
FcRn Blockade as a Targeted Therapeutic Strategy in Antibody-Mediated Autoimmune Diseases: A Focus on Warm Autoimmune Hemolytic Anemia

Abstract
1. Introduction
2. Warm Autoimmune Hemolytic Anemia
3. Current Warm Autoimmune Hemolytic Anemia Treatment
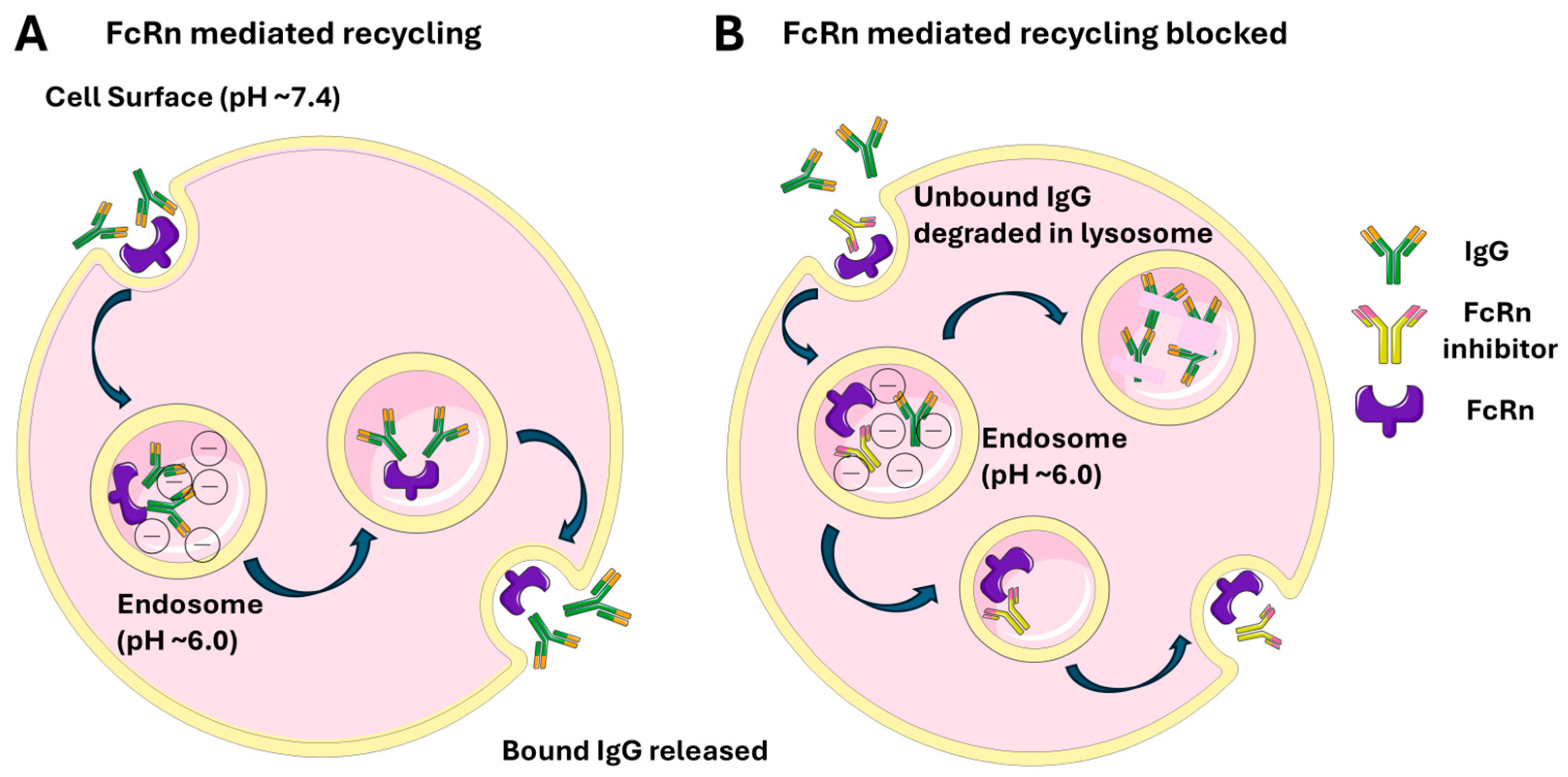
4. Neonatal Fragment Crystallizable Receptor (FcRn)
5. Nipocalimab
5.1. Nipocalimab in Other Indications
5.1.1. Myasthenia Gravis
5.1.2. Sjögren’s Disease
5.1.3. Hemolytic Disease of the Fetus and Newborn
6. Other FcRn Targeting Agents
6.1. Efgartigimod
6.2. Batoclimab
6.3. IMVT-1402
6.4. Rozanolixizumab
6.5. STSA-1301
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Conrad, N.; Misra, S.; Verbakel, J.Y.; Verbeke, G.; Molenberghs, G.; Taylor, P.N.; Mason, J.; Sattar, N.; McMurray, J.J.V.; McInnes, I.B.; et al. Incidence, prevalence, and co-occurrence of autoimmune disorders over time and by age, sex, and socioeconomic status: A population-based cohort study of 22 million individuals in the UK. Lancet Lond. Engl. 2023, 401, 1878–1890. [Google Scholar] [CrossRef]
- Ali, F.H.M.; Smatti, M.K.; Elrayess, M.A.; Al Thani, A.A.; Yassine, H.M. Role of genetics in eleven of the most common autoimmune diseases in the post genome-wide association studies era. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 8463–8485. [Google Scholar] [CrossRef]
- Shapira, Y.; Agmon-Levin, N.; Shoenfeld, Y. Defining and analyzing geoepidemiology and human autoimmunity. J. Autoimmun. 2010, 34, J168–J177. [Google Scholar] [CrossRef]
- Bieber, K.; Hundt, J.E.; Yu, X.; Ehlers, M.; Petersen, F.; Karsten, C.M.; Köhl, J.; Kridin, K.; Kalies, K.; Kasprick, A.; et al. Autoimmune pre-disease. Autoimmun. Rev. 2023, 22, 103236. [Google Scholar] [CrossRef] [PubMed]
- Eaton, W.W.; Rose, N.R.; Kalaydjian, A.; Pedersen, M.G.; Mortensen, P.B. Epidemiology of Autoimmune Diseases in Denmark. J. Autoimmun. 2007, 29, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; He, Y.-S.; Wang, Y.; Zha, C.-K.; Lu, J.-M.; Tao, L.-M.; Jiang, Z.-X.; Pan, H.-F. Global burden and cross-country inequalities in autoimmune diseases from 1990 to 2019. Autoimmun. Rev. 2023, 22, 103326. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.S.; Bynum, M.L.K.; Somers, E.C. Recent insights in the epidemiology of autoimmune diseases: Improved prevalence estimates and understanding of clustering of diseases. J. Autoimmun. 2009, 33, 197–207. [Google Scholar] [CrossRef]
- Bussel, J.B.; Cines, D.B.; Blumberg, R.S. Neonatal Fc Receptor—Biology and Therapeutics. N. Engl. J. Med. 2025, 392, 1621–1635. [Google Scholar] [CrossRef]
- Volkov, M.; Coppola, M.; Huizinga, R.; Eftimov, F.; Huizinga, T.W.J.; van der Kooi, A.J.; Oosten, L.E.M.; Raaphorst, J.; Rispens, T.; Sciarrillo, R.; et al. Comprehensive overview of autoantibody isotype and subclass distribution. J. Allergy Clin. Immunol. 2022, 150, 999–1010. [Google Scholar] [CrossRef]
- Karsten, C.M.; Köhl, J. The immunoglobulin, IgG Fc receptor and complement triangle in autoimmune diseases. Immunobiology 2012, 217, 1067–1079. [Google Scholar] [CrossRef]
- Sesarman, A.; Vidarsson, G.; Sitaru, C. The neonatal Fc receptor as therapeutic target in IgG-mediated autoimmune diseases. Cell. Mol. Life Sci. CMLS 2010, 67, 2533–2550. [Google Scholar] [CrossRef]
- Oganesyan, V.; Damschroder, M.M.; Cook, K.E.; Li, Q.; Gao, C.; Wu, H.; Dall’Acqua, W.F. Structural insights into neonatal Fc receptor-based recycling mechanisms. J. Biol. Chem. 2014, 289, 7812–7824. [Google Scholar] [CrossRef]
- Pyzik, M.; Rath, T.; Lencer, W.I.; Baker, K.; Blumberg, R.S. FcRn: The architect behind the immune and non-immune functions of IgG and albumin. J. Immunol. 2015, 194, 4595–4603. [Google Scholar] [CrossRef] [PubMed]
- Peter, H.-H.; Ochs, H.D.; Cunningham-Rundles, C.; Vinh, D.C.; Kiessling, P.; Greve, B.; Jolles, S. Targeting FcRn for immunomodulation: Benefits, risks, and practical considerations. J. Allergy Clin. Immunol. 2020, 146, 479–491.e5. [Google Scholar] [CrossRef]
- Ling, L.E.; Hillson, J.L.; Tiessen, R.G.; Bosje, T.; van Iersel, M.P.; Nix, D.J.; Markowitz, L.; Cilfone, N.A.; Duffner, J.; Streisand, J.B.; et al. M281, an Anti-FcRn Antibody: Pharmacodynamics, Pharmacokinetics, and Safety Across the Full Range of IgG Reduction in a First-in-Human Study. Clin. Pharmacol. Ther. 2019, 105, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Berentsen, S.; Barcellini, W. Autoimmune Hemolytic Anemias. N. Engl. J. Med. 2021, 385, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Immune Hemolytic Anemias. 2003. Available online: https://medlineplus.gov/ency/article/000576.htm (accessed on 25 May 2025).
- Loriamini, M.; Cserti-Gazdewich, C.; Branch, D.R. Autoimmune Hemolytic Anemias: Classifications, Pathophysiology, Diagnoses and Management. Int. J. Mol. Sci. 2024, 25, 4296. [Google Scholar] [CrossRef]
- Kalfa, T.A. Warm antibody autoimmune hemolytic anemia. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 690–697. [Google Scholar] [CrossRef]
- Barcellini, W.; Giannotta, J.; Fattizzo, B. Autoimmune hemolytic anemia in adults: Primary risk factors and diagnostic procedures. Expert. Rev. Hematol. 2020, 13, 585–597. [Google Scholar] [CrossRef]
- Barcellini, W.; Fattizzo, B. The Changing Landscape of Autoimmune Hemolytic Anemia. Front. Immunol. 2020, 11, 946. [Google Scholar] [CrossRef]
- Barcellini, W.; Fattizzo, B.; Zaninoni, A.; Radice, T.; Nichele, I.; Di Bona, E.; Lunghi, M.; Tassinari, C.; Alfinito, F.; Ferrari, A.; et al. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: A GIMEMA study of 308 patients. Blood 2014, 124, 2930–2936. [Google Scholar] [CrossRef]
- Garratty, G. The James Blundell Award Lecture 2007: Do we really understand immune red cell destruction? Transfus. Med. Oxf. Engl. 2008, 18, 321–334. [Google Scholar] [CrossRef]
- Jäger, U.; Barcellini, W.; Broome, C.M.; Gertz, M.A.; Hill, A.; Hill, Q.A.; Jilma, B.; Kuter, D.J.; Michel, M.; Montillo, M.; et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: Recommendations from the First International Consensus Meeting. Blood Rev. 2020, 41, 100648. [Google Scholar] [CrossRef] [PubMed]
- Barcellini, W.; Fattizzo, B. How I treat warm autoimmune hemolytic anemia. Blood 2021, 137, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Kuter, D.J. Warm autoimmune hemolytic anemia and the best treatment strategies. Hematol. Am. Soc. Hematol. Educ. Program 2022, 2022, 105–113. [Google Scholar] [CrossRef]
- Brodsky, R.A. Warm Autoimmune Hemolytic Anemia. N. Engl. J. Med. 2019, 381, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Go, R.S.; Winters, J.L.; Kay, N.E. How I treat autoimmune hemolytic anemia. Blood 2017, 129, 2971–2979. [Google Scholar] [CrossRef]
- Hill, Q.A.; Stamps, R.; Massey, E.; Grainger, J.D.; Provan, D.; Hill, A.; British Society for Haematology. The diagnosis and management of primary autoimmune haemolytic anaemia. Br. J. Haematol. 2017, 176, 395–411. [Google Scholar] [CrossRef]
- Meyer, O.; Stahl, D.; Beckhove, P.; Huhn, D.; Salama, A. Pulsed high-dose dexamethasone in chronic autoimmune haemolytic anaemia of warm type. Br. J. Haematol. 1997, 98, 860–862. [Google Scholar] [CrossRef]
- Mulder, F.V.M.; Evers, D.; de Haas, M.; Cruijsen, M.J.; Bernelot Moens, S.J.; Barcellini, W.; Fattizzo, B.; Vos, J.M.I. Severe autoimmune hemolytic anemia; epidemiology, clinical management, outcomes and knowledge gaps. Front. Immunol. 2023, 14, 1228142. [Google Scholar] [CrossRef]
- Murakhovskaya, I. Rituximab Use in Warm and Cold Autoimmune Hemolytic Anemia. J. Clin. Med. 2020, 9, 4034. [Google Scholar] [CrossRef]
- Birgens, H.; Frederiksen, H.; Hasselbalch, H.C.; Rasmussen, I.H.; Nielsen, O.J.; Kjeldsen, L.; Larsen, H.; Mourits-Andersen, T.; Plesner, T.; Rønnov-Jessen, D.; et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br. J. Haematol. 2013, 163, 393–399. [Google Scholar] [CrossRef]
- Maung, S.W.; Leahy, M.; O’Leary, H.M.; Khan, I.; Cahill, M.R.; Gilligan, O.; Murphy, P.; McPherson, S.; Jackson, F.; Ryan, M.; et al. A multi-centre retrospective study of rituximab use in the treatment of relapsed or resistant warm autoimmune haemolytic anaemia. Br. J. Haematol. 2013, 163, 118–122. [Google Scholar] [CrossRef]
- Michel, M.; Terriou, L.; Roudot-Thoraval, F.; Hamidou, M.; Ebbo, M.; Le Guenno, G.; Galicier, L.; Audia, S.; Royer, B.; Morin, A.-S.; et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the RAIHA study). Am. J. Hematol. 2017, 92, 23–27. [Google Scholar] [CrossRef]
- Peñalver, F.J.; Alvarez-Larrán, A.; Díez-Martin, J.L.; Gallur, L.; Jarque, I.; Caballero, D.; Díaz-Mediavilla, J.; Bustelos, R.; Fernández-Aceñero, M.J.; Cabrera, J.R.; et al. Rituximab is an effective and safe therapeutic alternative in adults with refractory and severe autoimmune hemolytic anemia. Ann. Hematol. 2010, 89, 1073–1080. [Google Scholar] [CrossRef]
- Bride, K.L.; Vincent, T.; Smith-Whitley, K.; Lambert, M.P.; Bleesing, J.J.; Seif, A.E.; Manno, C.S.; Casper, J.; Grupp, S.A.; Teachey, D.T. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: Results of a prospective multi-institutional trial. Blood 2016, 127, 17–28. [Google Scholar] [CrossRef]
- Zhang, Z.; Hu, Q.; Yang, C.; Chen, M.; Han, B. Sirolimus is effective for primary refractory/relapsed warm autoimmune haemolytic anaemia/Evans syndrome: A retrospective single-center study. Ann. Med. 2023, 55, 2282180. [Google Scholar] [CrossRef]
- Giudice, V.; Rosamilio, R.; Ferrara, I.; Seneca, E.; Serio, B.; Selleri, C. Efficacy and safety of splenectomy in adult autoimmune hemolytic anemia. Open Med. Wars. Pol. 2016, 11, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Maskal, S.; Al Marzooqi, R.; Fafaj, A.; Zolin, S.; Naples, R.; Iyer, A.; Petro, C.; Krpata, D.; Prabhu, A.; Rosen, M.; et al. Clinical and surgical outcomes of splenectomy for autoimmune hemolytic anemia. Surg. Endosc. 2022, 36, 5863–5872. [Google Scholar] [CrossRef] [PubMed]
- Akpek, G.; McAneny, D.; Weintraub, L. Comparative response to splenectomy in Coombs-positive autoimmune hemolytic anemia with or without associated disease. Am. J. Hematol. 1999, 61, 98–102. [Google Scholar] [CrossRef]
- Mahévas, M.; Michel, M.; Vingert, B.; Moroch, J.; Boutboul, D.; Audia, S.; Cagnard, N.; Ripa, J.; Menard, C.; Tarte, K.; et al. Emergence of long-lived autoreactive plasma cells in the spleen of primary warm auto-immune hemolytic anemia patients treated with rituximab. J. Autoimmun. 2015, 62, 22–30. [Google Scholar] [CrossRef]
- Xiao, Z.; Murakhovskaya, I. Development of New Drugs for Autoimmune Hemolytic Anemia. Pharmaceutics 2022, 14, 1035. [Google Scholar] [CrossRef]
- An Fc Receptor Structurally Related to MHC Class I Antigens|Nature. Available online: https://www-nature-com.elibrary.einsteinmed.edu/articles/337184a0 (accessed on 25 May 2025).
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Ward, E.S.; Zhou, J.; Ghetie, V.; Ober, R.J. Evidence to support the cellular mechanism involved in serum IgG homeostasis in humans. Int. Immunol. 2003, 15, 187–195. [Google Scholar] [CrossRef]
- Palmeira, P.; Quinello, C.; Silveira-Lessa, A.L.; Zago, C.A.; Carneiro-Sampaio, M. IgG placental transfer in healthy and pathological pregnancies. Clin. Dev. Immunol. 2012, 2012, 985646. [Google Scholar] [CrossRef]
- Patel, D.D.; Bussel, J.B. Neonatal Fc receptor in human immunity: Function and role in therapeutic intervention. J. Allergy Clin. Immunol. 2020, 146, 467–478. [Google Scholar] [CrossRef]
- Ulrichts, P.; Guglietta, A.; Dreier, T.; van Bragt, T.; Hanssens, V.; Hofman, E.; Vankerckhoven, B.; Verheesen, P.; Ongenae, N.; Lykhopiy, V.; et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J. Clin. Investig. 2018, 128, 4372–4386. [Google Scholar] [CrossRef]
- Murakhovskaya, I.; Fattizzo, B.; Ebrahim, T.; Sweet, K.; Shu, C. Energy Trial in Warm Autoimmune Hemolytic Anemia (wAIHA): Design of a Phase 2/3 Randomized, Double-Blind, Placebo-Controlled Study to Assess the Efficacy and Safety of Nipocalimab, an FcRn Blocker. Blood 2022, 140, 2443–2444. [Google Scholar] [CrossRef]
- Seth, N.P.; Xu, R.; DuPrie, M.; Choudhury, A.; Sihapong, S.; Tyler, S.; Meador, J.; Avery, W.; Cochran, E.; Daly, T.; et al. Nipocalimab, an immunoselective FcRn blocker that lowers IgG and has unique molecular properties. mAbs 2025, 17, 2461191. [Google Scholar] [CrossRef] [PubMed]
- Nipocalimab’s Selective Targeting of FcRn and IgG Clearance Preserves Key Immune Functions. ACR Meeting Abstracts. Available online: https://acrabstracts.org/abstract/nipocalimabs-selective-targeting-of-fcrn-and-igg-clearance-preserves-key-immune-functions/ (accessed on 1 June 2025).
- Taylor, P.C.; Schett, G.; Huizinga, T.W.; Wang, Q.; Ibrahim, F.; Zhou, B.; Liva, S.G.; Shaik, J.S.B.; Xiong, Y.; Leu, J.H.; et al. Nipocalimab, an anti-FcRn monoclonal antibody, in participants with moderate to severe active rheumatoid arthritis and inadequate response or intolerance to anti-TNF therapy: Results from the phase 2a IRIS-RA study. RMD Open 2024, 10, e004278. [Google Scholar] [CrossRef] [PubMed]
- Cossu, M.; Bobadilla Mendez, C.; Jackson, A.; Myshkin, E.; Liu, G.; Lam, E.; Beier, U.H.; Weisel, K.; Scott, B.; Leu, J.H.; et al. A randomized, open-label study on the effect of nipocalimab on vaccine responses in healthy participants. Hum. Vaccines Immunother. 2025, 21, 2491269. [Google Scholar] [CrossRef]
- Janssen Research & Development, LLC. Efficacy and Safety of M281 in Adults with Warm Autoim-Mune Hemolytic Anemia: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Study with a Long-Term Open-Label Extension. Clinicaltrials.gov, Clinical Trial Registration NCT04119050. May 2025. Available online: https://clinicaltrials.gov/study/NCT04119050 (accessed on 1 June 2025).
- Johnson & Johnson Receives FDA Approval for IMAAVYTM (Nipocalimab-Aahu), a New FcRn Blocker Offering Long-Lasting Disease Control in the Broadest Population of People Living with Generalized Myasthenia Gravis (gMG). Available online: https://www.jnj.com/media-center/press-releases/johnson-johnson-receives-fda-approval-for-imaavytm-nipocalimab-aahu-a-new-fcrn-blocker-offering-long-lasting-disease-control-in-the-broadest-population-of-people-living-with-generalized-myasthenia-gravis-gmg (accessed on 1 June 2025).
- Janssen Research & Development, LLC. A Phase 3 Randomized, Placebo-Controlled, Double-Blind, Mul-ticenter Study to Evaluate the Efficacy and Safety of Nipocalimab in Pregnancies at Risk for Severe Hemolytic Disease of the Fetus and Newborn (HDFN). Clinicaltrials.gov, Clinical Trial Registration NCT05912517, May 2025. Available online: https://clinicaltrials.gov/study/NCT05912517 (accessed on 1 June 2025).
- Janssen Research & Development, LLC. A Randomized, Placebo-controlled, Double-blind, Multicenter Phase 3 Protocol to Assess the Efficacy and Safety of Nipocalimab in Adults with Moderate to Severe Sjogren’s Disease (SjD). Clinicaltrials.gov, Clinical Trial Registration NCT06741969. May 2025. Available online: https://clinicaltrials.gov/study/NCT06741969 (accessed on 1 June 2025).
- Janssen Research & Development, LLC. Phase 2/3, Multistage, Multicenter, Randomized, Double-Blind, Placebo-Controlled Parallel Group Withdrawal Study to Evaluate the Efficacy and Safety of Nipocalimab Administered to Adults with Chronic Inflammatory Demye-linating Polyneuropathy (CIDP). clinicaltrials.gov, Clinical Trial Registration NCT05327114. April 2025. Available online: https://clinicaltrials.gov/study/NCT05327114 (accessed on 1 June 2025).
- Janssen Research & Development, LLC. Double-Blind, Randomized, Placebo-Controlled Study Evaluating the Safety and Efficacy of Nipocalimab in Reducing the Risk of Fetal and Neonatal Alloimmune Thrombocytopenia (FNAIT) in at-Risk Pregnancies. Clinicaltrials.gov, Clinical Trial Registration NCT06449651. May 2025. Available online: https://clinicaltrials.gov/study/NCT06449651 (accessed on 1 June 2025).
- Janssen Research & Development, LLC. A Multicenter, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Study of Nipocalimab in Adult Participants with Active Systemic Lupus Erythematosus. clinicaltrials.gov, Clinical Trial Registration NCT04882878. May 2025. Available online: https://clinicaltrials.gov/study/NCT04882878 (accessed on 1 June 2025).
- Janssen Research & Development, LLC. A Phase 2, Multicenter, Randomized, Double-Blind, Placebo-Controlled Study of Nipocalimab in Adult Participants with Active Lupus Nephritis. Clinicaltrials.gov, Clinical Trial Registration NCT04883619, May 2025. Available online: https://clinicaltrials.gov/study/NCT04883619 (accessed on 1 June 2025).
- Gilhus, N.E.; Tzartos, S.; Evoli, A.; Palace, J.; Burns, T.M.; Verschuuren, J.J.G.M. Myasthenia gravis. Nat. Rev. Dis. Primer 2019, 5, 30. [Google Scholar] [CrossRef]
- Antozzi, C.; Guptill, J.; Bril, V.; Gamez, J.; Meuth, S.G.; Nowak, R.J.; Quan, D.; Sevilla, T.; Jouvin, M.-H.; Jin, J.; et al. Safety and Efficacy of Nipocalimab in Patients with Generalized Myasthenia Gravis: Results From the Randomized Phase 2 Vivacity-MG Study. Neurology 2024, 102, e207937. [Google Scholar] [CrossRef]
- Antozzi, C.; Vu, T.; Ramchandren, S.; Nowak, R.J.; Farmakidis, C.; Bril, V.; De Bleecker, J.; Yang, H.; Minks, E.; Park, J.-S.; et al. Safety and efficacy of nipocalimab in adults with generalised myasthenia gravis (Vivacity-MG3): A phase 3, randomised, double-blind, placebo-controlled study. Lancet Neurol. 2025, 24, 105–116. [Google Scholar] [CrossRef]
- Antozzi, C.; Vu, T.; Ramchandren, S.; Nowak, R.; Farmakidis, C.; Bril, V.; De Bleecker, J.; Yang, H.; Minks, E.; Park, J.-S.; et al. Long-term Safety and Efficacy of Nipocalimab in Generalized Myasthenia Gravis: Vivacity-MG3 Open-label Extension Phase Results (P7-11.022). Neurology 2025, 104, 3608. [Google Scholar] [CrossRef]
- Akhtar, M.; Akhtar, M.; Farooqi, H.A.; Maryam, A.; Muzammil, A.; Hanif, U.; Athar, Z.; Hassan, S.M.; Khan, Z. Efficacy and safety of FcRn inhibitors in patients with Myasthenia gravis: An updated systematic review and meta-analysis. Clin. Neurol. Neurosurg. 2025, 254, 108910. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Emmi, G.; Greco, M.; Borro, M.; Sardanelli, F.; Murdaca, G.; Indiveri, F.; Puppo, F. Sjögren’s syndrome: A systemic autoimmune disease. Clin. Exp. Med. 2022, 22, 9–25. [Google Scholar] [CrossRef]
- Gottenberg, J.E.; Sivils, K.; Campbell, K.; Idokogi, J.; Lo, K.H.; Liva, S.; Dhatt, H.; Hubbard, J.; Noaiseh, G. LBA0010 Efficacy and safety of nipocalimab, an anti-FCRN monoclonal antibody, in primary sjogren’s disease: Results from a phase 2, multicenter, randomized, placebo-controlled, double-blind study (Dahlias). Ann. Rheum. Dis. 2024, 83, 240. [Google Scholar] [CrossRef]
- Nipocalimab, the First and Only Investigational Treatment to be Granted U.S. FDA Breakthrough Therapy Designation for the Treatment of Adults with Moderate-To-Severe Sjögren’s Disease, Has Now Received Fast Track Designation. Available online: https://www.jnj.com/media-center/press-releases/nipocalimab-the-first-and-only-investigational-treatment-to-be-granted-u-s-fda-breakthrough-therapy-designation-for-the-treatment-of-adults-with-moderate-to-severe-sjogrens-disease-has-now-received-fast-track-designation (accessed on 1 June 2025).
- Savoia, H.F.; Parakh, A.; Kane, S.C. How I manage pregnant patients who are alloimmunized to RBC antigens. Blood 2025, 145, 2275–2282. [Google Scholar] [CrossRef] [PubMed]
- Christensen, R.D.; Bahr, T.M.; Ilstrup, S.J.; Dizon-Townson, D.S. Alloimmune hemolytic disease of the fetus and newborn: Genetics, structure, and function of the commonly involved erythrocyte antigens. J. Perinatol. Off. J. Calif. Perinat. Assoc. 2023, 43, 1459–1467. [Google Scholar] [CrossRef]
- Canlorbe, G.; Macé, G.; Cortey, A.; Cynober, E.; Castaigne, V.; Larsen, M.; Mailloux, A.; Carbonne, B. Management of Very Early Fetal Anemia Resulting From Red-Cell Alloimmunization Before 20 Weeks of Gestation. Obstet. Gynecol. 2011, 118, 1323. [Google Scholar] [CrossRef] [PubMed]
- Kilby, M.D.; Bussel, J.B.; Moise, K.J. The contemporary management of haemolytic disease of the fetus and newborn. Vox Sang. 2025, 120, 644–652. [Google Scholar] [CrossRef]
- Lindenburg, I.; van Kamp, I.; van Zwet, E.; Middeldorp, J.; Klumper, F.; Oepkes, D. Increased perinatal loss after intrauterine transfusion for alloimmune anaemia before 20 weeks of gestation. BJOG Int. J. Obstet. Gynaecol. 2013, 120, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Moise, K.J.; Ling, L.E.; Oepkes, D.; Tiblad, E.; Verweij, E.J.T.J.; Lopriore, E.; Smoleniec, J.; Sachs, U.J.; Bein, G.; Kilby, M.D.; et al. Nipocalimab in Early-Onset Severe Hemolytic Disease of the Fetus and Newborn. N. Engl. J. Med. 2024, 391, 526–537. [Google Scholar] [CrossRef]
- Wolfe, G.I.; Ward, E.S.; de Haard, H.; Ulrichts, P.; Mozaffar, T.; Pasnoor, M.; Vidarsson, G. IgG regulation through FcRn blocking: A novel mechanism for the treatment of myasthenia gravis. J. Neurol. Sci. 2021, 430, 118074. [Google Scholar] [CrossRef]
- Howard, J.F.; Bril, V.; Vu, T.; Karam, C.; Peric, S.; Margania, T.; Murai, H.; Bilinska, M.; Shakarishvili, R.; Smilowski, M.; et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): A multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2021, 20, 526–536. [Google Scholar] [CrossRef]
- Dewilde, S.; Griffiths, A.; Qi, C.Z.; Phillips, G.; Gelinas, D.; Brauer, E.; Mantegazza, R.; Howard, J.F. Post-hoc analyses from the ADAPT clinical study demonstrate aggregate sustained benefit of Efgartigimod in generalized myasthenia gravis. J. Neurol. Sci. 2024, 466, 123264. [Google Scholar] [CrossRef]
- Nugent, D.; McMillan, R.; Nichol, J.L.; Slichter, S.J. Pathogenesis of chronic immune thrombocytopenia: Increased platelet destruction and/or decreased platelet production. Br. J. Haematol. 2009, 146, 585–596. [Google Scholar] [CrossRef]
- Newland, A.C.; Sánchez-González, B.; Rejtő, L.; Egyed, M.; Romanyuk, N.; Godar, M.; Verschueren, K.; Gandini, D.; Ulrichts, P.; Beauchamp, J.; et al. Phase 2 study of efgartigimod, a novel FcRn antagonist, in adult patients with primary immune thrombocytopenia. Am. J. Hematol. 2020, 95, 178–187. [Google Scholar] [CrossRef]
- Broome, C.M.; McDonald, V.; Miyakawa, Y.; Carpenedo, M.; Kuter, D.J.; Al-Samkari, H.; Bussel, J.B.; Godar, M.; Ayguasanosa, J.; De Beuf, K.; et al. Efficacy and safety of the neonatal Fc receptor inhibitor efgartigimod in adults with primary immune thrombocytopenia (ADVANCE IV): A multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Lond. Engl. 2023, 402, 1648–1659. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.F.; Vu, T.; Li, G.; Korobko, D.; Smilowski, M.; Liu, L.; Gistelinck, F.; Steeland, S.; Noukens, J.; Van Hoorick, B.; et al. Subcutaneous efgartigimod PH20 in generalized myasthenia gravis: A phase 3 randomized noninferiority study (ADAPT-SC) and interim analyses of a long-term open-label extension study (ADAPT-SC+). Neurotherapeutics 2024, 21, e00378. [Google Scholar] [CrossRef]
- argenx|argenx Announces U.S. Food and Drug Administration Approval of VYVGART Hytrulo (Efgartigimod Alfa and Hyaluronidase-Qvfc) Injection for Subcutaneous Use in Generalized Myasthenia Gravis. Available online: https://argenx.com/news/2023/argenx-announces-us-food-and-drug-administration-approval-vyvgart-hytrulo-efgartigimod-alfa (accessed on 25 May 2025).
- Allen, J.A.; Lin, J.; Basta, I.; Dysgaard, T.; Eggers, C.; Guptill, J.T.; Gwathmey, K.G.; Hewamadduma, C.; Hofman, E.; Hussain, Y.M.; et al. Safety, tolerability, and efficacy of subcutaneous efgartigimod in patients with chronic inflammatory demyelinating polyradiculoneuropathy (ADHERE): A multicentre, randomised-withdrawal, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2024, 23, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- argenx|argenx Announces Positive Phase 3 Data from ADVANCE Trial of VYVGART® (efgartigimod alfa-fcab) in Adults with Primary Immune Thrombocytopenia. Available online: https://argenx.com/news/2022/argenx-announces-positive-phase-3-data-advance-trial-vyvgartr-efgartigimod-alfa-fcab-adults (accessed on 25 May 2025).
- Yap, D.Y.H.; Hai, J.; Lee, P.C.H.; Zhou, X.; Lee, M.; Zhang, Y.; Wang, M.; Chen, X. Safety, tolerability, pharmacokinetics, and pharmacodynamics of HBM9161, a novel FcRn inhibitor, in a phase I study for healthy Chinese volunteers. Clin. Transl. Sci. 2021, 14, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- RVT-1401, A Novel Anti-FcRn Monoclonal Antibody, Is Well Tolerated in Healthy Subjects and Reduces Plasma IgG Following Subcutaneous or Intravenous Administration—UQ eSpace. Available online: https://espace.library.uq.edu.au/view/UQ:86edcae (accessed on 25 May 2025).
- Yan, C.; Yue, Y.; Guan, Y.; Bu, B.; Ke, Q.; Duan, R.; Deng, H.; Xue, Q.; Jiang, H.; Xiao, F.; et al. Batoclimab vs Placebo for Generalized Myasthenia Gravis: A Randomized Clinical Trial. JAMA Neurol. 2024, 81, 336–345. [Google Scholar] [CrossRef]
- Kahaly, G.J.; Dolman, P.J.; Wolf, J.; Giers, B.C.; Elflein, H.M.; Jain, A.P.; Srinivasan, A.; Hadjiiski, L.; Jordan, D.; Bradley, E.A.; et al. Proof-of-concept and Randomized, Placebo-controlled Trials of an FcRn Inhibitor, Batoclimab, for Thyroid Eye Disease. J. Clin. Endocrinol. Metab. 2023, 108, 3122–3134. [Google Scholar] [CrossRef]
- Wang, Y.; Zhong, X.; Wang, H.; Peng, Y.; Shi, F.; Jia, D.; Yang, H.; Zeng, Q.; Quan, C.; ZhangBao, J.; et al. Batoclimab as an add-on therapy in neuromyelitis optica spectrum disorder patients with acute attacks. Eur. J. Neurol. 2023, 30, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Single/Multiple Doses of Imvt-1402 in Healthy Participants and Participants with Autoimmune Diseases. Available online: https://www.isrctn.com/ISRCTN11659633 (accessed on 25 May 2025).
- Immunovant Announces Positive IMVT-1402 Initial 600 mg MAD Results that Confirm Best-in-Class Potential. Available online: https://www.immunovant.com/investors/news-events/press-releases/detail/57/immunovant-announces-positive-imvt-1402-initial-600-mg-mad (accessed on 25 May 2025).
- Smith, B.; Kiessling, A.; Lledo-Garcia, R.; Dixon, K.L.; Christodoulou, L.; Catley, M.C.; Atherfold, P.; D’Hooghe, L.E.; Finney, H.; Greenslade, K.; et al. Generation and characterization of a high affinity anti-human FcRn antibody, rozanolixizumab, and the effects of different molecular formats on the reduction of plasma IgG concentration. mAbs 2018, 10, 1111–1130. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, P.; Lledo-Garcia, R.; Watanabe, S.; Langdon, G.; Tran, D.; Bari, M.; Christodoulou, L.; Jones, E.; Price, G.; Smith, B.; et al. The FcRn inhibitor rozanolixizumab reduces human serum IgG concentration: A randomized phase 1 study. Sci. Transl. Med. 2017, 9, eaan1208. [Google Scholar] [CrossRef]
- Bril, V.; Drużdż, A.; Grosskreutz, J.; Habib, A.A.; Mantegazza, R.; Sacconi, S.; Utsugisawa, K.; Vissing, J.; Vu, T.; Boehnlein, M.; et al. Safety and efficacy of rozanolixizumab in patients with generalised myasthenia gravis (MycarinG): A randomised, double-blind, placebo-controlled, adaptive phase 3 study. Lancet Neurol. 2023, 22, 383–394. [Google Scholar] [CrossRef]
- Robak, T.; Kaźmierczak, M.; Jarque, I.; Musteata, V.; Treliński, J.; Cooper, N.; Kiessling, P.; Massow, U.; Woltering, F.; Snipes, R.; et al. Phase 2 multiple-dose study of an FcRn inhibitor, rozanolixizumab, in patients with primary immune thrombocytopenia. Blood Adv. 2020, 4, 4136–4146. [Google Scholar] [CrossRef]
- Cooper, N.; Bussel, J.B.; Kaźmierczak, M.; Miyakawa, Y.; Cluck, S.; Lledó García, R.; Haier, B.; Lavrov, A.; Singh, P.; Snipes, R.; et al. Inhibition of FcRn with rozanolixizumab in adults with immune thrombocytopenia: Two randomised, double-blind, placebo-controlled phase 3 studies and their open-label extension. Br. J. Haematol. 2025, 206, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Lledo-Garcia, R.; Dixon, K.; Shock, A.; Oliver, R. Pharmacokinetic-pharmacodynamic modelling of the anti-FcRn monoclonal antibody rozanolixizumab: Translation from preclinical stages to the clinic. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Gjølberg, T.T.; Mester, S.; Calamera, G.; Telstad, J.S.; Sandlie, I.; Andersen, J.T. Targeting the Neonatal Fc Receptor in Autoimmune Diseases: Pipeline and Progress. BioDrugs 2025, 39, 373–409. [Google Scholar] [CrossRef]
- Shu Taishen (300204.SZ): STSA-1301 Subcutaneous Injection (Primary Immune Thrombocytopenia ITP) Phase Ia Clinical Trial Completed the Administration of the First Case of Subjects. Available online: https://www.moomoo.com/news/post/30178441/shu-taishen-300204-sz-stsa-1301-subcutaneous-injection-primary (accessed on 25 May 2025).
- Jiangsu BioJeTay Biotechnology Co., Ltd. A Phase Ib/II Clinical Trial to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Multiple Doses of STSA-1301 Subcutaneous Injection in Healthy Subjects and Patients with Immune Thrombocytopenia (ITP). clinicaltrials.gov, Clinical Trial Registration NCT06929299. April 2025. Available online: https://clinicaltrials.gov/study/NCT06929299 (accessed on 25 May 2025).
- Ma, G.; Crowley, A.R.; Heyndrickx, L.; Rogiers, I.; Parthoens, E.; Van Santbergen, J.; Ober, R.J.; Bobkov, V.; de Haard, H.; Ulrichts, P.; et al. Differential effects of FcRn antagonists on the subcellular trafficking of FcRn and albumin. JCI Insight 2024, 9, e176166. [Google Scholar] [CrossRef]
- argenx|argenx Reports Topline Results from ADDRESS Study of Efgartigimod SC in Pemphigus. Available online: https://argenx.com/news/2023/argenx-reports-topline-results-address-study-efgartigimod-sc-pemphigus (accessed on 15 June 2025).
- Menon, D.; Bhandari, V. FcRn inhibitors in the context of myasthenia gravis. Expert Opin. Emerg. Drugs 2025, 30, 7–10. [Google Scholar] [CrossRef]
- Li, J.; Wu, X.; Chu, T.; Tan, X.; Wang, S.; Qu, R.; Chen, Z.; Wang, Z. The efficacy and safety of FcRn inhibitors in patients with myasthenia gravis: A systematic review and meta-analysis. J. Neurol. 2024, 271, 2298–2308. [Google Scholar] [CrossRef]
- Levine, T.; Muley, S. Early deterioration of CIDP following transition from IVIG to FcRn inhibitor treatment. J. Neurol. Sci. 2025, 468, 123313. [Google Scholar] [CrossRef]
- Jacobs, J.W.; Booth, G.S.; Raza, S.; Clark, L.M.; Fasano, R.M.; Gavriilaki, E.; Abels, E.A.; Binns, T.C.; Duque, M.A.; McQuilten, Z.K.; et al. Current state and potential applications of neonatal Fc receptor (FcRn) inhibitors in hematologic conditions. Am. J. Hematol. 2024, 99, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Fondazione Policlinico Universitario Agostino Gemelli IRCCS, “Markers of Favorable Response to FcRn Inhibitors (INFORM),” Clinicaltrials.gov, Clinical Trial Registration NCT06685055. November 2024. Available online: https://clinicaltrials.gov/study/NCT06685055 (accessed on 1 July 2025).

{kind=link}
| Therapy | Response Rate | Time to Response | Toxicities | Comments |
|---|---|---|---|---|
| Corticosteroids [16,24,26,27] | 70–80% | 2–3 weeks | Weight gain Hyperglycemia Peptic ulcer Adrenal insufficiency Delirium Myopathy Osteoporosis | To reduce risk of relapse, patients require long-term slow taper of steroids after reaching hemoglobin goal |
| Rituximab [33,34,35] | 80% | 3–6 weeks | Hypogammaglobulinemia Infection Impaired vaccine response | Can be used first-line in severe cases or second line for patients with steroid-refractory disease |
| Azathioprine [24,25,26,27] | 60% | 1–3 months | Increased infection risk Hepatotoxicity | |
| Cyclosporine [24,25,26] | 60% | 1–3 months | Increased infection risk Nephrotoxicity | |
| Cyclophosphamide [16,24,25,26] | 50–70% | 1–2 months | Increased infection risk Teratogen | |
| Splenectomy [39,40] | 80% | 1–2 weeks | Increased infection risk with encapsulated organism Thrombosis risk | Long-term efficacy unknown |
| Sirolimus [37] | 79.5% | 2 months | Increased infection risk Hepatotoxicity Mucositis | Particularly effective in autoimmune lymphoproliferative syndrome |
| Therapy | Disease Indication | Dosing Regimen | Toxicities | Trial Phases |
|---|---|---|---|---|
| Nipocalimab | wAIHA RA gMG HDFN Sjögren’s FNAIT | IV Every 2 weeks | Increased risk of infection Infusion reactions | Phase II/III—NCT04119050 Phase II—NCT04991753 Phase II—NCT04968912 Phase II—NCT03842189 Phase III—NCT06741969 Phase III—NCT04951622 |
| Efgartigimod | gMG ITP CIDP | IV, SQ Weekly or every 4–8 Weeks | Increased risk of infection Arthralgias Myalgias Injection-site reactions | Phase III—NCT03669588 Phase III—NCT04687072 Phase III—NCT04281472 |
| Batoclimab | NMOSD gMG Thyroid eye disease | SC weekly × 6 | Injection-site reactions Hypoalbuminemia Hypogammaglobulinemia Headache Hypercholesterolemia | Phase III—NCT05039190 Phase III—NCT05403541 |
| Rozanolixizumab | gMG ITP | SC weekly | Increased infection risk Headache Pyrexia Nausea | Phase III—NCT03971422 Phase II—NCT03052751 Phase I—NCT02220153 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandhu, M.; Murakhovskaya, I. FcRn Blockade as a Targeted Therapeutic Strategy in Antibody-Mediated Autoimmune Diseases: A Focus on Warm Autoimmune Hemolytic Anemia. Antibodies 2025, 14, 65. https://doi.org/10.3390/antib14030065
Sandhu M, Murakhovskaya I. FcRn Blockade as a Targeted Therapeutic Strategy in Antibody-Mediated Autoimmune Diseases: A Focus on Warm Autoimmune Hemolytic Anemia. Antibodies. 2025; 14(3):65. https://doi.org/10.3390/antib14030065
Chicago/Turabian StyleSandhu, Michael, and Irina Murakhovskaya. 2025. "FcRn Blockade as a Targeted Therapeutic Strategy in Antibody-Mediated Autoimmune Diseases: A Focus on Warm Autoimmune Hemolytic Anemia" Antibodies 14, no. 3: 65. https://doi.org/10.3390/antib14030065
APA StyleSandhu, M., & Murakhovskaya, I. (2025). FcRn Blockade as a Targeted Therapeutic Strategy in Antibody-Mediated Autoimmune Diseases: A Focus on Warm Autoimmune Hemolytic Anemia. Antibodies, 14(3), 65. https://doi.org/10.3390/antib14030065