.C.; Duso, D.K.; Eaton, S.M.; Lepak, N.M.; Johnson, L.L.; Swain, S.L.; Lund, F.E. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat. Immunol. 2000, 1, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Remus, N.; Reichenbach, J.; Picard, C.; Rietschel, C.; Wood, P.; Lammas, D.; Kumararatne, D.S.; Casanova, J.L. Impaired interferon gamma-mediated immunity and susceptibility to mycobacterial infection in childhood. Pediatr. Res. 2001, 50, 8–13. [Google Scholar] [CrossRef] [PubMed]
- McGarvey, J.; Bermudez, L.E. Pathogenesis of nontuberculous mycobacteria infections. Clin. Chest Med. 2002, 23, 569–583. [Google Scholar] [CrossRef]
- Griffith, D.E.; Aksamit, T.; Brown-Elliott, B.A.; Catanzaro, A.; Daley, C.; Gordin, F.; Holland, S.M.; Horsburgh, R.; Huitt, G.; Iademarco, M.F.; et al. An official ATS/IDSA statement: Diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am. J. Resp. Crit. Care Med. 2007, 175, 367–416. [Google Scholar] [CrossRef] [PubMed]
- Doffinger, R.; Helbert, M.R.; Barcenas-Morales, G.; Yang, K.; Dupuis, S.; Ceron-Gutierrez, L.; Espitia-Pinzon, C.; Barnes, N.; Bothamley, G.; Casanova, J.L.; et al. Autoantibodies to interferon-gamma in a patient with selective susceptibility to mycobacterial infection and organ-specific autoimmunity. Clin. Infect. Dis. 2004, 38, e10–e14. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.Y.; Chu, C.C.; Liu, J.P.; Lin, C.H.; Ho, M.W.; Lo, W.J.; Lin, P.C.; Chen, H.J.; Chou, C.H.; Feng, J.Y.; et al. Anti-IFN-gamma autoantibodies in adults with disseminated nontuberculous mycobacterial infections are associated with HLA-DRB1*16:02 and HLA-DQB1*05:02 and the reactivation of latent varicella-zoster virus infection. Blood 2013, 121, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Fujita-Suzuki, Y.; Yonemaru, M.; Ohkusu, K.; Sakagami, T.; Carpenter, S.M.; Otsuka, Y.; Namkoong, H.; Yano, I.; Hasegawa, N. Recurrence of disseminated Mycobacterium avium complex disease in a patient with anti-gamma interferon autoantibodies by reinfection. J. Clin. Microbiol. 2015, 53, 1436–1438. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, E.; Rosen, L.B.; LaRue, R.W.; Fabre, V.; Melia, M.T.; Auwaerter, P.G.; Holland, S.M.; Browne, S.K. The first US domestic report of disseminated Mycobacterium avium complex and anti-interferon-gamma autoantibodies. J. Clin. Immunol. 2014, 34, 928–932. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.Y.; Ding, L.; Brown, M.R.; Lantz, L.; Gay, T.; Cohen, S.; Martyak, L.A.; Kubak, B.; Holland, S.M. Anti-IFN-gamma autoantibodies in disseminated nontuberculous mycobacterial infections. J. Immunol. 2005, 175, 4769–4776. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 2009, 9, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Sparber, F.; LeibundGut-Landmann, S. Interleukin 17-Mediated Host Defense against Candida albicans. Pathogens 2015, 4, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, C.E.; Reich, K.; Lebwohl, M.; van de Kerkhof, P.; Paul, C.; Menter, A.; Cameron, G.S.; Erickson, J.; Zhang, L.; Secrest, R.J.; et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): Results from two phase 3 randomised trials. Lancet 2015, 386, 541–551. [Google Scholar] [CrossRef]
- Lubberts, E. The IL-23-IL-17 axis in inflammatory arthritis. Nat. Rev. Rheumatol. 2015, 11, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Rutz, S.; Eidenschenk, C.; Ouyang, W. IL-22, not simply a Th17 cytokine. Immunol. Rev. 2013, 252, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, C.H. Chronic mucocutaneous candidiasis. Pediatr. Infect. Dis. J. 2001, 20, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Lilic, D. New perspectives on the immunology of chronic mucocutaneous candidiasis. Curr. Opin. Infect. Dis. 2002, 15, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.D.; Brenchley, J.M.; Laurence, A.; Freeman, A.F.; Hill, B.J.; Elias, K.M.; Kanno, Y.; Spalding, C.; Elloumi, H.Z.; Paulson, M.L.; et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 2008, 452, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Glocker, E.O.; Hennigs, A.; Nabavi, M.; Schaffer, A.A.; Woellner, C.; Salzer, U.; Pfeifer, D.; Veelken, H.; Warnatz, K.; Tahami, F.; et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med. 2009, 361, 1727–1735. [Google Scholar] [CrossRef] [PubMed]
- Ferwerda, B.; Ferwerda, G.; Plantinga, T.S.; Willment, J.A.; van Spriel, A.B.; Venselaar, H.; Elbers, C.C.; Johnson, M.D.; Cambi, A.; Huysamen, C.; et al. Human dectin-1 deficiency and mucocutaneous fungal infections. N. Engl. J. Med. 2009, 361, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Cypowyj, S.; Marodi, L.; Abel, L.; Picard, C.; Casanova, J.L. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, E.S.; Handley, H.E. Chronic tetany and chronic mycelia stomatitis in a child aged four and one half years. Am. J. Dis. Child. 1929, 38, 228. [Google Scholar]
- Ahonen, P.; Myllarniemi, S.; Sipila, I.; Perheentupa, J. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N. Engl. J. Med. 1990, 322, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Finnish-German, A.C. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat. Genet. 1997, 17, 399–403. [Google Scholar]
- Strobel, P.; Murumagi, A.; Klein, R.; Luster, M.; Lahti, M.; Krohn, K.; Schalke, B.; Nix, W.; Gold, R.; Rieckmann, P.; et al. Deficiency of the autoimmune regulator AIRE in thymomas is insufficient to elicit autoimmune polyendocrinopathy syndrome type 1 (APS-1). J. Pathol. 2007, 211, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, J.; Golde, D.W.; Weisbart, R.H.; Gasson, J.C. Granulocyte-macrophage colony-stimulating factor enhances phagocytosis of bacteria by human neutrophils. Blood 1986, 68, 708–711. [Google Scholar] [PubMed]
- Dranoff, G.; Crawford, A.D.; Sadelain, M.; Ream, B.; Rashid, A.; Bronson, R.T.; Dickersin, G.R.; Bachurski, C.J.; Mark, E.L.; Whitsett, J.A.; et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science 1994, 264, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Stanley, E.; Lieschke, G.J.; Grail, D.; Metcalf, D.; Hodgson, G.; Gall, J.A.; Maher, D.W.; Cebon, J.; Sinickas, V.; Dunn, A.R. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc. Natl. Acad. Sci. USA 1994, 91, 5592–5596. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Ikegami, M.; Reed, J.A.; Chroneos, Z.C.; Whitsett, J.A. GM-CSF regulates protein and lipid catabolism by alveolar macrophages. Am. J. Physiol. Lung C. 2001, 280, L379–L386. [Google Scholar]
- Trapnell, B.C.; Whitsett, J.A.; Nakata, K. Pulmonary alveolar proteinosis. N. Engl. J. Med. 2003, 349, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Rosen, S.H.; Castleman, B.; Liebow, A.A. Pulmonary alveolar proteinosis. N. Engl. J. Med. 1958, 258, 1123–1142. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Tanaka, N.; Watanabe, J.; Uchida; Kanegasaki, S.; Yamada, Y.; Nakata, K. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1999, 190, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Carraway, M.S.; Ghio, A.J.; Carter, J.D.; Piantadosi, C.A. Detection of granulocyte-macrophage colony-stimulating factor in patients with pulmonary alveolar proteinosis. Am. J. Resp. Crit. Care Med. 2000, 161, 1294–1299. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, T.L.; Kavuru, M.S.; Thomassen, M.J. Anti-GM-CSF titer predicts response to GM-CSF therapy in pulmonary alveolar proteinosis. Clin. Immunol. 2002, 105, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Golde, D.W.; Territo, M.; Finley, T.N.; Cline, M.J. Defective lung macrophages in pulmonary alveolar proteinosis. Ann. Int. Med. 1976, 85, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rothi, R.J.; Harris, J.O. Pulmonary alveolar proteinosis. Further evaluation of abnormal alveolar macrophages. Chest 1986, 90, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.F.; Presneill, J.J. Pulmonary alveolar proteinosis: Progress in the first 44 years. Am. J. Resp. Crit. Care Med. 2002, 166, 215–235. [Google Scholar] [CrossRef] [PubMed]
- Behrens, F.; Tak, P.P.; Ostergaard, M.; Stoilov, R.; Wiland, P.; Huizinga, T.W.; Berenfus, V.Y.; Vladeva, S.; Rech, J.; Rubbert-Roth, A.; et al. MOR103, a human monoclonal antibody to granulocyte-macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: Results of a phase Ib/IIa randomised, double-blind, placebo-controlled, dose-escalation trial. Ann. Rheum. Dis. 2015, 74, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.J.; Hammacher, A.; Smith, D.K.; Matthews, J.M.; Ward, L.D. Interleukin-6: Structure-function relationships. Protein Sci. 1997, 6, 929–955. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.J.; Said, S.; Hernandez, G.T. Recurrent Skin and Lung Infections in Autosomal Dominant Hyper IgE Syndrome with Transactivation Domain STAT3 Mutation. Case Rep. Immunol. 2014, 2014, 136752. [Google Scholar] [CrossRef] [PubMed]
- Homann, C.; Hansen, M.B.; Graudal, N.; Hasselqvist, P.; Svenson, M.; Bendtzen, K.; Thomsen, A.C.; Garred, P. Anti-interleukin-6 autoantibodies in plasma are associated with an increased frequency of infections and increased mortality of patients with alcoholic cirrhosis. Scand. J. Immunol. 1996, 44, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Ogata, A.; Kishimoto, T. A new era for the treatment of inflammatory autoimmune diseases by interleukin-6 blockade strategy. Semin. Immunol. 2014, 26, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Tanaka, T.; Kishimoto, T. Therapeutic uses of anti-interleukin-6 receptor antibody. Int. Immunol. 2015, 27, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Emery, P.; Keystone, E.; Tony, H.P.; Cantagrel, A.; van Vollenhoven, R.; Sanchez, A.; Alecock, E.; Lee, J.; Kremer, J. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: Results from a 24-week multicentre randomised placebo-controlled trial. Ann. Rheum. Dis. 2008, 67, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, S.; Postacchini, L.; Manfredi, L.; Goteri, G.; Luchetti, M.M.; Festa, A.; Gabrielli, A.; Pomponio, G. Successful treatment of a Caucasian case of multifocal Castleman’s disease with TAFRO syndrome with a pathophysiology targeted therapy—A case report. Exp. Hematol. Oncol. 2015, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Goto, H.; Hirao, K.; Nakajima, A.; Origasa, H.; Tanaka, K.; Tomobe, M.; Totsuka, K. Longterm Safety of Tocilizumab: Results from 3 Years of Followup Postmarketing Surveillance of 5573 Patients with Rheumatoid Arthritis in Japan. J. Rheumatol. 2015, 42, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Horneff, G. Biologic-Associated Infections in Pediatric Rheumatology. Curr. Rheumatol. Rep. 2015, 17, 66. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. P. Roy. Soc. Lond. B Bio. 1957, 147, 258–267. [Google Scholar] [CrossRef]
- Decker, T.; Muller, M.; Stockinger, S. The yin and yang of type I interferon activity in bacterial infection. Nat. Rev. Immunol. 2005, 5, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Type I interferon: Friend or foe? J. Exp. Med. 2010, 207, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Fehr, T.; Schoedon, G.; Odermatt, B.; Holtschke, T.; Schneemann, M.; Bachmann, M.F.; Mak, T.W.; Horak, I.; Zinkernagel, R.M. Crucial role of interferon consensus sequence binding protein, but neither of interferon regulatory factor 1 nor of nitric oxide synthesis for protection against murine listeriosis. J. Exp. Med. 1997, 185, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Manca, C.; Tsenova, L.; Bergtold, A.; Freeman, S.; Tovey, M.; Musser, J.M.; Barry, C.E., 3rd; Freedman, V.H.; Kaplan, G. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha /beta. Proc. Natl. Acad. Sci. USA 2001, 98, 5752–5757. [Google Scholar] [CrossRef] [PubMed]
- Hooks, J.J.; Moutsopoulos, H.M.; Geis, S.A.; Stahl, N.I.; Decker, J.L.; Notkins, A.L. Immune interferon in the circulation of patients with autoimmune disease. N. Engl. J. Med. 1979, 301, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Ytterberg, S.R.; Schnitzer, T.J. Serum interferon levels in patients with systemic lupus erythematosus. Arthritis Rheum. 1982, 25, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Ronnblom, L.E.; Alm, G.V.; Oberg, K.E. Autoimmunity after alpha-interferon therapy for malignant carcinoid tumors. Ann. Int. Med. 1991, 115, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Sanchez Roman, J.; Castillo Palma, M.J.; Garcia Diaz, E.; Ferrer Ordinez, J.A. Systemic lupus erythematosus induced by recombinant alpha interferon treatment. Med. Clin. 1994, 102, 198. [Google Scholar]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Asano, S. Human granulocyte colony-stimulating factor: Its basic aspects and clinical applications. Am. J. Pediatr. Hematol. 1991, 13, 400–413. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Grail, D.; Hodgson, G.; Metcalf, D.; Stanley, E.; Cheers, C.; Fowler, K.J.; Basu, S.; Zhan, Y.F.; Dunn, A.R. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood 1994, 84, 1737–1746. [Google Scholar] [PubMed]
- Liu, F.; Wu, H.Y.; Wesselschmidt, R.; Kornaga, T.; Link, D.C. Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity 1996, 5, 491–501. [Google Scholar] [CrossRef]
- Huber, A.R.; Kunkel, S.L.; Todd, R.F., 3rd; Weiss, S.J. Regulation of transendothelial neutrophil migration by endogenous interleukin-8. Science 1991, 254, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M.; Appella, E.; Yamada, M.; Matsushima, K.; Gronenborn, A.M. Three-dimensional structure of interleukin 8 in solution. Biochemistry 1990, 29, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Baggiolini, M.; Walz, A.; Kunkel, S.L. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J. Clin. Investig. 1989, 84, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Detmers, P.A.; Lo, S.K.; Olsen-Egbert, E.; Walz, A.; Baggiolini, M.; Cohn, Z.A. Neutrophil-activating protein 1/interleukin 8 stimulates the binding activity of the leukocyte adhesion receptor CD11b/CD18 on human neutrophils. J. Exp. Med. 1990, 171, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Moser, B.; Clark-Lewis, I.; Zwahlen, R.; Baggiolini, M. Neutrophil-activating properties of the melanoma growth-stimulatory activity. J. Exp. Med. 1990, 171, 1797–1802. [Google Scholar] [CrossRef] [PubMed]
- Paccaud, J.P.; Schifferli, J.A.; Baggiolini, M. NAP-1/IL-8 induces up-regulation of CR1 receptors in human neutrophil leukocytes. Biochem. Biophys. Res. Commun. 1990, 166, 187–192. [Google Scholar] [CrossRef]
- Ashbaugh, D.G.; Bigelow, D.B.; Petty, T.L.; Levine, B.E. Acute respiratory distress in adults. Lancet 1967, 2, 319–323. [Google Scholar] [CrossRef]
- Fudala, R.; Krupa, A.; Stankowska, D.; Allen, T.C.; Kurdowska, A.K. Anti-IL-8 autoantibody:IL-8 immune complexes suppress spontaneous apoptosis of neutrophils. Am. J. Physiol. Lung C. 2007, 293, L364–L374. [Google Scholar] [CrossRef] [PubMed]
- Bux, J.; Sachs, U.J. The pathogenesis of transfusion-related acute lung injury (TRALI). Br. J. Haematol. 2007, 136, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Triulzi, D.J. Transfusion-related acute lung injury: An update. Hematology 2006, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.W. Landmark advances in the development of erythropoietin. Exp. Biol. Med. 2010, 235, 1398–1411. [Google Scholar] [CrossRef] [PubMed]
- Melli, G.; Keswani, S.; Höke, A. History and Biology of Erythropoietin in Hematopoietic and Non-Neural Tissues. In Erythropoietin and the Nervous System; Höke, A., Ed.; Springer: New York, NY, USA, 2006; pp. 1–13. [Google Scholar]
- Kuhrt, D.; Wojchowski, D.M. Emerging EPO and EPO receptor regulators and signal transducers. Blood 2015, 125, 3536–3541. [Google Scholar] [CrossRef] [PubMed]
- Larpthaveesarp, A.; Ferriero, D.M.; Gonzalez, F.F. Growth factors for the treatment of ischemic brain injury (growth factor treatment). Brain Sci. 2015, 5, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Chen, W.; Sinha, B.; Tu, Y.; Manning, S.; Thomas, N.; Zhou, S.; Jiang, H.; Ma, H.; Kroessler, D.A.; et al. Neuroprotective agents for neonatal hypoxic-ischemic brain injury. Drug Discov. Today 2015, 20, 1372–1381. [Google Scholar] [CrossRef] [PubMed]
- Juul, S.E.; Pet, G.C. Erythropoietin and Neonatal Neuroprotection. Clin. Perinatol. 2015, 42, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Macdougall, I.C.; Roger, S.D.; de Francisco, A.; Goldsmith, D.J.; Schellekens, H.; Ebbers, H.; Jelkmann, W.; London, G.; Casadevall, N.; Horl, W.H.; et al. Antibody-mediated pure red cell aplasia in chronic kidney disease patients receiving erythropoiesis-stimulating agents: New insights. Kidney Int. 2012, 81, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Pollock, C.; Johnson, D.W.; Horl, W.H.; Rossert, J.; Casadevall, N.; Schellekens, H.; Delage, R.; De Francisco, A.; Macdougall, I.; Thorpe, R.; et al. Pure red cell aplasia induced by erythropoiesis-stimulating agents. Clin. J. Am. Soc. Nephrol. 2008, 3, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, K.U.; Casadevall, N. Pure red-cell aplasia due to anti-erythropoietin antibodies. Nephrol. Dial. Transpl. 2003, 18, 865–869. [Google Scholar] [CrossRef]
- Casadevall, N.; Dupuy, E.; Molho-Sabatier, P.; Tobelem, G.; Varet, B.; Mayeux, P. Autoantibodies against erythropoietin in a patient with pure red-cell aplasia. N. Engl. J. Med. 1996, 334, 630–633. [Google Scholar] [CrossRef] [PubMed]
- Peschle, C.; Marmont, A.M.; Marone, G.; Genovese, A.; Sasso, G.F.; Condorelli, M. Pure red cell aplasia: Studies on an IgG serum inhibitor neutralizing erythropoietin. Br. J. Haematol. 1975, 30, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Linardaki, G.D.; Boki, K.A.; Fertakis, A.; Tzioufas, A.G. Pure red cell aplasia as presentation of systemic lupus erythematosus: Antibodies to erythropoietin. Scand. J. Rheumatol. 1999, 28, 189–191. [Google Scholar] [PubMed]
- Casadevall, N. Pure red cell aplasia and anti-erythropoietin antibodies in patients treated with epoetin. Nephrol. Dial. Transpl. 2003, 18 (Suppl. 8), viii37–viii41. [Google Scholar] [CrossRef]
- Rossert, J.; Casadevall, N.; Eckardt, K.U. Anti-erythropoietin antibodies and pure red cell aplasia. J. Am. Soc. Nephrol. 2004, 15, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Wish, J.B. Erythropoiesis-stimulating agents and pure red-cell aplasia: You can’t fool Mother Nature. Kidney Int. 2011, 80, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Wu, U.I.; Holland, S.M. Host susceptibility to non-tuberculous mycobacterial infections. Lancet Infect. Dis. 2015, 15, 968–980. [Google Scholar] [CrossRef]
- Seymour, J.F.; Presneill, J.J.; Schoch, O.D.; Downie, G.H.; Moore, P.E.; Doyle, I.R.; Vincent, J.M.; Nakata, K.; Kitamura, T.; Langton, D.; et al. Therapeutic efficacy of granulocyte-macrophage colony-stimulating factor in patients with idiopathic acquired alveolar proteinosis. Am. J. Resp. Crit. Care Med. 2001, 163, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Venkateshiah, S.B.; Yan, T.D.; Bonfield, T.L.; Thomassen, M.J.; Meziane, M.; Czich, C.; Kavuru, M.S. An open-label trial of granulocyte macrophage colony stimulating factor therapy for moderate symptomatic pulmonary alveolar proteinosis. Chest 2006, 130, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Tazawa, R.; Trapnell, B.C.; Inoue, Y.; Arai, T.; Takada, T.; Nasuhara, Y.; Hizawa, N.; Kasahara, Y.; Tatsumi, K.; Hojo, M.; et al. Inhaled granulocyte/macrophage-colony stimulating factor as therapy for pulmonary alveolar proteinosis. Am. J. Resp. Crit. Care Med. 2010, 181, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Di Michele, D.M. Immune tolerance induction in haemophilia: Evidence and the way forward. J. Thromb. Haemost. 2011, 9 (Suppl. 1), 216–225. [Google Scholar] [CrossRef] [PubMed]
- Gilhus, N.E.; Verschuuren, J.J. Myasthenia gravis: Subgroup classification and therapeutic strategies. Lancet Neurol. 2015, 14, 1023–1036. [Google Scholar] [CrossRef]
- Akasaka, K.; Tanaka, T.; Kitamura, N.; Ohkouchi, S.; Tazawa, R.; Takada, T.; Ichiwata, T.; Yamaguchi, E.; Hirose, M.; Arai, T.; et al. Outcome of corticosteroid administration in autoimmune pulmonary alveolar proteinosis: A retrospective cohort study. BMC Pulm. Med. 2015, 15, 88. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, T.L.; Russell, D.; Burgess, S.; Malur, A.; Kavuru, M.S.; Thomassen, M.J. Autoantibodies against granulocyte macrophage colony-stimulating factor are diagnostic for pulmonary alveolar proteinosis. Am. J. Resp. Cell Mol. Biol. 2002, 27, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Luisetti, M.; Rodi, G.; Perotti, C.; Campo, I.; Mariani, F.; Pozzi, E.; Trapnell, B.C. Plasmapheresis for treatment of pulmonary alveolar proteinosis. Eur. Resp. J. 2009, 33, 1220–1222. [Google Scholar] [CrossRef] [PubMed]
- Kavuru, M.S.; Malur, A.; Marshall, I.; Barna, B.P.; Meziane, M.; Huizar, I.; Dalrymple, H.; Karnekar, R.; Thomassen, M.J. An open-label trial of rituximab therapy in pulmonary alveolar proteinosis. Eur. Resp. J. 2011, 38, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.K.; Zaman, R.; Sampaio, E.P.; Jutivorakool, K.; Rosen, L.B.; Ding, L.; Pancholi, M.J.; Yang, L.M.; Priel, D.L.; Uzel, G.; et al. Anti-CD20 (rituximab) therapy for anti-IFN-gamma autoantibody-associated nontuberculous mycobacterial infection. Blood 2012, 119, 3933–3939. [Google Scholar] [CrossRef] [PubMed]
- Behler, C.M.; Terrault, N.A.; Etzell, J.E.; Damon, L.E. Rituximab therapy for pure red cell aplasia due to anti-epoetin antibodies in a woman treated with epoetin-alfa: A case report. J. Med. Case Rep. 2009, 3, 7335. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knight, V.; Merkel, P.A.; O’Sullivan, M.D. Anticytokine Autoantibodies: Association with Infection and Immune Dysregulation. Antibodies 2016, 5, 3. https://doi.org/10.3390/antib5010003
Knight V, Merkel PA, O’Sullivan MD. Anticytokine Autoantibodies: Association with Infection and Immune Dysregulation. Antibodies. 2016; 5(1):3. https://doi.org/10.3390/antib5010003
Chicago/Turabian StyleKnight, Vijaya, Patricia A. Merkel, and Michael D. O’Sullivan. 2016. "Anticytokine Autoantibodies: Association with Infection and Immune Dysregulation" Antibodies 5, no. 1: 3. https://doi.org/10.3390/antib5010003
APA StyleKnight, V., Merkel, P. A., & O’Sullivan, M. D. (2016). Anticytokine Autoantibodies: Association with Infection and Immune Dysregulation. Antibodies, 5(1), 3. https://doi.org/10.3390/antib5010003