


A Comprehensive Review About the Use of Monoclonal Antibodies in Cancer Therapy

 , ,
, ,
Abstract
1. Introduction
2. Immunomodulatory Approaches (Checkpoint Inhibitors, Bispecific Antibodies, ADCs with Immune Modulation)
2.1. Immune Checkpoint Pathway
2.1.1. Mechanism of Action
2.1.2. The Future of Immune Checkpoint Inhibitors
2.1.3. Use of Bispecific Antibodies: Mechanisms of Action
2.1.4. The Action of T-Cell Engagers (TCEs)
2.2. Antibody–Drug Conjugates (ADCs) with Immune Modulation
- Inducing Immunogenic Cell Death (ICD): Some ADC payloads, such as microtubule inhibitors (e.g., MMAE) or DNA-damaging agents (e.g., PBD dimers), can trigger ICD, leading to the release of damage-associated molecular patterns (DAMPs) that activate dendritic cells and enhance antigen presentation;
- Targeting the Tumour Microenvironment (TME): Some ADCs are designed to deplete regulatory T cells (Tregs) or myeloid-derived suppressor cells (MDSCs), by targeting specific markers like CD25 or CCR8;
- Enhancing T-cell Infiltration: ADCs can modify the tumour microenvironment by reducing stromal barriers or disrupting tumour-associated vasculature, thereby facilitating better immune cell infiltration;
- Activating Innate Immunity: ADCs can engage immune cells through Fcγ receptor interactions, promoting phagocytosis or antibody-dependent cellular cytotoxicity (ADCC);
- Combining with Immune Checkpoint Inhibitors (ICIs): ADCs that induce immune responses may synergise with ICIs (e.g., anti-PD-1/PD-L1) to enhance anti-tumour immunity.
- Examples of ADCs with immune modulation include the following:
- ▪
- Enfortumab vedotin (EV, targeting Nectin-4): Induces ICD and enhances the efficacy of checkpoint inhibitors in urothelial carcinoma.
3. Receptor-Targeted Therapies (HER2, EGFR, IGF-1R Inhibitors)
3.1. HER2 Inhibition and Receptor Tyrosine Kinase Signalling
3.2. Trastuzumab, Pertuzumab, and Margetuximab Are Monoclonal Antibodies Targeting HER2-Positive Cancers
3.3. Epidermal Growth Factor Receptor (EGFR) in Cancer Therapy
3.3.1. Mechanism of Action
3.3.2. Anti-EGFR Antibody Characteristics
3.3.3. FOLFIRI and FOLFOX
4. VEGF/VEGFR Inhibition Cancer Therapy
4.1. Targeting Angiogenesis in Cancer: VEGF/VEGFR Pathway
4.2. Angiogenesis and the VEGF/VEGFR Pathway
4.3. Indications for VEGF/VEGFR Inhibitors
5. Emerging Strategies (Trop-2 ADCs, Combinatorial Approaches, Next-Generation Abs)
5.1. Tumour Cell Surface Marker: Trop-2
5.2. Mechanism of Action: Trop-2 ADCC
- ▪
- Metastatic triple-negative breast cancer (mTNBC): Approved in 2020 for individuals who have previously undergone at least two treatment regimens for metastatic disease;
- ▪
- HR-positive/HER2-negative metastatic breast cancer: Approved in 2023 for patients who have received prior endocrine therapy along with at least two systemic treatments;
- ▪
- Locally advanced or metastatic urothelial carcinoma: Approved in 2021 for patients who have been previously treated with platinum-based chemotherapy and a PD-1/PD-L1 inhibitor.
5.3. Other Trop-2 ADCs in Development
5.4. Next Generation Antibodies
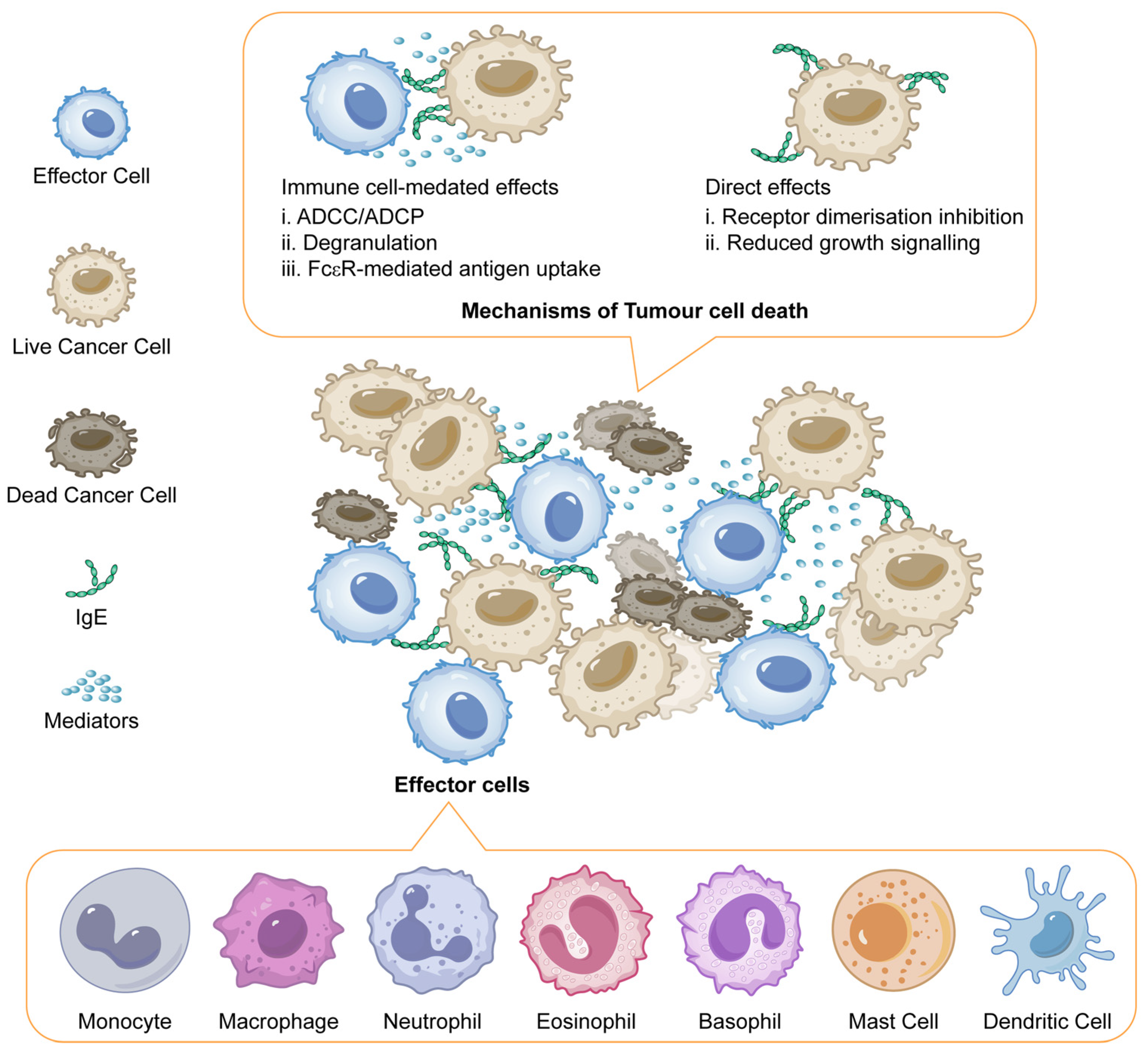
5.5. IgE Antibodies Against Tumours
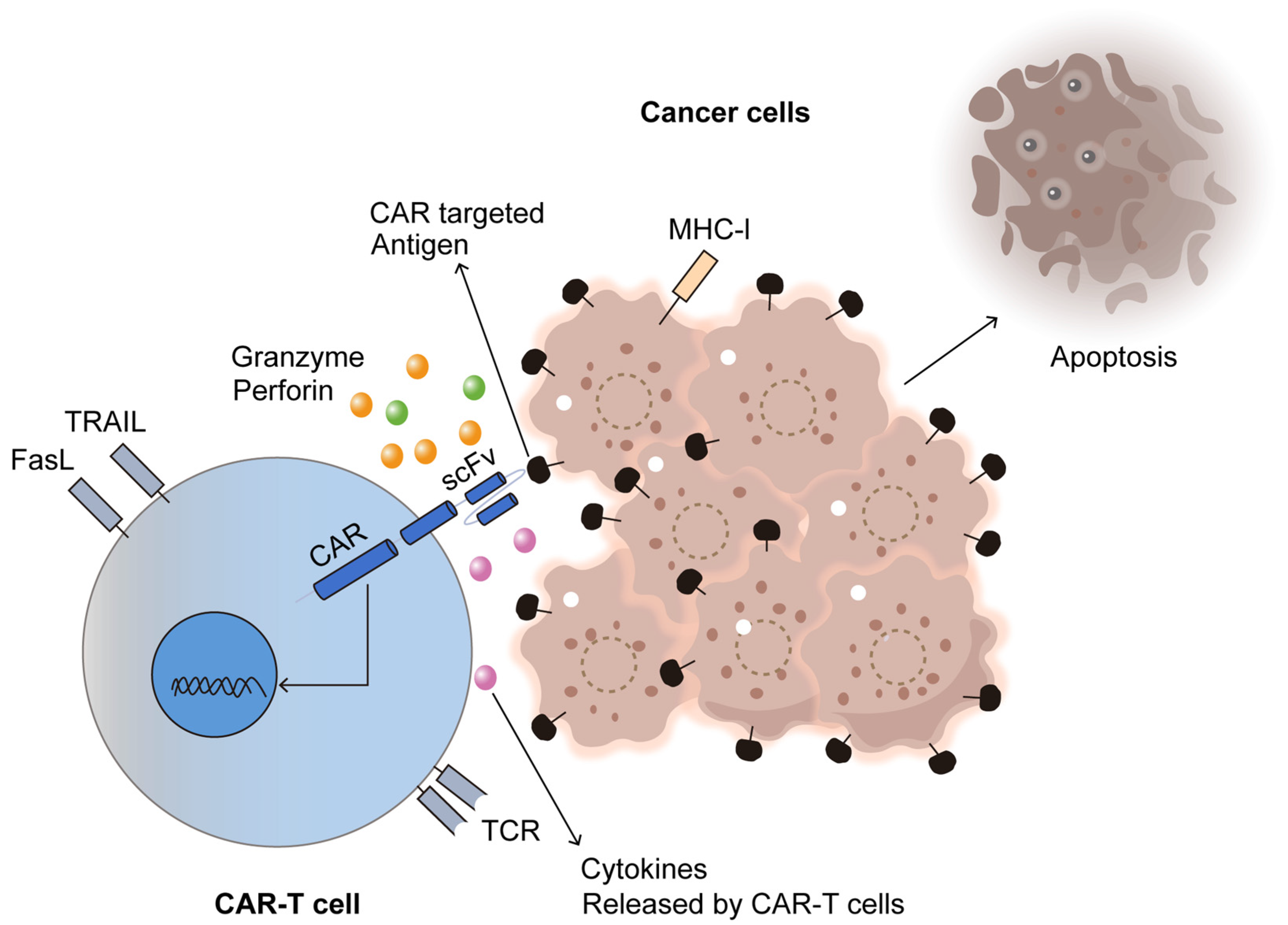
6. Integration of Monoclonal Antibodies with Chimeric Antigen Receptor (CAR) T-Cell Therapy
7. Other Issues in Cancer Immunotherapy
7.1. Recent FDA Approval of Next-Generation Antibodies for Cancer
7.2. Exploring IgA Antibodies in Cancer Therapy: A Focus on Mucosal Surface Tumour
7.3. Engineered IgA for Cancer Immunotherapy: Targeting Tumours in Mucosal Tissues
- Direct Tumour Cell Lysis: If complement activation overcomes regulatory mechanisms, membrane attack complex (MAC) formation can lead to tumour cell death;
- Opsonization and Phagocytosis: C3b deposition enhances recognition by immune cells, facilitating phagocytosis and antigen presentation;
- Pro-Inflammatory Responses: Complement components (e.g., C5a, C3a) can recruit immune cells to the tumour microenvironment, promoting an anti-tumour response;
- Immune Evasion: Many tumour cells resist complement attack by overexpressing complement inhibitors, limiting complement-mediated destruction.
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ghemrawi, R.; Abuamer, L.; Kremesh, S.; Hussien, G.; Ahmed, R.; Mousa, W.; Khair, M. Revolutionizing Cancer Treatment: Recent Advances in Immunotherapy. Biomedicines 2024, 12, 2158. [Google Scholar] [CrossRef] [PubMed]
- Raghani, N.R.; Chorawala, M.R.; Mahadik, M.; Patel, R.B.; Prajapati, B.G.; Parekh, P.S. Revolutionizing Cancer Treatment: Comprehensive Insights into Immunotherapeutic Strategies. Med. Oncol. 2024, 41, 51. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Kankala, R.K.; Yang, Z.; Li, W.; Xie, S.; Li, H.; Chen, A.Z.; Zou, L. Antibody-based drug delivery systems for cancer therapy: Mechanisms, challenges, and prospects. Theranostics 2022, 12, 3719. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Li, J.; Xu, X.; Ye, Y.; Wang, C.; Pang, G.; Liu, W.; Liu, A.; Zhao, C.; Hao, X. Strategies to enhance the therapeutic efficacy of anti-PD-1 antibody, anti-PD-L1 antibody and anti-CTLA-4 antibody in cancer therapy. J. Transl. Med. 2024, 22, 751. [Google Scholar] [CrossRef]
- Jia, G.; Jiang, Y.; Li, X. Targeted Drug Conjugates in Cancer Therapy: Challenges and Opportunities. Pharm. Sci. Adv. 2024, 2, 100048. [Google Scholar] [CrossRef]
- Ponziani, S.; Di Vittorio, G.; Pitari, G.; Cimini, A.M.; Ardini, M.; Gentile, R.; Giansanti, F. Antibody-Drug Conjugates: The New Frontier of Chemotherapy. Int. J. Mol. Sci. 2020, 21, 5510. [Google Scholar] [CrossRef]
- Lan, H.R.; Chen, M.; Yao, S.Y.; Chen, J.X.; Jin, K.T. Bispecific Antibodies Revolutionizing Breast Cancer Treatment: A Comprehensive Overview. Front. Immunol. 2023, 14, 1266450. [Google Scholar] [CrossRef]
- Caers, J.; Duray, E.; Vrancken, L.; Marcion, G.; Bocuzzi, V.; De Veirman, K.; D’Huyvetter, M. Radiotheranostic Agents in Hematological Malignancies. Front. Immunol. 2022, 13, 911080. [Google Scholar] [CrossRef]
- Fourie Zirkelbach, J.; Shah, M.; Vallejo, J.; Cheng, J.; Ayyoub, A.; Liu, J.; Hudson, R.; Sridhara, R.; Ison, G.; Amiri-Kordestani, L.; et al. Improving dose-optimization processes used in oncology drug development to minimize toxicity and maximize benefit to patients. J. Clin. Oncol. 2022, 40, 3489–3500. [Google Scholar] [CrossRef]
- Mohite, P.; Yadav, V.; Pandhare, R.; Maitra, S.; Saleh, F.M.; Saleem, R.M.; Al-Malky, H.S.; Kumarasamy, V.; Subramaniyan, V.; Abdel-Daim, M.M.; et al. Revolutionizing Cancer Treatment: Unleashing the Power of Viral Vaccines, Monoclonal Antibodies, and Proteolysis-Targeting Chimeras in the New Era of Immunotherapy. ACS Omega 2024, 9, 7277–7295. [Google Scholar] [CrossRef]
- Carbone, F.; Russo, C.; Colamatteo, A.; La Rocca, C.; Fusco, C.; Matarese, A.; Procaccini, C.; Matarese, G. Cellular and molecular signaling towards T cell immunological self-tolerance. J. Biol. Chem. 2024, 300, 107134. [Google Scholar] [CrossRef] [PubMed]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dai, Z.; Wu, W.; Wang, Z.; Zhang, N.; Zhang, L.; Zeng, W.J.; Liu, Z.; Cheng, Q. Regulatory mechanisms of immune checkpoints PD-L1 and CTLA-4 in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 184. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.K.; Wong, B.H.; Poh, Z.S.; Udayakumar, A.; Verma, R.; Goh, R.K.; Duggan, S.P.; Shelat, V.G.; Chandy, K.G.; Grigoropoulos, N.F. Obstacles for T-lymphocytes in the tumour microenvironment: Therapeutic challenges, advances and opportunities beyond immune checkpoint. eBioMedicine 2022, 83, 104216. [Google Scholar] [CrossRef]
- Chang, C.Y.; Park, H.; Malone, D.C.; Wang, C.Y.; Wilson, D.L.; Yeh, Y.M.; Lo-Ciganic, W.H. Immune Checkpoint Inhibitors and Immune-Related Adverse Events in Patients with Advanced Melanoma: A Systematic Review and Network Meta-Analysis. JAMA Netw. Open 2020, 3, e201611. [Google Scholar] [CrossRef]
- Wang, D.D.; Shaver, L.G.; Shi, F.Y.; Wei, J.J.; Qin, T.Z.; Wang, S.Z.; Kong, Y.J. Comparative efficacy and safety of PD-1/PD-L1 immunotherapies for non-small cell lung cancer: A network meta-analysis. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 2866–2884. [Google Scholar]
- Chen, L.; Mo, D.C.; Hu, M.; Zhao, S.J.; Yang, Q.W.; Huang, Z.L. PD-1/PD-L1 inhibitor monotherapy in recurrent or metastatic squamous cell carcinoma of the head and neck: A meta-analysis. Am. J. Otolaryngol. 2022, 43, 103324. [Google Scholar] [CrossRef]
- Galicia-Carmona, T.; Arango-Bravo, E.A.; Coronel-Martínez, J.A.; Cetina-Perez, L.; Vanoye-Carlo, E.G.; Villalobos-Valencia, R.; García-Pacheco, J.A.; Cortés-Esteban, P. Advanced, recurrent, and persistent cervical cancer management: In the era of immunotherapy. Front. Oncol. 2024, 14, 1392639. [Google Scholar] [CrossRef]
- Martini, A.; Raggi, D.; Fallara, G.; Nocera, L.; Schultz, J.G.; Belladelli, F.; Marandino, L.; Salonia, A.; Briganti, A.; Montorsi, F.; et al. Immunotherapy versus chemotherapy as first-line treatment for advanced urothelial cancer: A systematic review and meta-analysis. Cancer Treat. Rev. 2022, 104, 102360. [Google Scholar] [CrossRef]
- Lin, Q.; Wang, X.; Hu, Y. The opportunities and challenges in immunotherapy: Insights from the regulation of PD-L1 in cancer cells. Cancer Lett. 2023, 569, 216318. [Google Scholar] [CrossRef]
- Skafi, N.; Fayyad-Kazan, M.; Badran, B. Immunomodulatory Role for MicroRNAs: Regulation of PD-1/PD-L1 and CTLA-4 Immune Checkpoints Expression. Gene 2020, 754, 144888. [Google Scholar] [CrossRef]
- Garg, P.; Malhotra, J.; Kulkarni, P.; Horne, D.; Salgia, R.; Singhal, S.S. Emerging Therapeutic Strategies to Overcome Drug Resistance in Cancer Cells. Cancers 2024, 16, 2478. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, C.D.; Pulanco, M.C.; Cui, W.; Lu, L.; Zang, X. PD-L1 and B7-1 cis-interaction: New mechanisms in immune checkpoints and immunotherapies. Trends Mol. Med. 2021, 27, 207–219. [Google Scholar] [CrossRef]
- Korman, A.J.; Garrett-Thomson, S.C.; Lonberg, N. The Foundations of Immune Checkpoint Blockade and the Ipilimumab Approval Decennial. Nat. Rev. Drug Discov. 2022, 21, 509–528. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Khan, F.; Qari, H.A.; Upadhyay, T.K.; Alkhateeb, A.F.; Oves, M. Revolutionization in Cancer Therapeutics via Targeting Major Immune Checkpoints PD-1, PD-L1 and CTLA-4. Pharmaceuticals 2022, 15, 335. [Google Scholar] [CrossRef] [PubMed]
- Budczies, J.; Kazdal, D.; Menzel, M.; Beck, S.; Kluck, K.; Altbürger, C.; Schwab, C.; Allgäuer, M.; Ahadova, A.; Kloor, M.; et al. Tumour mutational burden: Clinical utility, challenges and emerging improvements. Nat. Rev. Clin. Oncol. 2024, 21, 725–742. [Google Scholar] [CrossRef]
- Sacchi de Camargo Correia, G.; Zhao, Y.; Manochakian, R.; Lou, Y. The Role of Immunotherapy Sensitizers and Novel Immunotherapy Modalities in the Treatment of Cancer. Front. Oncol. 2024, 14, 1336546. [Google Scholar] [CrossRef]
- He, P.; Ma, L.; Xu, B.; Wang, Y.; Li, X.; Chen, H.; Li, Y. Research Progress and Future Directions of Immune Checkpoint Inhibitor Combination Therapy in Advanced Gastric Cancer. Ther. Adv. Med. Oncol. 2024, 16, 17588359241266156. [Google Scholar] [CrossRef]
- Cai, L.; Li, Y.; Tan, J.; Xu, L.; Li, Y. Targeting LAG-3, TIM-3, and TIGIT for cancer immunotherapy. J. Hematol. Oncol. 2023, 16, 101. [Google Scholar] [CrossRef]
- Kong, X.; Zhang, J.; Chen, S.; Wang, X.; Xi, Q.; Shen, H.; Zhang, R. Immune checkpoint inhibitors: Breakthroughs in cancer treatment. Cancer Biol. Med. 2024, 21, 451–472. [Google Scholar] [CrossRef]
- Shukla, N.; Hanna, N. Neoadjuvant and Adjuvant Immunotherapy in Early-Stage Non-Small Cell Lung Cancer. Lung Cancer Targets Ther. 2021, 12, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Luo, Q. When artificial intelligence meets PD-1/PD-L1 inhibitors: Population screening, response prediction and efficacy evaluation. Comput. Biol. Med. 2022, 145, 105499. [Google Scholar] [CrossRef]
- Gupta, R.; Srivastava, D.; Sahu, M.; Tiwari, S.; Ambasta, R.K.; Kumar, P. Artificial Intelligence to Deep Learning: Machine Intelligence Approach for Drug Discovery. Mol. Divers. 2021, 25, 1315–1360. [Google Scholar] [CrossRef]
- Wang, Y.; Tong, Z.; Zhang, W.; Zhang, W.; Buzdin, A.; Mu, X.; Li, X. FDA-Approved and Emerging Next-Generation Predictive Biomarkers for Immune Checkpoint Inhibitors in Cancer Patients. Front. Oncol. 2021, 11, 683419. [Google Scholar] [CrossRef]
- Marei, H.E.; Hasan, A.; Pozzoli, G.; Cenciarelli, C. Cancer Immunotherapy with Immune Checkpoint Inhibitors (ICIs): Potential, Mechanisms of Resistance, and Strategies for Reinvigorating T Cell Responsiveness When Resistance Is Acquired. Cancer Cell Int. 2023, 23, 64. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, J.W.; Lorentzen, C.L.; Martinenaite, E.; Ellebaek, E.; Donia, M.; Holmstroem, R.B.; Klausen, T.W.; Madsen, C.O.; Ahmed, S.M.; Weis-Banke, S.E.; et al. A phase 1/2 trial of an immune-modulatory vaccine against IDO/PD-L1 in combination with nivolumab in metastatic melanoma. Nat. Med. 2021, 27, 2212–2223. [Google Scholar] [CrossRef]
- Yi, M.; Zheng, X.; Niu, M.; Zhu, S.; Ge, H.; Wu, K. Combination strategies with PD-1/PD-L1 blockade: Current advances and future directions. Mol. Cancer 2022, 21, 28. [Google Scholar] [CrossRef] [PubMed]
- Marcus, L.; Fashoyin-Aje, L.A.; Donoghue, M.; Yuan, M.; Rodriguez, L.; Gallagher, P.S.; Philip, R.; Ghosh, S.; Theoret, M.R.; Beaver, J.A.; et al. FDA approval summary: Pembrolizumab for the treatment of tumour mutational burden–high solid tumours. Clin. Cancer Res. 2021, 27, 4685–4689. [Google Scholar] [CrossRef]
- Liu, S.V.; Reck, M.; Mansfield, A.S.; Mok, T.; Scherpereel, A.; Reinmuth, N.; Garassino, M.C.; De Castro Carpeno, J.; Califano, R.; Nishio, M.; et al. Updated overall survival and PD-L1 subgroup analysis of patients with extensive-stage small-cell lung cancer treated with atezolizumab, carboplatin, and etoposide (IMpower133). J. Clin. Oncol. 2021, 39, 619–630. [Google Scholar] [CrossRef]
- Catalano, M.; Shabani, S.; Venturini, J.; Ottanelli, C.; Voltolini, L.; Roviello, G. Lung cancer immunotherapy: Beyond common immune checkpoints inhibitors. Cancers 2022, 14, 6145. [Google Scholar] [CrossRef]
- Hu, P.; Dai, H.I.; Bourdage, J.; Zhou, D.; Trang, K.; Kowalski, K.; Bello, C.; Hibma, J.; Khandelwal, A.; Cowan, K.; et al. Immunogenicity of avelumab in patients with metastatic Merkel cell carcinoma or advanced urothelial carcinoma. Clin. Transl. Sci. 2024, 17, e13730. [Google Scholar] [CrossRef] [PubMed]
- Gui, T.; Ma, L. The Evaluation of Effects, Mechanisms, and Clinical Data of Avelumab. Highlights Sci. Eng. Technol. 2023, 36, 23–28. [Google Scholar] [CrossRef]
- Adams, S.; Othus, M.; Patel, S.P.; Miller, K.D.; Chugh, R.; Schuetze, S.M.; Chamberlin, M.D.; Haley, B.J.; Storniolo, A.M.; Reddy, M.P.; et al. A multicenter phase II trial of ipilimumab and nivolumab in unresectable or metastatic metaplastic breast cancer: Cohort 36 of dual anti–CTLA-4 and anti–PD-1 blockade in rare tumours (DART, SWOG S1609). Clin. Cancer Res. 2022, 28, 271–278. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus ipilimumab in advanced non–small-cell lung cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Liu, M.; Zhang, Y.; Wang, X. Bispecific T cell engagers: An emerging therapy for management of hematologic malignancies. J. Hematol. Oncol. 2021, 14, 75. [Google Scholar] [CrossRef]
- Aamir, S.; Anwar, M.Y.; Khalid, F.; Khan, S.I.; Ali, M.A.; Khattak, Z.E. Systematic Review and Meta-analysis of CD19-Specific CAR-T Cell Therapy in Relapsed/Refractory Acute Lymphoblastic Leukemia in the Pediatric and Young Adult Population: Safety and Efficacy Outcomes. Clin. Lymphoma Myeloma Leuk 2021, 21, e334–e347. [Google Scholar] [CrossRef]
- Subklewe, M.; Magno, G.; Gebhardt, C.; Bücklein, V.; Szelinski, F.; Arévalo, H.J.; Hänel, G.; Dörner, T.; Zugmaier, G.; von Bergwelt-Baildon, M.; et al. Application of blinatumomab, a bispecific anti-CD3/CD19 T-cell engager, in treating severe systemic sclerosis: A case study. Eur. J. Cancer 2024, 204, 114071. [Google Scholar] [CrossRef]
- Lantz, J.; Pham, N.; Jones, C.; Reed, D.; El Chaer, F.; Keng, M. Blinatumomab in Practice. Curr. Hematol. Malig. Rep. 2024, 19, 1–8. [Google Scholar] [CrossRef]
- Cech, P.; Skórka, K.; Dziki, L.; Giannopoulos, K. T-Cell Engagers-The Structure and Functional Principle and Application in Hematological Malignancies. Cancers 2024, 16, 1580. [Google Scholar] [CrossRef]
- Gholap, A.D.; Gupta, J.S.; Kamandar, P.A.; Banchhod, G.V.; Hatvate, N.T. Antibody-drug conjugates for cancer therapy: An up-to-date review on the chemistry and pharmacology. In Comprehensive Analytical Chemistry; Elsevier: Amsterdam, The Netherlands, 2023; Volume 103, pp. 105–190. [Google Scholar]
- Elshazly, A.M.; Gewirtz, D.A. An overview of resistance to Human epidermal growth factor receptor 2 (Her2) targeted therapies in breast cancer. Cancer Drug Resist. 2022, 5, 472. [Google Scholar] [CrossRef]
- Talukdar, S.; Emdad, L.; Das, S.K.; Fisher, P.B. EGFR: An essential receptor tyrosine kinase-regulator of cancer stem cells. Adv. Cancer Res. 2020, 147, 161–188. [Google Scholar] [PubMed]
- Cheng, X. A comprehensive review of HER2 in cancer biology and therapeutics. Genes 2024, 15, 903. [Google Scholar] [CrossRef]
- Pan, L.; Li, J.; Xu, Q.; Gao, Z.; Yang, M.; Wu, X.; Li, X. HER2/PI3K/AKT pathway in HER2-positive breast cancer: A review. Medicine 2024, 103, e38508. [Google Scholar] [CrossRef]
- Oh, D.Y.; Bang, Y.J. HER2-targeted therapies—A role beyond breast cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef]
- Singh, D.D.; Lee, H.J.; Yadav, D.K. Clinical updates on tyrosine kinase inhibitors in HER2-positive breast cancer. Front. Pharmacol. 2022, 13, 1089066. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, P.; Tolaney, S.M. The dawn of the antibody–drug conjugates era: How T-DM1 reinvented the future of chemotherapy for solid tumours. Cancer Res. 2022, 82, 3659–3661. [Google Scholar] [CrossRef] [PubMed]
- Kronig, M.N.; Wehrli, M.; Salas-Benito, D.; Maus, M.V. Hurdles race for CAR T-cell therapy in digestive tract cancer. Immunol. Rev. 2023, 320, 100–119. [Google Scholar] [CrossRef]
- Gámez-Chiachio, M.; Sarrió, D.; Moreno-Bueno, G. Novel therapies and strategies to overcome resistance to anti-HER2-targeted drugs. Cancers 2022, 14, 4543. [Google Scholar] [CrossRef]
- Swain, S.M.; Shastry, M.; Hamilton, E. Targeting HER2-positive breast cancer: Advances and future directions. Nat. Rev. Drug Discov. 2023, 22, 101–126. [Google Scholar] [CrossRef]
- Swain, S.M.; Miles, D.; Kim, S.B.; Im, Y.H.; Im, S.A.; Semiglazov, V.; Ciruelos, E.; Schneeweiss, A.; Loi, S.; Monturus, E.; et al. CLEOPATRA study group Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): End-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 519–530. [Google Scholar] [CrossRef]
- Alasmari, M.M. A Review of Margetuximab-Based Therapies in Patients with HER2-Positive Metastatic Breast Cancer. Cancers 2022, 15, 38. [Google Scholar] [CrossRef]
- Zhou, G.; Fu, S.; Zhang, Y.; Li, S.; Guo, Z.; Ouyang, D.; Ying, T.; Lu, Y.; Zhao, Q. Antibody recognition of human epidermal growth factor receptor-2 (HER2) juxtamembrane domain enhances anti-tumour response of chimeric antigen receptor (CAR)-T cells. Antibodies 2024, 13, 45. [Google Scholar] [CrossRef] [PubMed]
- Orofiamma, L.A.; Vural, D.; Antonescu, C.N. Control of cell metabolism by the epidermal growth factor receptor. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119359. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, B.; Sun, Z. Spectrum of EGFR aberrations and potential clinical implications: Insights from integrative pan-cancer analysis. Cancer Commun. 2020, 40, 43–59. [Google Scholar] [CrossRef]
- Murphrey, M.B.; Quaim, L.; Rahimi, N.; Varacallo, M.A. Biochemistry, Epidermal Growth Factor Receptor. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK482459/ (accessed on 9 March 2025).
- Li, Q.; Li, Z.; Luo, T.; Shi, H. Targeting the PI3K/AKT/mTOR and RAF/MEK/ERK pathways for cancer therapy. Mol. Biomed. 2022, 3, 47. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.L.; Feng, P.H.; Lee, K.Y.; Chen, K.Y.; Sun, W.L.; Van Hiep, N.; Luo, C.S.; Wu, S.M. The role of EREG/EGFR pathway in tumour progression. Int. J. Mol. Sci. 2021, 22, 12828. [Google Scholar] [CrossRef]
- Esteban-Villarrubia, J.; Soto-Castillo, J.J.; Pozas, J.; San Román-Gil, M.; Orejana-Martín, I.; Torres-Jiménez, J.; Carrato, A.; Alonso-Gordoa, T.; Molina-Cerrillo, J. Tyrosine kinase receptors in oncology. Int. J. Mol. Sci. 2020, 21, 8529. [Google Scholar] [CrossRef]
- Cai, W.Q.; Zeng, L.S.; Wang, L.F.; Wang, Y.Y.; Cheng, J.T.; Zhang, Y.; Han, Z.W.; Zhou, Y.; Huang, S.L.; Wang, X.W.; et al. The latest battles between EGFR monoclonal antibodies and resistant tumour cells. Front. Oncol. 2020, 10, 1249. [Google Scholar] [CrossRef]
- Uribe, M.L.; Marrocco, I.; Yarden, Y. EGFR in cancer: Signaling mechanisms, drugs, and acquired resistance. Cancers 2021, 13, 2748. [Google Scholar] [CrossRef]
- Liu, C.Y.; Lin, H.F.; Lai, W.Y.; Lin, Y.Y.; Lin, T.W.; Yang, Y.P.; Tsai, F.T.; Wang, C.L.; Luo, Y.H.; Chen, Y.M.; et al. Molecular target therapeutics of EGF-TKI and downstream signaling pathways in non-small cell lung cancers. J. Chin. Med. Assoc. 2022, 85, 409–413. [Google Scholar] [CrossRef]
- Abourehab, M.A.; Alqahtani, A.M.; Youssif, B.G.; Gouda, A.M. Globally approved EGFR inhibitors: Insights into their syntheses, target kinases, biological activities, receptor interactions, and metabolism. Molecules 2021, 26, 6677. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, S.; Wang, Y.; Zhao, Y.; Li, Q. Protein tyrosine kinase inhibitor resistance in malignant tumours: Molecular mechanisms and future perspective. Signal Transduct. Target. Ther. 2022, 7, 329. [Google Scholar] [CrossRef] [PubMed]
- Batra, U.; Biswas, B.; Prabhash, K.; Krishna, M.V. Differential clinicopathological features, treatments and outcomes in patients with Exon 19 deletion and Exon 21 L858R EGFR mutation-positive adenocarcinoma non-small-cell lung cancer. BMJ Open Respir. Res. 2023, 10, e001492. [Google Scholar] [CrossRef] [PubMed]
- Passaro, A.; Mok, T.; Peters, S.; Popat, S.; Ahn, M.J.; De Marinis, F. Recent advances on the role of EGFR tyrosine kinase inhibitors in the management of NSCLC with uncommon, non exon 20 insertions, EGFR mutations. J. Thorac. Oncol. 2021, 16, 764–773. [Google Scholar] [CrossRef]
- Ferrara, M.G.; Di Noia, V.; D’Argento, E.; Vita, E.; Damiano, P.; Cannella, A.; Ribelli, M.; Pilotto, S.; Milella, M.; Tortora, G.; et al. Oncogene-addicted non-small-cell lung cancer: Treatment opportunities and future perspectives. Cancers 2020, 12, 1196. [Google Scholar] [CrossRef]
- Graham, J.; Muhsin, M.; Kirkpatrick, P. Cetuximab. Nat. Rev. Drug Discov. 2004, 3, 549–550. [Google Scholar]
- Kast, J.; Dutta, S.; Upreti, V.V. Panitumumab: A review of clinical pharmacokinetic and pharmacology properties after over a decade of experience in patients with solid tumours. Adv. Ther. 2021, 38, 3712–3723. [Google Scholar] [CrossRef]
- Liu, T.; Jiang, S.; Teng, X.; Zhong, L.; Liu, M.; Jin, Y.; Dong, M. A comparison of panitumumab and cetuximab in the treatment of KRAS wild-type metastatic colorectal cancer: A systematic review and meta-analysis. Immunopharmacol. Immunotoxicol. 2023, 45, 1–9. [Google Scholar] [CrossRef]
- John, A.; Noronha, V.; Singh, A.; Menon, N.; Prabhash, K. Amivantamab: A narrative drug review. Cancer Res. Stat. Treat. 2023, 6, 261–271. [Google Scholar] [CrossRef]
- Papassotiriou, I.; Kapogiannatos, A.; Makatsoris, C.; Bakogeorgou, S.; Mantogiannakou, I.; Roussou, E.; Souras, G.; Liakas, D.; Sergentanis, T.N.; Gavriatopoulou, M.; et al. Efficacy and safety of amivantamab in advanced or metastatic EGFR-mutant non-small cell lung cancer: A systematic review. J. Clin. Med. 2024, 13, 5489. [Google Scholar] [CrossRef]
- Mohelnikova-Duchonova, B.; Melichar, B.; Soucek, P. FOLFOX/FOLFIRI pharmacogenetics: The call for a personalized approach in colorectal cancer therapy. World J. Gastroenterol. WJG 2014, 20, 10316. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bove, A.M.; Simone, G.; Ma, B. Molecular bases of VEGFR-2-mediated physiological function and pathological role. Front. Cell Dev. Biol. 2020, 8, 599281. [Google Scholar] [CrossRef]
- Yang, Y.; Cao, Y. The impact of VEGF on cancer metastasis and systemic disease. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2022; Volume 86, pp. 251–261. [Google Scholar]
- Al-Ostoot, F.H.; Salah, S.; Khamees, H.A.; Khanum, S.A. Tumour angiogenesis: Current challenges and therapeutic opportunities. Cancer Treat. Res. Commun. 2021, 28, 100422. [Google Scholar] [CrossRef]
- Liu, Y.; Li, H.M.; Wang, R. Effectiveness and safety of adding bevacizumab to platinum-based chemotherapy as first-line treatment for advanced non-small-cell lung cancer: A meta-analysis. Front. Med. 2021, 8, 616380. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Visseren-Grul, C.; Rizzo, M.T.; Puri, T.; Chenji, S.; Reck, M. Clinical outcomes of ramucirumab plus docetaxel in the treatment of patients with non-small cell lung cancer after immunotherapy: A systematic literature review. Front. Oncol. 2023, 13, 1247879. [Google Scholar] [CrossRef]
- Patel, S.A.; Nilsson, M.B.; Le, X.; Cascone, T.; Jain, R.K.; Heymach, J.V. Molecular mechanisms and future implications of VEGF/VEGFR in cancer therapy. Clin. Cancer Res. 2023, 29, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, Z.; Auvray, M.; Vano, Y.; Oudard, S.; Helley, D.; Mauge, L. Renal Carcinoma and Angiogenesis: Therapeutic Target and Biomarkers of Response in Current Therapies. Cancers 2022, 14, 6167. [Google Scholar] [CrossRef]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef]
- Bennouna, J.; Falchero, L.; Schott, R.; Bonnetain, F.; Coudert, M.; Ben Hadj Yahia, B.; Chouaid, C. Bevacizumab in combination with platinum-based chemotherapy in patients with advanced non-squamous non-small cell lung cancer with or without brain metastases: A French cohort study (EOLE). Oncology 2018, 94, 55–64. [Google Scholar] [CrossRef]
- Tong, Y.; Fan, X.; Liu, H.; Liang, T. Advances in Trop-2 targeted antibody-drug conjugates for breast cancer: Mechanisms, clinical applications, and future directions. Front. Immunol. 2024, 15, 1495675. [Google Scholar] [CrossRef]
- Pramanik, D. Development of Antibody-Drug Conjugates: Future Perspective Towards Solid Tumour Treatment. Anti-Cancer Agents Med. Chem. 2023, 23, 642–657. [Google Scholar] [CrossRef] [PubMed]
- Okajima, D.; Yasuda, S.; Suzuki, T.; Kitamura, M.; Yamaguchi, J.; Maejima, T.; Karibe, T.; Toki, T.; Phillips, P.; Agatsuma, T. Datopotamab deruxtecan (Dato-DXd) enhances antitumour response to PD-1/PD-L1 inhibitors in TROP2-expressing tumours in mice. Cancer Res. 2023, 83 (Suppl. 7), 2932. [Google Scholar] [CrossRef]
- Mode, D. Dato-DXd Approved in US for HR+ Breast Cancer. 2025. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-datopotamab-deruxtecan-dlnk-unresectable-or-metastatic-hr-positive-her2-negative-breast (accessed on 9 March 2025).
- Jiang, Y.; Zhou, H.; Liu, J.; Ha, W.; Xia, X.; Li, J.; Chao, T.; Xiong, H. Progress and Innovative Combination Therapies in Trop-2-Targeted ADCs. Pharmaceuticals 2024, 17, 652. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Chandley, P.; Rohatgi, S. Recent advances in the development of monoclonal antibodies and next-generation antibodies. Immunohorizons 2023, 7, 886–897. [Google Scholar] [CrossRef]
- Lou, H.; Cao, X. Antibody variable region engineering for improving cancer immunotherapy. Cancer Commun. 2022, 42, 804–827. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Dixit, S.; Srinivasan, K.; Vincent, P.D.R. Personalized cancer vaccine design using AI-powered technologies. Front. Immunol. 2024, 15, 1357217. [Google Scholar] [CrossRef]
- Raja, A.; Kasana, A.; Verma, V. Next-generation therapeutic antibodies for cancer treatment: Advancements, applications, and challenges. Mol. Biotechnol. 2024, 1–21. [Google Scholar] [CrossRef]
- Zhang, C.; Zhuang, Q.; Liu, J.; Liu, X. Synthetic biology in chimeric antigen receptor T (CAR T) cell engineering. ACS Synth. Biol. 2022, 11, 1–15. [Google Scholar] [CrossRef]
- Dent, M.; Mayer, K.L.; Verjan Garcia, N.; Guo, H.; Kajiura, H.; Fujiyama, K.; Matoba, N. Impact of glycoengineering and antidrug antibodies on the anticancer activity of a plant-made lectin-Fc fusion protein. Plant Biotechnol. J. 2022, 20, 2217–2230. [Google Scholar] [CrossRef]
- Karagiannis, S.N.; Josephs, D.H.; Bax, H.J.; Spicer, J.F. Therapeutic IgE antibodies: Harnessing a macrophage-mediated immune surveillance mechanism against cancer. Cancer Res. 2017, 77, 2779–2783. [Google Scholar] [CrossRef]
- Bergamini, A.; Ferrero, S.; Leone Roberti Maggiore, U.; Scala, C.; Pella, F.; Vellone, V.G.; Petrone, M.; Rabaiotti, E.; Cioffi, R.; Candiani, M.; et al. Folate receptor alpha antagonists in preclinical and early stage clinical development for the treatment of epithelial ovarian cancer. Expert Opin. Investig. Drugs 2016, 25, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Spicer, J.; Basu, B.; Montes, A.; Banerji, U.; Kristeleit, R.; Miller, R.; Veal, G.J.; Corrigan, C.J.; Till, S.J.; Figini, M.; et al. Safety and anti-tumour activity of the IgE antibody MOv18 in patients with advanced solid tumours expressing folate receptor-alpha: A phase I trial. Nat. Commun. 2023, 14, 4180. [Google Scholar] [CrossRef]
- Sutton, B.J.; Davies, A.M.; Bax, H.J.; Karagiannis, S.N. IgE Antibodies: From Structure to Function and Clinical Translation. Antibodies 2019, 8, 19. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L. Monoclonal antibodies in cancer therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Damián-Blanco, P.; Ahuexoteco-Sánchez, S.; Carbajal-Gallardo, A.A.; Coctecon-Chávelas, F.C.; Rodríguez-Nava, C.; Vences-Velázquez, A.; Medina-Flores, Y.; Mata-Ruíz, O.; Lloret-Sánchez, L.; Cortés-Sarabia, K. Use of monoclonal antibodies in cancer immunotherapy: Types and mechanisms of action. Boletín Médico Hosp. Infant. México 2023, 80, 153–164. [Google Scholar] [CrossRef]
- Sheykhhasan, M.; Manoochehri, H.; Dama, P. Use of CAR T-cell for acute lymphoblastic leukemia (ALL) treatment: A review study. Cancer Gene Ther. 2022, 29, 1080–1096. [Google Scholar] [CrossRef] [PubMed]
- Haslauer, T.; Greil, R.; Zaborsky, N.; Geisberger, R. CAR T-cell therapy in hematological malignancies. Int. J. Mol. Sci. 2021, 22, 8996. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Muñoz, R.J.; Grupp, S.A. CAR-T in the clinic: Drive with care. Gene Ther. 2018, 25, 157–161. [Google Scholar] [CrossRef]
- Subklewe, M.; von Bergwelt-Baildon, M.; Humpe, A. Chimeric antigen receptor T cells: A race to revolutionize cancer therapy. Transfus. Med. Hemotherapy 2019, 46, 15–24. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Khosravi, G.R.; Mostafavi, S.; Bastan, S.; Ebrahimi, N.; Gharibvand, R.S.; Eskandari, N. Immunologic tumour microenvironment modulators for turning cold tumours hot. Cancer Commun. 2024, 44, 521–553. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Rauth, S.; Aithal, A.; Kaur, S.; Ganguly, K.; Orzechowski, C.; Varshney, G.C.; Jain, M.; Batra, S.K. The current landscape of antibody-based therapies in solid malignancies. Theranostics 2021, 11, 1493. [Google Scholar] [CrossRef] [PubMed]
- Flugel, C.L.; Majzner, R.G.; Krenciute, G.; Dotti, G.; Riddell, S.R.; Wagner, D.L.; Abou-el-Enein, M. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat. Rev. Clin. Oncol. 2023, 20, 49–62. [Google Scholar] [CrossRef]
- Huang, Y.; Qin, Y.; He, Y.; Qiu, D.; Zheng, Y.; Wei, J.; Zhang, L.; Yang, D.H.; Li, Y. Advances in molecular targeted drugs in combination with CAR-T cell therapy for hematologic malignancies. Drug Resist Updat 2024, 74, 101082. [Google Scholar] [CrossRef]
- Sharma, R.; Suravarjhula, L.; Banerjee, M.; Kumar, G.; Kumar, N. Chimeric antigen receptor T-cell therapy in cancer: A critical review. Curr. Drug Res. Rev. 2023, 15, 241–261. [Google Scholar] [CrossRef]
- Ebrahimi, N.; Akbari, M.; Ghanaatian, M.; Roozbahani Moghaddam, P.; Adelian, S.; Borjian Boroujeni, M.; Yazdani, E.; Ahmadi, A.; Hamblin, M.R. Development of neoantigens: From identification in cancer cells to application in cancer vaccines. Expert Rev. Vaccines 2022, 21, 941–955. [Google Scholar] [CrossRef]
- Liu, Z.; Lei, W.; Wang, H.; Liu, X.; Fu, R. Challenges and strategies associated with CAR-T cell therapy in blood malignancies. Exp. Hematol. Oncol. 2024, 13, 22. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A. Evolving CAR-T-cell therapy for cancer treatment: From scientific discovery to cures. Cancers 2023, 16, 39. [Google Scholar] [CrossRef]
- Blair, H.A. Zenocutuzumab: First Approval. Drugs 2025, 85, 591–597. [Google Scholar] [CrossRef]
- Rais, T.; Khan, A.; Riaz, R. Elrexfio™(elranatamab-bcmm): The game-changer in treatment of multiple myeloma. Rare Tumours 2023, 15, 20363613231207483. [Google Scholar] [CrossRef]
- Shaver, J.; Horton, D.; Halford, Z. Targeting GPRC5D With Talquetamab: A New Frontier in Bispecific Antibody Therapy for Relapsed/Refractory Multiple Myeloma. Ann. Pharmacother. 2024, 59, 10600280241271192. [Google Scholar] [CrossRef] [PubMed]
- Pelosci, A. European Commission Approves Glofitamab in Relapsed/Refractory DLBCL. Cancer Netw. 2023. [Google Scholar]
- Riaz, R.; Khan, A.; Siddiqui, T. Epcoritamab-bysp (Epkinly)–A phenomenal breakthrough in the treatment of diffuse large B-cell lymphoma. Rare Tumours 2023, 15, 20363613231193566. [Google Scholar] [CrossRef]
- Parvez, S.; Prakash, K.; Soni, D. A Review on Chemistry, Mechanism, Clinical Trials Studies of ELAHERE (Mirvetuximab Soravtansine-Gynx): A Novel FDA Approved Drug to Treat Ovarian Cancer. World J. Pharm. Pharm. Sci. 2023, 12, 891–898. [Google Scholar]
- Zhou, X.; Wu, Y.; Zhu, Z.; Lu, C.; Zhang, C.; Zeng, L.; Xie, F.; Zhang, L.; Zhou, F. Mucosal immune response in biology, disease prevention and treatment. Signal Transduct. Target. Ther. 2025, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Ye, H.; Lin, T.; Chen, Z.; Xu, X.; Zhuang, J.; Yang, Y.; Chen, X.; Chen, C.; Lin, M.; et al. The association between Chlamydia pneumoniae infection and prognosis in lung cancer patients: A prospective study. BMC Infect. Dis. 2025, 25, 119. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Wang, Z.; Wei, Y.; Wang, M.; Li, K.; Chen, X.; Huang, X.; Zhou, L.; Gan, Q.; Xu, X.; et al. Dynamic changes in peripheral blood immunophenotyping and its prognostic value in cervical cancer patients undergoing immune checkpoint blockade therapy. Discov. Oncol. 2025, 16, 167. [Google Scholar] [CrossRef]
- Jackson, S.S.; Francis, J.; Pfeiffer, R.M.; Proietti, C.; Coghill, A.E.; Yu, K.J.; Sarathkumara, Y.D.; Hsu, W.-L.; Argirion, I.; Wang, C.-P.; et al. Sex differences in anti-EBV antibody responses. J. Infect. Dis. 2025, jiaf067. [Google Scholar] [CrossRef]
- van Tetering, G.; Evers, M.; Chan, C.; Stip, M.; Leusen, J. Fc Engineering Strategies to Advance IgA Antibodies as Therapeutic Agents. Antibodies 2020, 9, 70. [Google Scholar] [CrossRef]
- Meyer, S.; Leusen, J.H.; Boross, P. Regulation of complement and modulation of its activity in monoclonal antibody therapy of cancer. In MAbs; Taylor & Francis: Abingdon, UK, 2014; Volume 6, pp. 1133–1144. [Google Scholar]
- Dobó, J.; Kocsis, A.; Farkas, B.; Demeter, F.; Cervenak, L.; Gál, P. The Lectin Pathway of the Complement System-Activation, Regulation, Disease Connections and Interplay with Other (Proteolytic) Systems. Int. J. Mol. Sci. 2024, 25, 1566. [Google Scholar] [CrossRef]
- Cedzyński, M.; Świerzko, A.S. Components of the Lectin Pathway of Complement in Solid Tumour Cancers. Cancers 2022, 14, 1543. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Indication | Comments | Reference |
|---|---|---|
| Melanoma | These therapies revolutionised treatment by significantly improving survival rates | [15] |
| Non-Small Cell Lung Cancer (NSCLC) | PD-1/PD-L1 inhibitors are standard for advanced or metastatic cases | [16] |
| Head and Neck Cancer | Immune checkpoint inhibitors have shown efficacy in recurrent or metastatic disease | [17] |
| Cervical Cancer | These treatments address advanced or recurrent cervical carcinoma, particularly in PD-L1-positive patients | [18] |
| Urothelial Carcinoma | Effective for metastatic urothelial cancers, especially in patients who are ineligible for chemotherapy | [19] |
| Antibody | Remarks | Reference |
|---|---|---|
| Nivolumab | Enhances immune activity against malignant cells by blocking PD-1 on T-cells. | [36,37] |
| Pembrolizumab | Another PD-1 inhibitor with broad indications, including melanoma and lung cancer. | [38] |
| Atezolizumab | PD-L1 inhibitor used in bladder cancer and non-small cell lung cancer. | [39,40] |
| Avelumab | PD-L1 inhibitor, approved for Merkel cell carcinoma and urothelial carcinoma. | [41,42] |
| Ipilimumab | First approved CTLA-4 inhibitor, used for melanoma and in combination with PD-1 inhibitors like nivolumab for other cancers including lung and breast cancer. | [43,44] |
| Antibodies | Remarks | Reference |
|---|---|---|
| Cetuximab | “Cetuximab is a chimeric monoclonal antibody that targets the epidermal growth factor receptor (EGFR), a transmembrane protein frequently overexpressed in several cancers”. By binding to EGFR, it disrupts downstream signalling pathways involved in cell proliferation, survival, and angiogenesis. Cetuximab is primarily indicated for metastatic colorectal cancer (mCRC) and squamous cell carcinoma of the head and neck (SCCHN). Indications and dosage:
| [79] |
| Panitumumab | Panitumumab is a fully human monoclonal antibody that also targets EGFR, inhibiting ligand binding and subsequent activation of cancer-promoting pathways. It is primarily employed in the treatment of NRAS mCRC or wild-type KRAS. Being fully human, panitumumab has a reduced risk of immunogenicity compared to chimeric antibodies like cetuximab. Indications and Dosages
Side Effects Panitumumab’s adverse effects resemble those of cetuximab, including acneiform dermatitis, hypomagnesaemia, and infusion-related reactions. Rare but serious side effects include pulmonary fibrosis and severe diarrhoea. | [80,81] |
| Amivantamab | Amivantamab is a bispecific monoclonal antibody that simultaneously targets EGFR and MET, two proteins involved in oncogenic signalling. This dual action is particularly beneficial for tumours with EGFR exon 20 insertions, frequently observed in non-small cell lung cancer (NSCLC). In addition to inhibiting these pathways, amivantamab also triggers antibody-dependent cellular cytotoxicity (ADCC). Indications and Dosage
| [82] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Justiz-Vaillant, A.; Pandit, B.R.; Unakal, C.; Vuma, S.; Akpaka, P.E. A Comprehensive Review About the Use of Monoclonal Antibodies in Cancer Therapy. Antibodies 2025, 14, 35. https://doi.org/10.3390/antib14020035
Justiz-Vaillant A, Pandit BR, Unakal C, Vuma S, Akpaka PE. A Comprehensive Review About the Use of Monoclonal Antibodies in Cancer Therapy. Antibodies. 2025; 14(2):35. https://doi.org/10.3390/antib14020035
Chicago/Turabian StyleJustiz-Vaillant, Angel, Bijay Raj Pandit, Chandrashekhar Unakal, Sehlule Vuma, and Patrick Eberechi Akpaka. 2025. "A Comprehensive Review About the Use of Monoclonal Antibodies in Cancer Therapy" Antibodies 14, no. 2: 35. https://doi.org/10.3390/antib14020035
APA StyleJustiz-Vaillant, A., Pandit, B. R., Unakal, C., Vuma, S., & Akpaka, P. E. (2025). A Comprehensive Review About the Use of Monoclonal Antibodies in Cancer Therapy. Antibodies, 14(2), 35. https://doi.org/10.3390/antib14020035