
Opportunities for Conformation-Selective Antibodies in Amyloid-Related Diseases


Abstract
:
1. Introduction
2. Alzheimer’s Disease
2.1. Targeting Aβ Fibrils: Lessons Learnt
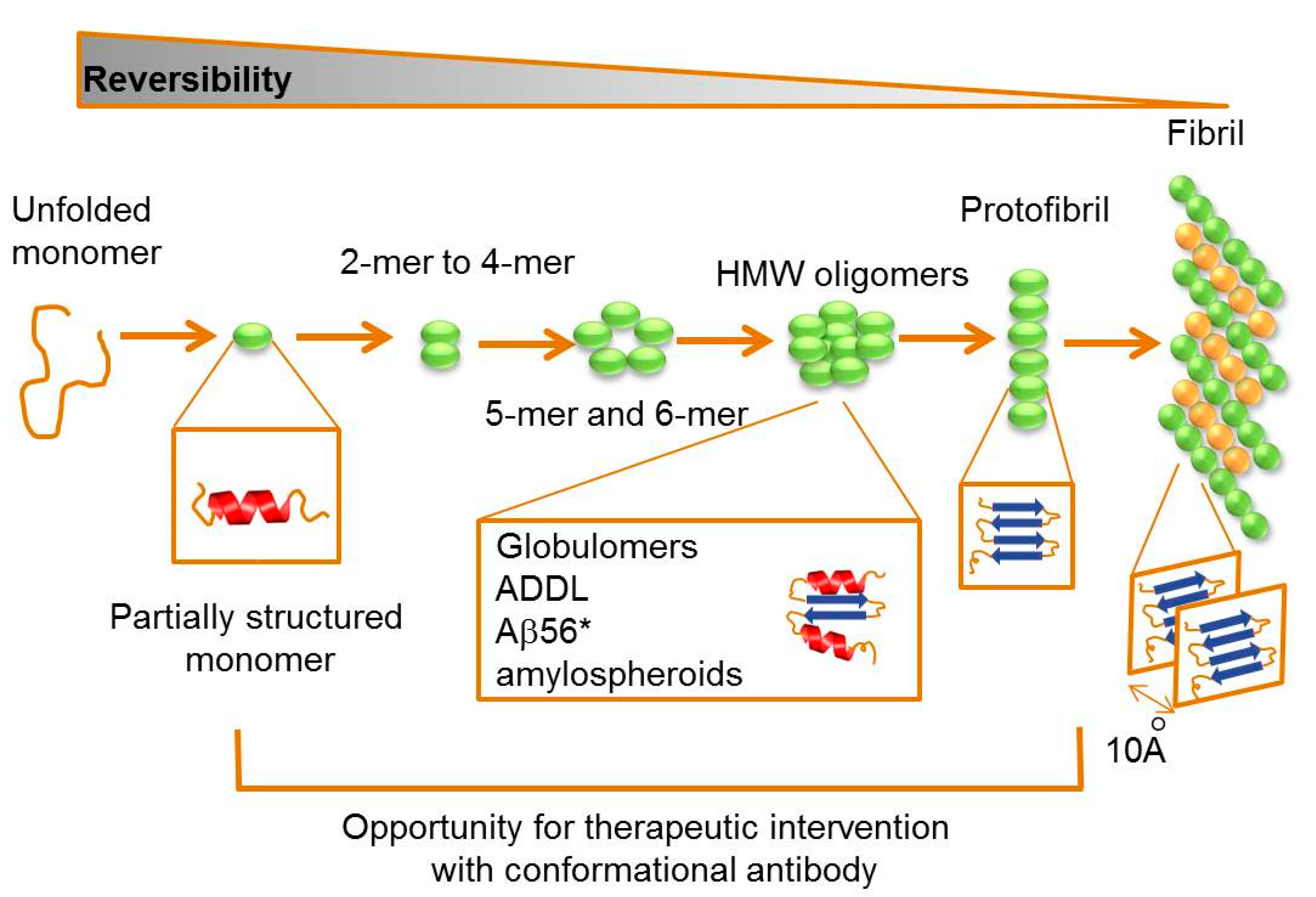
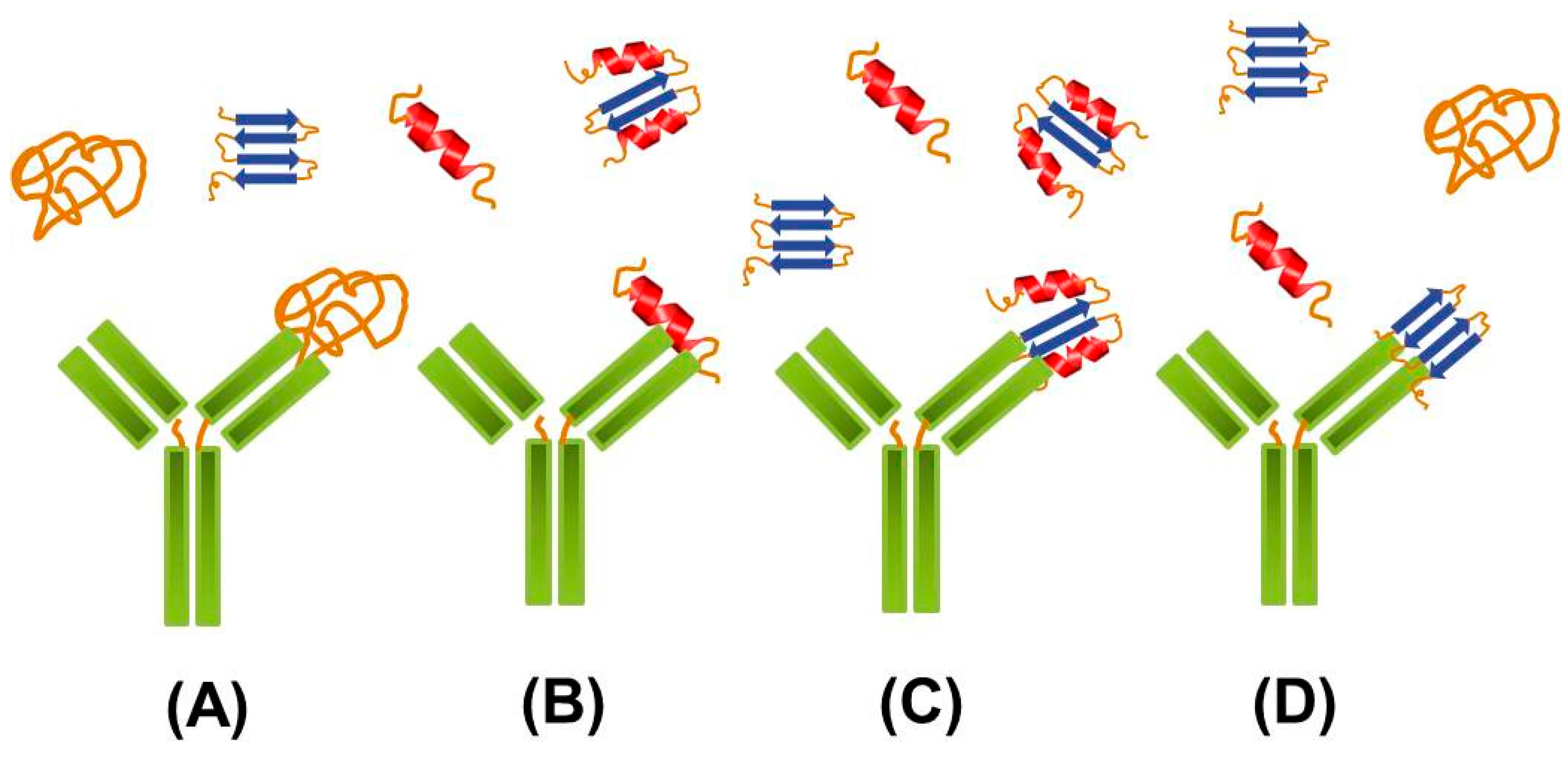
2.2. Targeting Intermediate Conformations

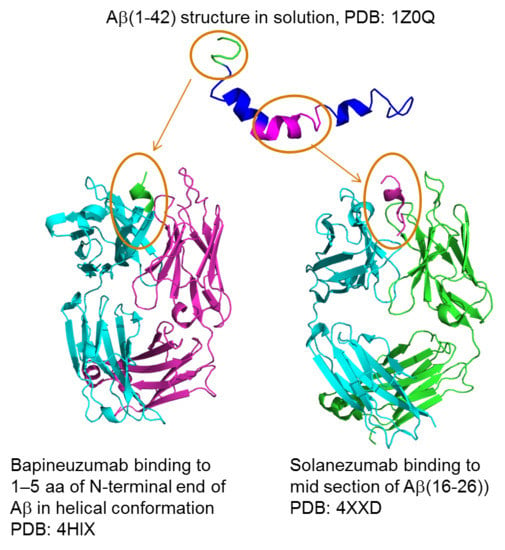
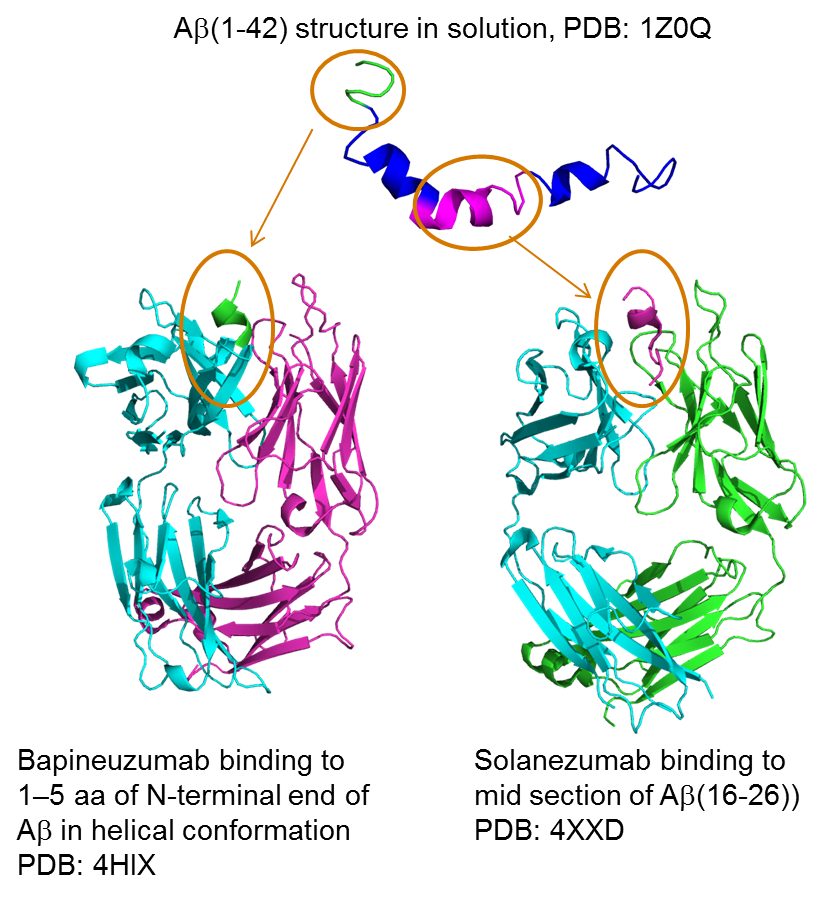{kind=link}
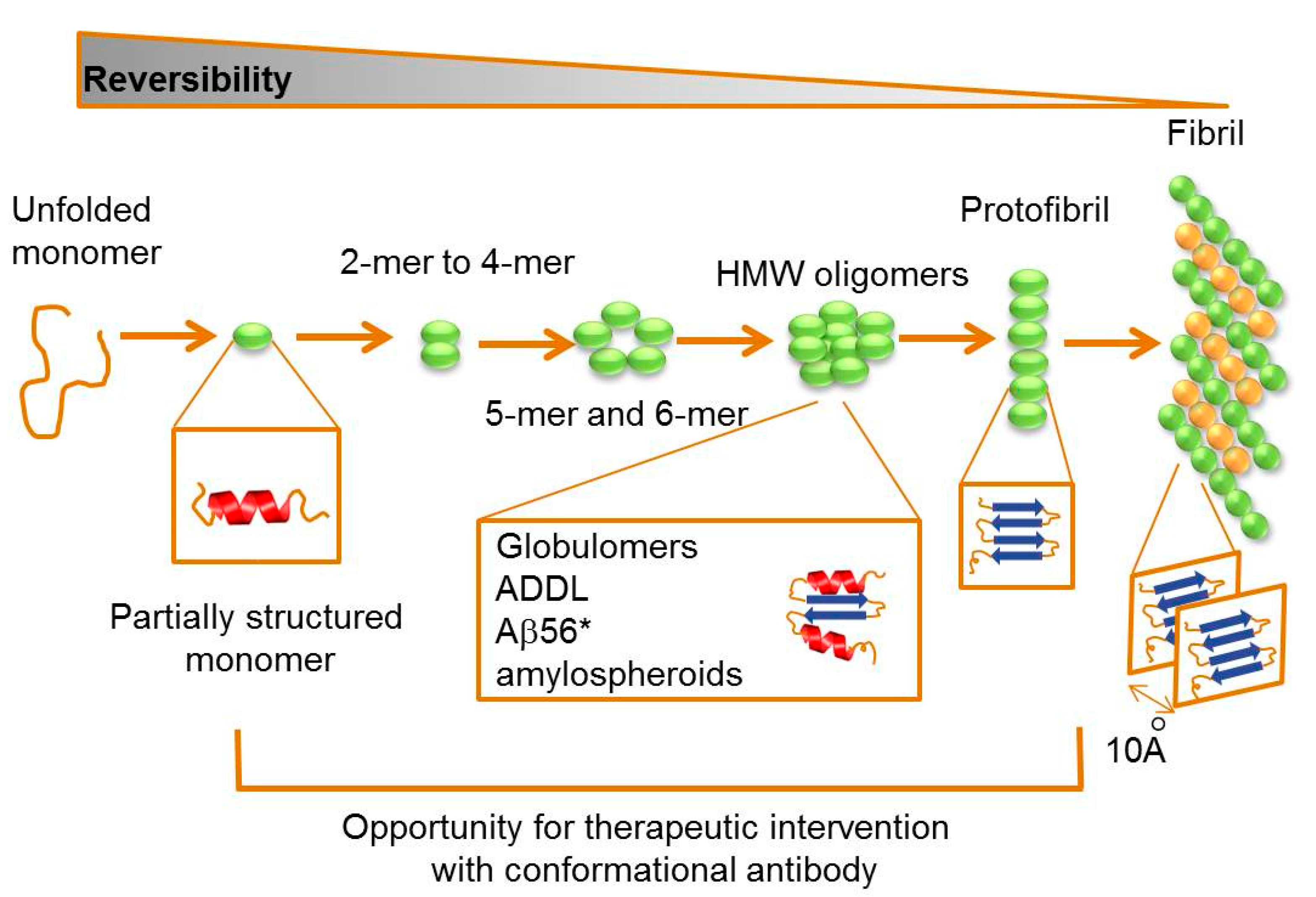{kind=link}
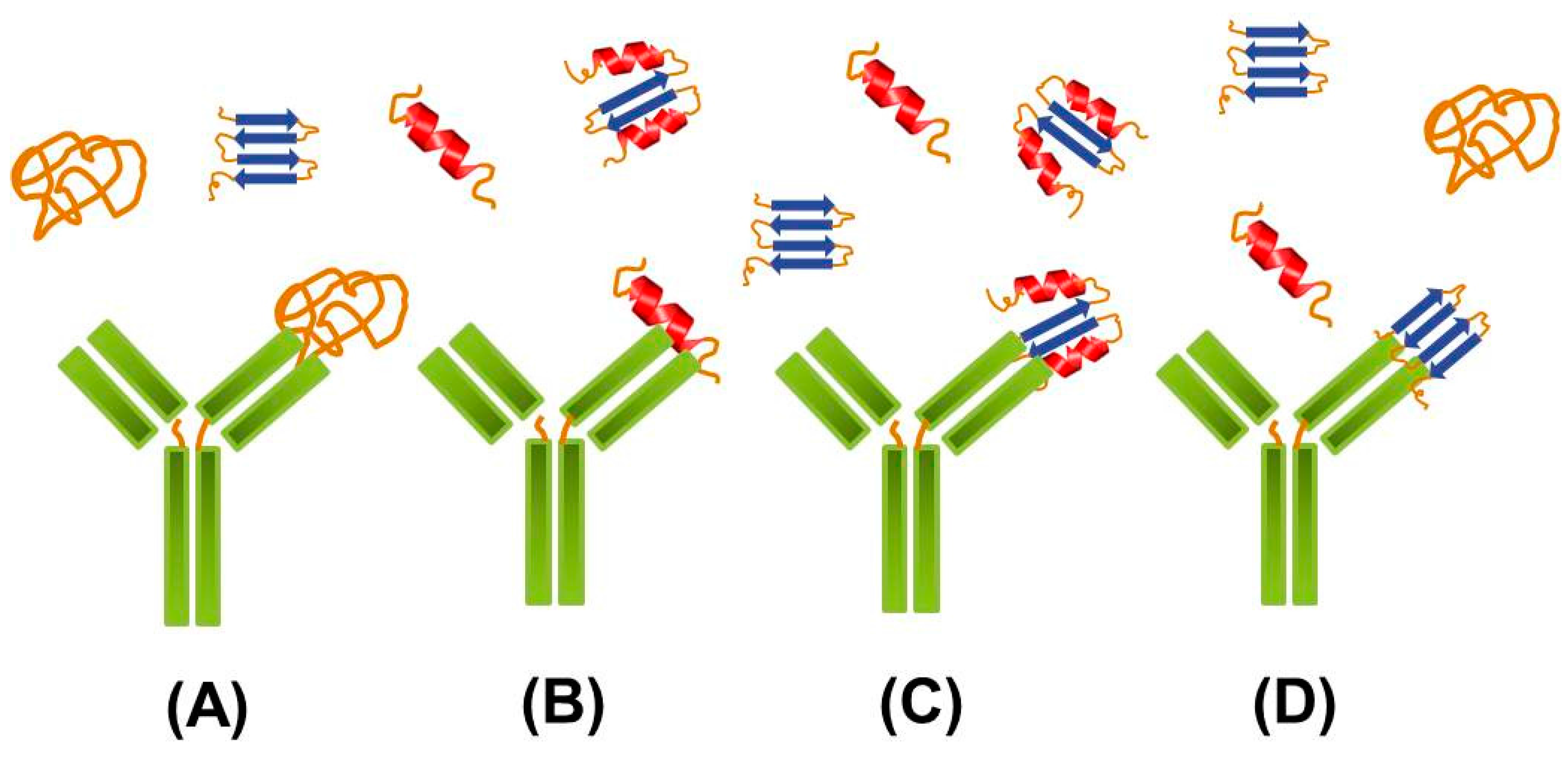{kind=link}
| Antibody | Selective for | Recognition mechanism and mode of action | Reference |
|---|---|---|---|
| Amy-33 | Fibrils | Aβ 1–28 aa residues, preventing self-association and disintegrating fibrils into a nonfibrilar conformation | [25] |
| 6F/3 | Fibrils | 8–17 aa residues, preventing self-association and disintegrating fibrils into a nonfibrilar conformation | [25] |
| Bapineuzumab | Fibrils | 1–5 aa of N-terminal end of Aβ in helical conformation | [34–36] |
| Gantenerumab | Fibrils | Targeting 1–10 aa and 19–26 aa | [38] |
| B10 | Fibrils, Protofibrils | Pattern-recognition mechanism, binding to an anionic surface moiety | [24] |
| KW1 | Oligomers | Hydrophobic and aromatic surface motif of Aβ residues 18–20 | [21] |
| A11 | Prefibrillar oligomers | A generic epitope common to prefibrillar oligomers | [51,53] |
| Crenezumab | Protofibrils, oligomers | Recognition of multiple conformational epitopes (aa13–14 relevant) and promoting disintegration of Aβ | [54] |
| SAR228810 | Protofibrils and LMW Aβ | Binds to congophilic amyloid plaques (present only in AD patients) | [55] |
| A-887755 | Aβ20-43 globulomers, condensed and hydrophobic oligomers | Recognition of the relevant structural motif located in the C-terminal part of the Aβ sequence between amino acids 20 and 42Recognises N-terminally truncated Aβ oligomers | [56] |
| Solanezumab | Intermediate conformations of soluble, monomeric Aβ | Targeting mid region of Aβ | [57] |
| BAN2401 | soluble Aβ protofibrils | It is thought to either enhance clearance of Aβ protofibrils and/or to neutralize toxic effects on neurons in the brain | [57] |
| Grafted AMyloid-Motif AntiBODIES, ‘Gammabodies’ | Aβ, α-synuclein and islet amyloid polypeptide | Prevents amyloid formation and further aggregation by stabilising benign intermediates | [60] |
| Aducanumab (BIIB037) | Soluble and insoluble Aβ | Preferentially binds parenchymal over vascular Aβ | [61] |
2.3. Natural Anti-Aβ Antibodies
2.4. Targeting Tau
3. Lewy Body Diseases: Targeting α-synuclein
4. Huntington’s Disease: Targeting Huntingtin
5. Transmissible Spongiform Encephalopathy: Targeting Prion
6. Concluding Remarks
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Dobson, C. Principles of protein folding, misfolding and aggregation. Semin. Cell Develop. Biol. 2004, 15, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, J.S.; Lindquist, S. Mechanisms of protein-folding diseases at a glance. Dis. Model. Mechan. 2014, 7, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Herczenik, E.; Gebbink, M.F.B.G. Molecular and cellular aspects of protein misfolding and disease. FASEB J. 2008, 22, 2115–2133. [Google Scholar] [CrossRef] [PubMed]
- Vanderhaegen, S.; Fislage, M.; Domanska, K.; Versees, W.; Pardon, E.; Bellotti, V.; Steyaert, J. Structure of an early native-like intermediate of b2-microglobulin amyloidogenesis. Protein Sci. 2013, 22, 1349–1357. [Google Scholar] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Calela, A.M. Prions: Protein aggregation and infectious diseases. Physiol. Rev. 2009, 89, 1105–1152. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; O’Connor, T. Protein aggregation diseases: Pathogenicity and therapeutic perspectives. Nat. Rev. Drug Discov. 2010, 9, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α‑synuclein: From structure and toxicity to therapeutic target. Nature 2013, 14, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Breydo, L.; Wu, J.W.; Uversky, V.N. α-Synuclein misfolding and Parkinson's disease. Biochim. Biophys. Acta 2012, 1822, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. Huntington’s disease: Underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci. 2013, 38, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s association. Alzheimer’s Disease and Dementia. Available online: http://www.alz.org (accessed on 28 December 2014).
- Small, D.H. Acetylcholinasterase inhibitors for the treatment of dementia in Alzheimer’s disease: Do we need ne inhibitors? Exp. Opin. Emerg. Drugs 2005, 10, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Yacoubian, T.A.; Standaert, D.G. Targets for neuroprotection in Parkinson’s disease. Biochim. Biophys. Acta 2009, 1792, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S.; Oakes, D.; Shoulson, I.; Kieburtz, K.; Rudolph, A.; Lang, A.; Olanow, C.W.; Tanner, C.; Marek, K. Levodopa and the progression of Parkinson’s disease. N. Eng. J. Med. 2004, 351, 2498–2508. [Google Scholar]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Debelouchina, G.T.; Bayro, M.J.; Clare, D.K.; Caporini, M.A.; Bajaj, V.S.; Jaroniec, C.P.; Wang, L.; Ladizhansky, V.; Müller, S.A.; et al. Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc. Natl. Acad. Sci. USA 2013, 110, 5468–5473. [Google Scholar] [CrossRef] [PubMed]
- Haas, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Moreth, J.; Mavoungou, C.; Schindowski, K. Possitive anti-amyloid immunotheraphy in Alzheimer’s disease: What are the most promising targets? Immun. Ageing 2013, 10, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Sarsoza, F.; Saing, T.; Cotman, C.W.; Necula, M.; Margol, L.; Wu, J.; Breydo, L.; Thompson, J.L.; et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegen. 2007, 2, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Morgado, I.; Wieligman, K.; Bereza, M.; Rönicke, R.; Meinhardt, K.; Annamalai, K.; Baumann, M.; Wacker, J.; Hortschansky, P.; Malešević, M.; et al. Molecular basis of β-amyloid oligomer recognition with a conformational antibody fragment. Proc. Natl. Acad. Sci. USA 2012, 109, 12503–12508. [Google Scholar] [CrossRef] [PubMed]
- Perchiacca, J.M.; Reza, A.; Ladiwala, A.; Bhattacharya, M.; Tessier, P.M. Structure-based design of conformation- and sequence-specific antibodies against amyloid β. Proc. Natl. Acad. Sci. USA 2012, 109, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, T.; El-Turk, F.; Buell, A.K.; O’Day, E.M.; Aprile, F.A.; Esbjörner, E.K.; Vendruscolo, M.; Cremades, N.; Pardon, E.; Wyns, L.; et al. Nanobodies raised against monomeric α-synuclein distinguish between fibrils at different maturation stages. J. Mol. Biol. 2013, 425, 2397–2411. [Google Scholar] [CrossRef] [PubMed]
- Habicht, G.; Haupt, C.; Friedrich, R.P.; Hortschansky, P.; Sachse, C.; Meinhardt, J.; Wieligmann, K.; Gellerman, G.P.; Brodhun, M.; Götz, J.; et al. Directed selection of a conformational antibody domain that prevents mature amyloid fibril formation by stabilizing Aβ profibrils. Proc. Natl. Acad. Sci. USA 2007, 104, 19232–19237. [Google Scholar] [CrossRef] [PubMed]
- Salomon, B.; Koppel, R.; Hanan, E.; Katzav, T. Monoclonal antibodies inhibit in vitro fibrillar aggregation of the Alzheimer 83-amyloid peptide. Proc. Natl. Acad. Sci. USA 1995, 93, 452–455. [Google Scholar] [CrossRef]
- Weggen, S.; Beher, D. Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer's disease. Alzheimers Res. Ther. 2012, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule associated protein (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 93, 4913–4917. [Google Scholar] [CrossRef]
- Goedert, M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Ann. NY Acad. Sci. 1996, 777, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Duering, M.; Grimm, M.O.; Grimm, H.S.; Schroder, J.; Hartmann, T. Mean age of onset in familial Alzheimer’s disease is determined by amyloid beta42. Neurobiol. Aging 2005, 26, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Pimplikar, S.W.; Nixon, R.A.; Robakis, N.K.; Shen, J.; Tsai, L. Amyloid-Independent Mechanisms in Alzheimer’s Disease Pathogenesis. J. Neurosci. 2010, 30, 14946–14954. [Google Scholar] [CrossRef] [PubMed]
- Gilman, S.; Koller, M.; Black, R.; Jenkins, L.; Griffith, S.; Fox, N.; Eisner, L.; Kirby, L.; Rovira, M.; Forette, F.; et al. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005, 64, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, J.A.; Wilkinson, D.; Holmes, C.; Steart, P.; Markham, H.; Weller, R.O. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: A case report. Nat. Med. 2003, 9, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Vellas, B.; Black, R.; Thal, L.J.; Fox, N.C.; Daniels, M.; McLennan, G.; Tompkins, C.; Leibman, C.; Pomfret, M.; Grundman, M. Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Curr. Alzheimer Res. 2009, 6, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.A.; Crespi, G.A.N.; Doughty, L.; Parker, M.W. Bapineuzumab captures the N-terminus of the Alzheimer’s disease amyloid-beta peptide in a helical conformation. Nat. Sci. Rep. 2013, 3, 1303. [Google Scholar] [CrossRef] [PubMed]
- Seubert, P.; Barbour, R.; Khan, K.; Motter, R.; Tang, P.; Kholodenko, D.; Kling, K.; Schenk, D.; Johnson-Wood, K.; Schroeter, S.; et al. Antibody capture of soluble Abeta does not reduce cortical Abeta amyloidosis in the PDPAPP mouse. Neurodegener. Dis. 2008, 5, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Colletier, J.P.; Laganowsky, A.; Landau, M.; Zhao, M.; Soriaga, A.B.; Goldschmidt, L.; Flot, D.; Cascio, D.; Sawaya, M.R.; Eisengerg, D. Molecular basis for amyloid-beta polymorphism. Proc. Natl. Acad. Sci. USA 2011, 108, 16938–16943. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.; Salloway, S.; Brooks, D.J.; Tampieri, D.; Barakos, J.; Fox, N.C.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; et al. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 2012, 11, 241–249. [Google Scholar] [CrossRef]
- Bohrmann, B.; Baumann, K.; Benz, J.; Gerber, F.; Huber, W.; Knoflach, F.; Messer, J.; Oroszlan, K.; Rauchenberger, R.; Richter, W.F.; et al. Gantenerumab: A novel human anti-Aβ antibody demonstrates sustained vertebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J. Alzheimer’s Dis. 2012, 28, 49–69. [Google Scholar]
- End of the RoAD for Gantenerumab? Roche Declares Prodromal Alzheimer's Trial Futile. Available online: http://www.alzforum.org/news/research-news/end-road-gantenerumab-roche-declares-prodromal-alzheimers-trial-futile (accessed on 27 June 2015).
- Lemere, C.A. Immunotherapy for Alzheimer’s disease: Hoops and hurdles. Mol. Neurodegener. 2013, 8, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Clinical Trials. Available online: www.clinicalcrials.gov (accessed on 27 June 2015).
- Dickson, D.W.; Chrystal, H.A.; Bevona, C.; Honer, W.; Vincent, I.; Davis, P. Correlations of synaptic and pathological markers with cognition of the eldery. Neurobiol. Aging 1995, 16, 285–298. [Google Scholar] [CrossRef]
- Naslund, J.; Haroutunian, V.; Mohs, R.; Davis, K.L.; Davis, P.; Greengard, P.; Buxbaum, J. Correlation between elevated levels if amyloid β-peptide in the brain and cognitive decline. JAMA 2000, 283, 1571–1577. [Google Scholar] [CrossRef] [PubMed]
- Busciglio, J.; Lorenzo, A.; Yankner, B. Methological variables in the assessment of β-amyloid neurotoxicity. Neurobiol. Aging 1992, 13, 609–612. [Google Scholar] [CrossRef]
- Pryor, N.E.; Moss, M.A.; Hestekin, C.N. Unravelling the early events of amyloid-β Protein (Aβ) aggregation: Techniques for the determination of Aβ aggregate size. Int. J. Mol. Sci. 2012, 13, 3038–3072. [Google Scholar] [CrossRef] [PubMed]
- Broersen, K.; Rousseau, F.; Schymkowitz, J. The culprit behind amyloid beta peptide related neurotoxicity in Alzheimer’s disease: oligomer size or conformation? Alzheimer’s Res. Ther. 2010, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Glabe, C.G. Structural classification of toxic amyloid oligomers. J. Biol. Chem. 2008, 283, 29639–29643. [Google Scholar] [CrossRef] [PubMed]
- Streltsov, V.A.; Varghese, J.N.; Masters, C.L.; Nuttall, S.D. Crystal structure of the amyloid-βp3 fragment provides a model for oligomer formation in Alzheimer’s disease. J. Neurosci. 2011, 31, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Zameer, A.; Kasturirangan, S.; Emadi, S.; Nimmagadda, S.V.; Sierks, M.R. Anti-oligomeric Aβ single chain variable domain antibody blocks Aβ-induced toxicity against human neuroblastoma cells. J. Mol. Biol. 2008, 384, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Lafaye, P.; Achour, I.; England, P.; Duyckaerts, C.; Rougeon, F. Single-domain antibodies recognize selectively small oligomeric forms of amyloid β, prevent Aβ-induced neurotoxicity and inhibit fibril formation. Mol. Immunol. 2009, 46, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Kasturirangan, S.; Li, L.; Emadi, S.; Boddapati, S.; Schulz, P.; Sierks, M.R. Nanobody specific for oligomeric beta-amyloid stabilizes nontoxic form. Neurobiol. Aging 2012, 33, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Adolfsson, O.; Pihlgren, M.; Toni, N.; Varisco, Y.; Buccarello, A.L.; Antoniello, K.; Lohmann, S.; Piorkowska, K.; Gafner, V.; Arwal, J.K.; et al. An effector-reduced anti- β-amyloid (Aβ) antibody with unique Aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J. Neurosci. 2012, 32, 9677–9689. [Google Scholar]
- Dunstan, R.; Bussiere, T.; Rhodes, K.; Engber, T.; Maier, M.; Weinreb, P.; Grimm, J.; Nitsch, R.; Arustu, M.; Qian, F.; et al. Molecular characterisation and preclinical efficacy. Alzheimer’s Dement. 2011, 7, S457. [Google Scholar] [CrossRef]
- Relo, A.; Barghorn, S.; Ebert, U.; Hillen, H.; Gross, G.; Schoemaker, H.; Bespalov, A. Restoration of home cage activity in Tg2576 mice by immunotherapy with the Ab-oligomer selective antibody A-887755. Alzheimer’s Dement. 2011, 7, S772. [Google Scholar] [CrossRef]
- Lannfelt, L.; Moller, C.; Basun, H.; Osswald, G.; Sehlin, D.; Satlin, A.; Logovinsky, V.; Gellerfors, P. Perspectives on future Alzheimer’s therapies: amyloid-β protofibrils-a new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Meli, G.; Visintin, M.; Cannistraci, I.; Cattaneo, A. Directin vivo intracellular selection of conformation-sensitive antibody domains targeting Alzheimer’s amyloid-beta oligomers. J. Mol. Biol. 2009, 387, 584–606. [Google Scholar] [CrossRef] [PubMed]
- Meli, G.; Lecci, A.; Manca, A.; Krako, N.; Albertini, V.; Benussi, L.; Ghidoni, R.; Cattaneo, A. Conformational targeting of intracellular Aβ oligomers demonstrates their pathological oligomerization inside the endoplasmic reticulum. Nat. Commun. 2014, 3, 3867–3878. [Google Scholar] [CrossRef] [PubMed]
- Ladiwala, A.R.A.; Bhattacharya, M.; Perchiacca, J.M.; Cao, P.; Raleigh, D.P.; Abedini, A.; Schmidt, A.M.; Varkey, J.; Langen, R.; Tessier, P.M. Rational design of potent domain antibody inhibitors of amyloid fibril assembly. Proc. Natl. Acad. Sci. USA 2012, 109, 19965–19970. [Google Scholar] [CrossRef] [PubMed]
- Biogen Idec Presents Positive Interim Results from Phase 1B Study of Investigational Alzheimer’s Disease Treatment Aducanumab (BIIB037) at 2015 AD/PD™ Conference | Biogen Media. Available online: http://media.biogen.com/press-release/corporate/biogen-idec-presents-positive-interim-results-phase-1b-study-investigational (accessed on 20 April 2015).
- Panza, F.; Solfrizzi, V.; Imbimbo, B.P.; Tortelli, R.; Santamato, A.; Logroscino, G. Amyloid- based immunotherapy for Alzheimer’s disease in the time of prevention trials: the way forward. Expert Rev. Clin. Immunol. 2014, 10, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Lopera, F.; Ardilla, A.; Martínez, A.; Madrigal, L.; Arango-Viana, J.; Lemere, C.A.; Arango-Lasprilla, J.; Hincapié, L.; Arcos-Burgos, M.; Ossa, J.E.; et al. Clinical Features of Early-Onset Alzheimer Disease in a Large Kindred With an E280A Presenilin-1 Mutation. JAMA 1997, 277, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, Y.; Wang, Z.; Jiang, Y. Clinical trials of amyloid-based immunotherapy for Alzheimer’s disease: end of beginning or beginning of end? Expert Rev. Clin. Immunol. 2013, 13, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Weksler, M.E.; Relkin, N.; Turkenich, R.; LaRusse, S.; Zhou, L.; Szabo, P. Patients with Alzheimer disease have lower levels of serum anti-amyloid peptide antibodies than healthy elderly individuals. Exp. Gerontol. 2002, 37, 943–948. [Google Scholar] [CrossRef]
- Fillit, H.; Hess, G.; Hill, J.; Bonnet, P.; Toso, C. IV immunoglobulin is associated with a reduced risk of Alzheimer disease and related disorders. Neurology 2009, 73, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Britschgi, M.; Olin, C.E.; Johns, H.T.; Takeda-Uchimura, Y.; LeMieux, M.C.; Rufibach, K.; Rajadas, J.; Zhang, H.; Tomooka, B.; Robinson, W.H.; et al. Neuroprotective natural antibodies to assemblies of amyloidogenic peptides decrease with normal aging and advancing Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 12145–12150. [Google Scholar] [CrossRef] [PubMed]
- Mengel, D.; Roskam, S.; Neff, F.; Balakrishnan, K.; Deuster, O.; Gold, M.; Oertel, W.H.; Bacher, M.; Bach, J.-P.; Dodel, R. Naturally occurring autoantibodies interfere with [beta]-amyloid metabolism and improve cognition in a transgenic mouse model of Alzheimer/’s disease 24h after single treatment. Transl. Psychiat. 2013, 3, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Dodel, R.; Balakrishnan, K.; Keyvani, K.; Deuster, O.; Neff, F.; Andrei-Selmer, L.C.; Roskam, S.; Stuer, C.; Al-Abed, Y.; Noelker, C.; et al. Naturally occurring autoantibodies against beta-amyloid: investigating their role in transgenic animal and in vitro models of Alzheimer’s disease. J. Neurosci. 2011, 31, 5847–5854. [Google Scholar] [CrossRef] [PubMed]
- Kfpke, E.; Tung, Y.C.; Shaikh, S.; Alonso, A.C.; Iqbal, K.; Grundke-Iqbal, I. Microtubule associated protein tau: abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar]
- Augustinack, J.C.; Schneider, A.; Mandelkow, E.M.; Hyman, B.T. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. 2002, 103, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Sarmiento, J.; Troncoso, J.; Jackson, G.R.; Kayed, R. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. 2012, 26, 1946–1959. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, E.M. Immunotherapy Targeting Pathological Tau Protein in Alzheimer's Disease and Related Tauopathies. J. Alzheimers Dis. 2008, 15, 157–168. [Google Scholar] [PubMed]
- Krishnamurthy, P.K.; Deng, Y.; Sigurdsson, E.M. Mechanistic studies of antibody-mediated clearance of Tau aggregates using an ex vivo brain slice model. Front. Psychiat. 2011, 2, 59. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Congdon, E.E.; Sigurdsson, E.M. Two Novel Tau Antibodies Targeting the 396/404 Region Are Primarily Taken Up by Neurons and Reduce Tau Protein Pathology. J. Biol. Chem. 2013, 288, 33081–33091. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Gu, J.; Sait, H.B.R.; Sigurdsson, E.M. Antibody Uptake into Neurons Occurs Primarily via Clathrin-dependent Fc_ Receptor Endocytosis and Is a Prerequisite for Acute Tau Protein Clearance. J. Biol. Chem. 2013, 288, 35452–35465. [Google Scholar] [CrossRef] [PubMed]
- Boutajangout, A.; Wisniewski, T. Tau-based therapeutic approaches for Alzheimer’s disease—A mini-review. Gerontology 2014, 60, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.; Lee, D.; Brownlow, M.; Selenica, M.-L.; Reid, P.; Alvarez, J.; Gordon, M.N. Opposing roles of microglial activation in amyloid depositing and tau depositing transgenic mice. Neurodegen. Dis. 2011, 8 (Suppl. 1), 1. [Google Scholar]
- d’Abramo, C.; Acker, C.M.; Jimenez, H.T.; Davies, P. Tau Passive Immunotherapy in Mutant P301L Mice: Antibody Affinity versus Specificity. PLoS One 2013, 8, e62402. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of Tau Misfolding from the Outside to the Inside of a Cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Lee, V.M. Seeding of Normal Tau by Pathological Tau Conformers Drives Pathogenesis of Alzheimer-like Tangles. J. Biol. Chem. 2011, 286, 15317–15331. [Google Scholar] [CrossRef] [PubMed]
- de Calignon, A.; Polydoro, M.; Suarez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of Tau Pathology in a Model of Early Alzheimer’s Disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Yanamandra, K.; Kfoury, N.; Jiang, H.; Mahan, T.E.; Ma, S.; Maloney, S.E.; Wozniak, D.F.; Diamond, M.I.; Holtzman, D.M. Anti-Tau Antibodies that Block Tau Aggregate Seeding In Vitro Markedly Decrease Pathology and Improve Cognition In Vivo. Neuron 2013, 80, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Kfoury, N.; Holmes, B.B.; Jiang, H.; Holtzman, D.M.; Diamond, M.I. Trans-cellular propagation of Tau aggregation by fibrillar species. J. Biol. Chem. 2012, 287, 19440–19451. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Carranza, D.L.; Sengupta, U.; Guerrero-Muñoz, M.J.; Lasagna-Reeves, C.A.; Gerso, J.E.; Singh, G.; Estes, D.M.; Barrett, A.D.; Dineley, K.T.; Jackson, G.R.; et al. Passive immunization with Tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J. Neurosci. 2014, 34, 4260–4272. [Google Scholar] [CrossRef] [PubMed]
- Valera, E.; Masliah, E. Immunotherapy for neurodegenerative diseases: Focus on α-synucleinopathies. Pharmacol. Therap. 2013, 138, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Visanji, N.P.; Brooks, P.L.; Hazrati, L.; Lang, A.E. The prion hypothesis in Parkinson's disease: Braak to the future. Acta Neuropathol. Commun. 2013, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.; Angot, E.; Bergström, A.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Invest. 2011, 121, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Dodiya, H.B.; Kordower, A.M.; Terpstra, B.; Paumier, K.; Madhavan, L.; Sortwell, C.; Steece-Collier, K.; Collier, T.J. Transfer of host-derived α synuclein to grafted dopaminergic neurons in rat. Neurobiol. Dis. 2011, 43, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Luk, K.L.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meany, D.F.; Trojanowski, J.Q.; Lee, V.M.-Y. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [PubMed]
- Mougenot, A.L.; Nicot, S.; Bencsik, A.; Morignat, E.; Verchere, J.; Lakhdar, L.; Legastelios, S.; Baron, T. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol. Aging 2012, 33, 2225–2228. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Hu, D.; Han, S.; Hong, D.P.; Fink, A.L. Role of different regions of α-synuclein in the assembly of fibrils. Biochemistry 2007, 46, 13322–13330. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.T.; Chung, C.H.; Iba, M.; Zhang, B.; Trojanowski, J.Q.; Luk, K.C.; Lee, V.M.Y. α-synuclein immunotherapy blocks uptake and template propagation of misfolded α-synuclein and neurodegeneration. Cell Rep. 2014, 7, 2054–2065. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Rockenstein, E.; Mante, M.; Crews, L.; Spencer, B.; Adame, A.; Patrick, C.; Trejo, M.; Ubhi, K.; Rohn, T.T.; et al. Passive immunisation reduces behavioural and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One 2011, 6, e19338. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J. Biol. Chem. 2001, 276, 10737–10744. [Google Scholar] [CrossRef] [PubMed]
- Fagerqvist, T.; Lindstrom, V.; Nordstrom, E.; Lord, A.; Tucker, S.M.E.; Su, X.; Sahlin, C.; Kasrayan, A.; Andersson, J.; Welander, H.; et al. Monoclonal antibodies selective for α-synuclein oligomers/protofibrils recognise brain pathology in Lewy body disorders and α-synuclein transgenic mice with the disease-causing A30P mutation. J. Neurochem. 2013, 126, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Emadi, S.; Kasturirangan, S.; Wang, M.S.; Schulz, P.; Sierks, M. Detecting morphologically distinct oligomeric forms of α-synuclein. J. Biol. Chem. 2009, 284, 11048–11058. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, V.; Fagerqvist, T.; Nordstrom, E.; Eriksson, F.; Lord, A.; Tucker, S.M.E.; Andersson, J.; Johannesson, M.; Schell, P.J.; Moller, C.; et al. Immunotherapy targeting α-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] α-synuclein mice. Neurobiol. Dis. 2014, 69, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Breydo, L.; Green, R.; Kis, V.; Puska, G.; Lorincz, P.; Perju-Dumbrava, L.; Giera, R.; Pirker, W.; Lutz, M.; et al. Intracellular processing of disease-associated α-synuclein in the human brain suggest prion-like cell-to-cell spread. Neurobiol. Dis. 2014, 69, 76–92. [Google Scholar] [CrossRef] [PubMed]
- De Genst, E.J.; Guilliams, T.; Wellens, J.; O’Day, E.M.; Waudby, C.A.; Meehan, S.; Dumoulin, M.; Hsu, A.D.; Cremades, N.; Verschueren, K.H.G.; et al. Structure and properties of a complex of α-synuclein and single-domain camelid antibody. J. Biol. Chem. 2010, 402, 326–343. [Google Scholar] [CrossRef] [PubMed]
- Volles, M.J.; Lansbury, P.T., Jr. Relationships between the sequence of alpha-synuclein and its membrane affinity, fibrillization propensity, and yeast toxicity. J. Mol. Biol. 2007, 366, 1510–1522. [Google Scholar] [CrossRef] [PubMed]
- Auluck, P.K.; Caraveo, G.; Lindquist, S. α-Synuclein: membrane interactions and toxicity in Parkinson’s disease. Ann. Rev. Cell Develop. Biol. 2010, 26, 211–233. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wmadi, S.; Sierks, M.R.; Messer, A. A human single-chain Fv intrabody blocks aberrant cellular effects of overexpressed alpha-synuclein. Mol. Ther. 2004, 10, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.M.; Zhou, C.; Messer, A. An scFv intrabody against the nonamyloid component of alpha-synuclein reduces intracellular aggregation and toxicity. J. Mol. Biol. 2008, 377, 136–147. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Butler, D.C.; McLear, J.A.; Messer, A. Engineered antibody therapies to counteract mutant huntingtin and related toxic intracellular proteins. Prog. Neurobiol. 2012, 97, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Schiefner, A.; Chatwell, L.; Korner, J.; Neumaier, I.; Colby, D.W.; Volkmer, R.; Wittrup, K.D.; Skerra, A. A disulfide-free single-domain V(L) intrabody with blocking activity towards huntingtin reveals a novel mode of epitope recognition. J. Mol. Biol. 2011, 414, 337–355. [Google Scholar] [CrossRef] [PubMed]
- Southwell, A.L.; Khoshnan, A.; Dunn, D.E.; Bugg, C.W.; Lo, D.C.; Patterson, P.H. Intrabodies binding the proline-rich domains of mutant huntingtin increase its turnover and reduce neurotoxicity. J. Neurosc. 2008, 28, 9013–9020. [Google Scholar] [CrossRef] [PubMed]
- Lecerf, J.M.; Shirley, T.L.; Zhu, Q.; Kazantsev, A.; Amersdorfer, P.; Housman, D.E.; Messer, A.; Huston, J.S. Human single-chain Fv intrabodies counteract in situ huntingtin aggregation in cellular models of Huntington's disease. Proc. Natl. Acad. Sci. USA 2001, 98, 4764–4769. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Zhou, C.; Gines, S.; MacDonald, M.E.; Mazarakis, N.D.; Bates, G.P.; Huston, J.S.; Messer, A. A human single-chain Fv intrabody preferentially targets amino-terminal Huntingtin's fragments in striatal models of Huntington's disease. Neurobiol. Dis. 2005, 19, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Butler, D.C.; Messer, A. Bifunctional anti-huntingtin proteasome-directed intrabodies mediate efficient degradation of mutant huntingtin exon 1 protein fragments. PLoS One 2011, 6, e29199. [Google Scholar] [CrossRef] [PubMed]
- Khoshnan, A.; Ko, J.; Patterson, P.H. Effects of intracellular expression of antihuntingtin antibodies of various specificities on mutant huntingtin aggregation and toxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.; Saftig, P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 2009, 1793, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Southwell, A.L.; Bugg, C.W.; Kaltenbach, L.S.; Dunn, D.E.; Butland, S.; Weiss, A.; Paganetti, P.; Lo, D.C.; Patterson, P.H. Perturbation with intrabodies reveals that calpain cleavage is required for degradation of huntingtin exon 1. PLoS One 2011, 6, e16676. [Google Scholar] [CrossRef] [PubMed]
- Polymenidou, M.; Moos, R.; Scott, M.; Sigurdson, C.; Shi, Y.; Yajima, B.; Hafner-Bratkovic, I.; Jerala, R.; Hornemann, S.; Wuthrich, K.; et al. The POM Monoclonals: A Comprehensive Set of Antibodies to Non-Overlapping Prion Protein Epitopes. PLoS One 2008, 3, e3872. [Google Scholar] [CrossRef] [PubMed]
- Sonati, T.; Reimann, R.R.; Falsig, J.; Baral, P.K.; O’Connor, T.; Hornemann, S.; Yaganoglu, S.; Li, B.; Herrmann, U.S.; Wieland, B.; et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 2013, 501, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Abskharon, R.N.N.; Giachin, G.; Wohlkonig, A.; Soror, S.H.; Pardon, E.; Legname, G.; Steyaert, J. Probing the N-terminal β-sheet conversion in the crystal structure of the human prion protein bound to a nanobody. J. Am. Chem. Soc. 2014, 136, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Roettger, Y.; Tan, B.; He, Y.; Dodel, R.; Hampel, H.; Wei, G.; Haney, J.; Gu, H.; Johnstone, B.H.; et al. Human anti-prion antibodies block prion peptide fibril formation and neurotoxicity. J. Biol. Chem. 2012, 287, 12858–12866. [Google Scholar] [CrossRef] [PubMed]
- Rovis, T.L.; Legname, G. Prion protein-specific antibodies-development, modes of action and therapeutics applications. Viruses 2014, 6, 3719–3737. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Musahl, C.; Arrighi, I.; Klein, M.A.; Rulicke, T.; Oesch, B.; Zinkernagel, R.M.; Kalinke, U.; Aguzzi, A. Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science 2001, 294, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Moda, F.; Vimercati, C.; Campagnani, I.; Ruggerone, M.; Giaccone, G.; Morbin, M.; Zentilin, L.; Giacca, M.; Zucca, I.; Legname, G.; et al. Brain delivery of aav9 expressing an anti-prp monovalent antibody delays prion disease in mice. Prion 2012, 6, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Wuertzer, C.A.; Sullivan, M.A.; Qiu, X.; Federoff, H.J. CNS delivery of vectored prion-specific single-chain antibodies delays disease onset. Mol. Ther. 2008, 16, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, A.; Biocca, S. Gene-based antibody strategies for prion diseases. Int. J. Cell Biol. 2013, 2013, 710406. [Google Scholar] [CrossRef] [PubMed]
- Skrlj, N.; Drevensek, G.; Hudoklin, S.; Romih, R.; Curin Serbec, V.; Dolinar, M. Recombinant single-chain antibody with the trojan peptide penetratin positioned in the linker region enables cargo transfer across the blood-brain barrier. Appl. Biochem. Biotechnol. 2013, 169, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Joy, Y.Y.; Watts, R.J. Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics 2013, 10, 459–472. [Google Scholar]
- Lawson, A.D.G. Antibody-enabled small molecule drug discovery. Nat. Rev. Drug Discov. 2012, 11, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Kokjohn, T.A.; Roher, E.A. Amyloid precursor protein transgenic mouse models and Alzheimer’s disease: Understanding the paradigms, limitations, and contributions. Alzheimer’s Dement. 2009, 5, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Rockenstein, E.; Crews, L.; Masliah, E. Transgenic animal models of neurodegenerative diseases and their application to treatment development. Adv. Drug Deliv. Rev. 2007, 59, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Elder, G.A.; Gama Sosa, M.A.; De Gaspert, R. Transgenic mouse models of Alzheimer’s disease. Mt. Sinai J. Med. 2010, 77, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Götz, J.; Gladbach, A.; Pennanen, L.; van Eersel, J.; Schild, A.; David, D.; Ittner, L.M. Animal models reveal role for tau phosphorylation in human disease. Biochim. Biophys. Acta 2010, 1802, 860–871. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Westwood, M.; Lawson, A.D.G. Opportunities for Conformation-Selective Antibodies in Amyloid-Related Diseases. Antibodies 2015, 4, 170-196. https://doi.org/10.3390/antib4030170
Westwood M, Lawson ADG. Opportunities for Conformation-Selective Antibodies in Amyloid-Related Diseases. Antibodies. 2015; 4(3):170-196. https://doi.org/10.3390/antib4030170
Chicago/Turabian StyleWestwood, Marta, and Alastair D. G. Lawson. 2015. "Opportunities for Conformation-Selective Antibodies in Amyloid-Related Diseases" Antibodies 4, no. 3: 170-196. https://doi.org/10.3390/antib4030170
APA StyleWestwood, M., & Lawson, A. D. G. (2015). Opportunities for Conformation-Selective Antibodies in Amyloid-Related Diseases. Antibodies, 4(3), 170-196. https://doi.org/10.3390/antib4030170