
New Insights into Pathogenesis and Treatment of ANCA-Associated Vasculitis: Autoantibodies and Beyond


Abstract
1. Introduction
2. Epidemiology
Epigenetics and Environmental Factors
3. Classification and Diagnostic Criteria
4. Pathogenesis
4.1. The Role of Genetic
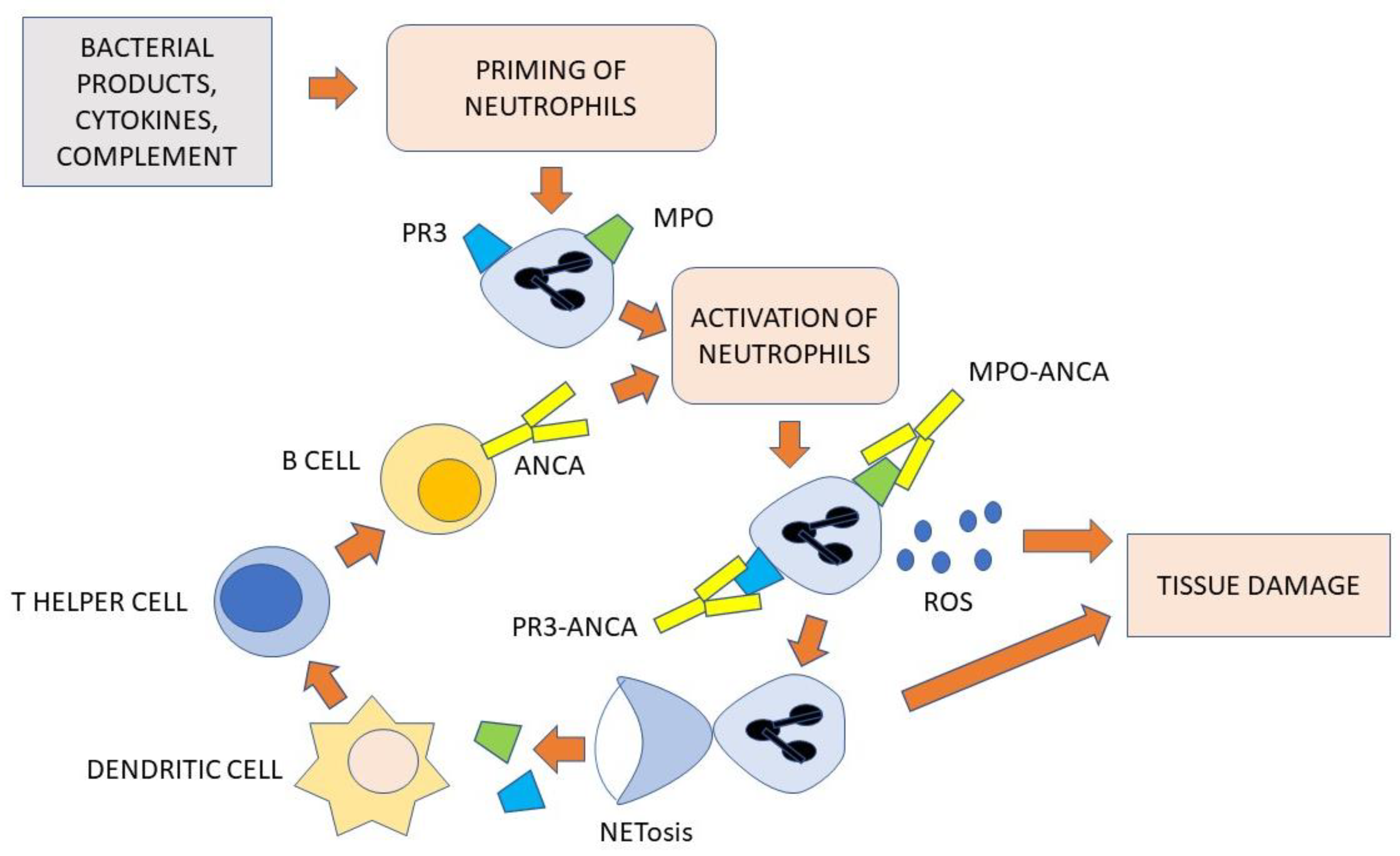
4.2. The Role of ANCA and Neutrophils
4.3. The Role of B- and T-Cells
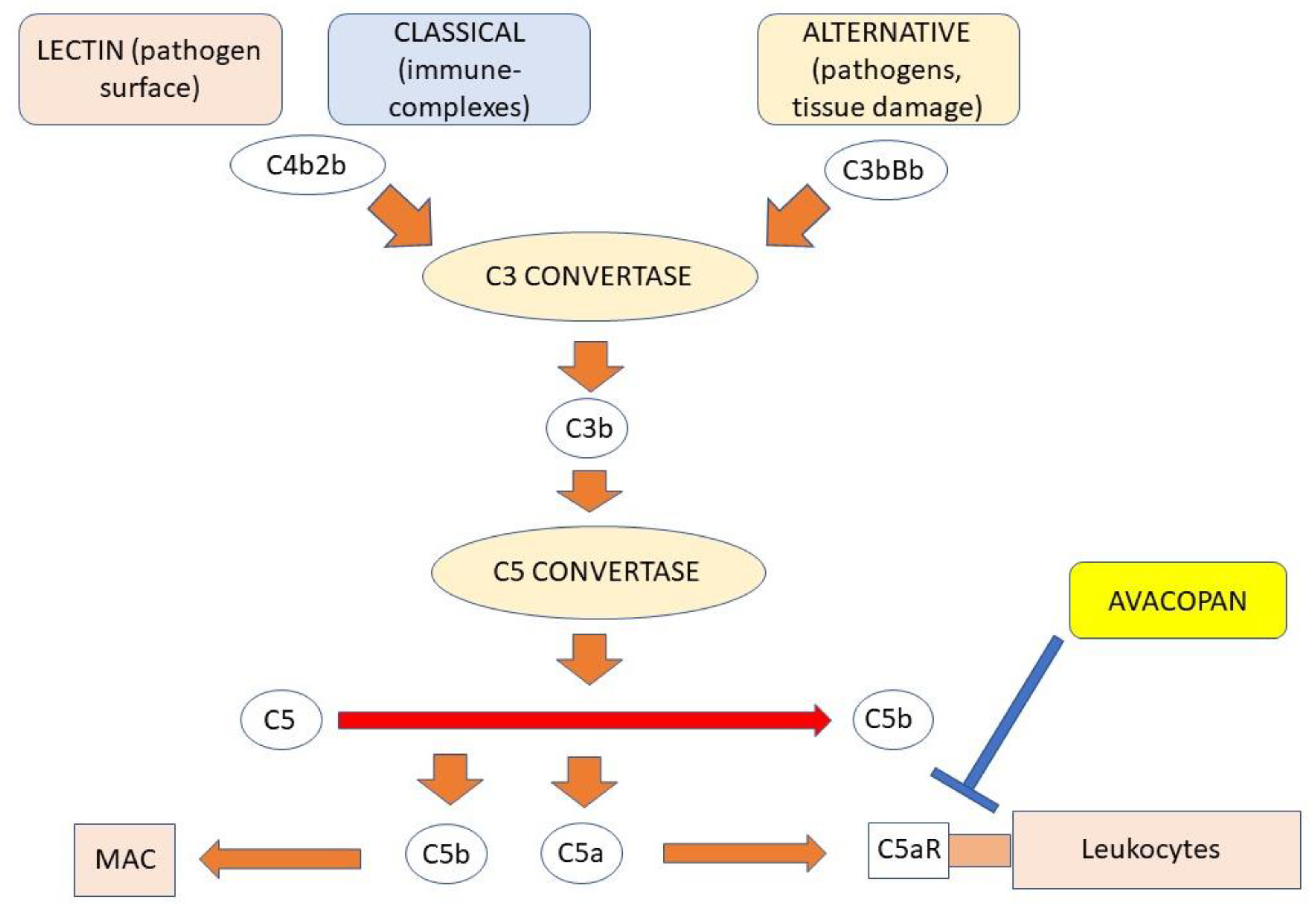
4.4. The Role of Complement
4.5. The Role of Eosinophils
5. Clinical Presentation
6. Disease Activity
7. Treatment of AAV
7.1. Treatment with Conventional Immunosuppressants
7.2. Rituximab
7.3. C5aR Antagonist Avacopan
7.4. The Blockage of Eosinophils in EGPA
8. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Jennette, J.C.; Falk, R.J. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat. Rev. Rheumatol. 2014, 10, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013, 65, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Radic, M.; Martinovic Kaliterna, D.; Radic, J. Drug-induced vasculitis: A clinical and pathological review. Neth. J. Med. 2012, 70, 12–17. [Google Scholar] [PubMed]
- Geetha, D.; Jefferson, J.A. ANCA-Associated Vasculitis: Core Curriculum 2020. Am. J. Kidney Dis. 2020, 75, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Kitching, A.R.; Anders, H.J.; Basu, N.; Brouwer, E.; Gordon, J.; Jayne, D.R.; Kullman, J.; Lyons, P.A.; Merkel, P.A.; Savage, C.O.S.; et al. ANCA-associated vasculitis. Nat. Rev. Dis. Prim. 2020, 6, 71. [Google Scholar] [CrossRef]
- Guchelaar, N.A.D.; Waling, M.M.; Adhin, A.A.; van Daele, P.L.A.; Schreurs, M.W.J.; Rombach, S.M. The value of anti-neutrophil cytoplasmic antibodies (ANCA) testing for the diagnosis of ANCA-associated vasculitis, a systematic review and meta-analysis. Autoimmun. Rev. 2021, 20, 102716. [Google Scholar] [CrossRef]
- Falk, R.J.; Hogan, S.; Carey, T.S.; Jennette, J.C. Clinical course of anti-neutrophil cytoplasmic autoantibody-associated glomerulonephritis and systemic vasculitis. The Glomerular Disease Collaborative Network. Ann. Intern. Med. 1990, 113, 656–663. [Google Scholar] [CrossRef]
- Cartin-Ceba, R.; Diaz-Caballero, L.; Al-Qadi, M.O.; Tryfon, S.; Fervenza, F.C.; Ytterberg, S.R.; Specks, U. Diffuse Alveolar Hemorrhage Secondary to Antineutrophil Cytoplasmic Antibody-Associated Vasculitis: Predictors of Respiratory Failure and Clinical Outcomes. Arthritis Rheumatol. 2016, 68, 1467–1476. [Google Scholar] [CrossRef]
- Watts, R.; Lane, S.; Hanslik, T.; Hauser, T.; Hellmich, B.; Koldingsnes, W.; Mahr, A.; Segelmark, M.; Cohen-Tervaert, J.W.; Scott, D. Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann. Rheum. Dis. 2007, 66, 222–227. [Google Scholar] [CrossRef]
- Grayson, P.C.; Ponte, C.; Suppiah, R.; Robson, J.C.; Craven, A.; Judge, A.; Khalid, S.; Hutchings, A.; Luqmani, R.A.; Watts, R.A.; et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology Classification Criteria for Eosinophilic Granulomatosis with Polyangiitis. Ann. Rheum. Dis. 2022, 81, 309–314. [Google Scholar] [CrossRef]
- Robson, J.C.; Grayson, P.C.; Ponte, C.; Suppiah, R.; Craven, A.; Judge, A.; Khalid, S.; Hutchings, A.; Watts, R.A.; Merkel, P.A.; et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology Classification Criteria for Granulomatosis with Polyangiitis. Arthritis Rheumatol. 2022, 74, 393–399. [Google Scholar] [CrossRef]
- Suppiah, R.; Robson, J.C.; Grayson, P.C.; Ponte, C.; Craven, A.; Khalid, S.; Judge, A.; Hutchings, A.; Merkel, P.A.; Luqmani, R.A.; et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology classification criteria for microscopic polyangiitis. Ann. Rheum. Dis. 2022, 81, 321–326. [Google Scholar] [CrossRef]
- Li, W.; Huang, H.; Cai, M.; Yuan, T.; Sheng, Y. Antineutrophil Cytoplasmic Antibody-Associated Vasculitis Update: Genetic Pathogenesis. Front. Immunol. 2021, 12, 624848. [Google Scholar] [CrossRef]
- Nilsen, A.T.; Karlsen, C.; Bakland, G.; Watts, R.; Luqmani, R.; Koldingsnes, W. Increasing incidence and prevalence of ANCA-associated vasculitis in Northern Norway. Rheumatology 2020, 59, 2316–2324. [Google Scholar] [CrossRef]
- Watts, R.A.; Hatemi, G.; Burns, J.C.; Mohammad, A.J. Global epidemiology of vasculitis. Nat. Rev. Rheumatol. 2022, 18, 22–34. [Google Scholar] [CrossRef]
- Mohammad, A.J.; Jacobsson, L.T.; Westman, K.W.; Sturfelt, G.; Segelmark, M. Incidence and survival rates in Wegener’s granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome and polyarteritis nodosa. Rheumatology 2009, 48, 1560–1565. [Google Scholar] [CrossRef]
- Hellmich, B.; Lamprecht, P.; Spearpoint, P.; Gotte, D.; Deichmann, A.; Buchholz, I.; Schonermark, M.P.; Rutherford, P. New insights into the epidemiology of ANCA-associated vasculitides in Germany: Results from a claims data study. Rheumatology 2021, 60, 4868–4873. [Google Scholar] [CrossRef]
- Pagnoux, C. Churg-Strauss syndrome: Evolving concepts. Discov. Med. 2010, 9, 243–252. [Google Scholar]
- Watts, R.A.; Lane, S.E.; Bentham, G.; Scott, D.G. Epidemiology of systemic vasculitis: A ten-year study in the United Kingdom. Arthritis Rheum. 2000, 43, 414–419. [Google Scholar] [CrossRef]
- Pearce, F.A.; Grainge, M.J.; Lanyon, P.C.; Watts, R.A.; Hubbard, R.B. The incidence, prevalence and mortality of granulomatosis with polyangiitis in the UK Clinical Practice Research Datalink. Rheumatology 2017, 56, 589–596. [Google Scholar] [CrossRef]
- Watts, R.A.; Lane, S.E.; Scott, D.G.; Koldingsnes, W.; Nossent, H.; Gonzalez-Gay, M.A.; Garcia-Porrua, C.; Bentham, G.A. Epidemiology of vasculitis in Europe. Ann. Rheum. Dis. 2001, 60, 1156–1157. [Google Scholar] [CrossRef]
- Pamuk, O.N.; Donmez, S.; Calayir, G.B.; Pamuk, G.E. The epidemiology of antineutrophil cytoplasmic antibody-associated vasculitis in northwestern Turkey. Clin. Rheumatol. 2016, 35, 2063–2071. [Google Scholar] [CrossRef]
- Berti, A.; Cornec, D.; Crowson, C.S.; Specks, U.; Matteson, E.L. The Epidemiology of Antineutrophil Cytoplasmic Autoantibody-Associated Vasculitis in Olmsted County, Minnesota: A Twenty-Year US Population-Based Study. Arthritis Rheumatol. 2017, 69, 2338–2350. [Google Scholar] [CrossRef]
- Gonzalez-Gay, M.A.; Garcia-Porrua, C.; Guerrero, J.; Rodriguez-Ledo, P.; Llorca, J. The epidemiology of the primary systemic vasculitides in northwest Spain: Implications of the Chapel Hill Consensus Conference definitions. Arthritis Rheum. 2003, 49, 388–393. [Google Scholar] [CrossRef]
- Salvador, F. ANCA associated vasculitis. Eur. J. Intern. Med. 2020, 74, 18–28. [Google Scholar] [CrossRef]
- Popa, E.R.; Stegeman, C.A.; Abdulahad, W.H.; van der Meer, B.; Arends, J.; Manson, W.M.; Bos, N.A.; Kallenberg, C.G.; Tervaert, J.W. Staphylococcal toxic-shock-syndrome-toxin-1 as a risk factor for disease relapse in Wegener’s granulomatosis. Rheumatology 2007, 46, 1029–1033. [Google Scholar] [CrossRef]
- Ooi, J.D.; Jiang, J.H.; Eggenhuizen, P.J.; Chua, L.L.; van Timmeren, M.; Loh, K.L.; O’Sullivan, K.M.; Gan, P.Y.; Zhong, Y.; Tsyganov, K.; et al. A plasmid-encoded peptide from Staphylococcus aureus induces anti-myeloperoxidase nephritogenic autoimmunity. Nat. Commun. 2019, 10, 3392. [Google Scholar] [CrossRef]
- Stegeman, C.A.; Tervaert, J.W.; de Jong, P.E.; Kallenberg, C.G. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener’s granulomatosis. Dutch Co-Trimoxazole Wegener Study Group. N. Engl. J. Med. 1996, 335, 16–20. [Google Scholar] [CrossRef]
- Salmela, A.; Rasmussen, N.; Tervaert, J.W.C.; Jayne, D.R.W.; Ekstrand, A.; European Vasculitis Study, G. Chronic nasal Staphylococcus aureus carriage identifies a subset of newly diagnosed granulomatosis with polyangiitis patients with high relapse rate. Rheumatology 2017, 56, 965–972. [Google Scholar] [CrossRef]
- Gomez-Puerta, J.A.; Gedmintas, L.; Costenbader, K.H. The association between silica exposure and development of ANCA-associated vasculitis: Systematic review and meta-analysis. Autoimmun. Rev. 2013, 12, 1129–1135. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Saito, A.; Ojima, Y.; Kagaya, S.; Fukami, H.; Sato, H.; Matsuda, K.; Nagasawa, T. The influence of the Great East Japan earthquake on microscopic polyangiitis: A retrospective observational study. PLoS ONE 2017, 12, e0177482. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, M.; Muso, E.; Itoh-Ihara, T.; Oyama, A.; Hashimoto, K.; Kawamura, T.; Ono, T.; Sasayama, S. Significantly high regional morbidity of MPO-ANCA-related angitis and/or nephritis with respiratory tract involvement after the 1995 great earthquake in Kobe (Japan). Am. J. Kidney Dis. 2000, 35, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, H.J.; McGettigan, B.; Chapman, P.T.; O’Donnell, J.L.; Frampton, C.; Stamp, L.K. Incidence of anti-neutrophil cytoplasmic antibody-associated vasculitis before and after the February 2011 Christchurch Earthquake. Intern. Med. J. 2017, 47, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Hunder, G.G.; Arend, W.P.; Bloch, D.A.; Calabrese, L.H.; Fauci, A.S.; Fries, J.F.; Leavitt, R.Y.; Lie, J.T.; Lightfoot, R.W., Jr.; Masi, A.T.; et al. The American College of Rheumatology 1990 criteria for the classification of vasculitis. Introduction. Arthritis Rheum. 1990, 33, 1065–1067. [Google Scholar] [CrossRef]
- Rasmussen, N.; Wiik, A. Indirect immunofluorescence examination for IgG-ANCA in sera submitted for the 1st international workshop on ANCA, 1988. APMIS Suppl. 1989, 6, 16–20. [Google Scholar]
- Schulte-Pelkum, J.; Radice, A.; Norman, G.L.; Lomicronpez Hoyos, M.; Lakos, G.; Buchner, C.; Musset, L.; Miyara, M.; Stinton, L.; Mahler, M. Novel clinical and diagnostic aspects of antineutrophil cytoplasmic antibodies. J. Immunol. Res. 2014, 2014, 185416. [Google Scholar] [CrossRef]
- Savige, J.; Gillis, D.; Benson, E.; Davies, D.; Esnault, V.; Falk, R.J.; Hagen, E.C.; Jayne, D.; Jennette, J.C.; Paspaliaris, B.; et al. International Consensus Statement on Testing and Reporting of Antineutrophil Cytoplasmic Antibodies (ANCA). Am. J. Clin. Pathol. 1999, 111, 507–513. [Google Scholar] [CrossRef]
- Lutalo, P.M.; D’Cruz, D.P. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis). J. Autoimmun. 2014, 48–49, 94–98. [Google Scholar] [CrossRef]
- Chung, S.A.; Seo, P. Microscopic polyangiitis. Rheum. Dis. Clin. N. Am. 2010, 36, 545–558. [Google Scholar] [CrossRef]
- Gioffredi, A.; Maritati, F.; Oliva, E.; Buzio, C. Eosinophilic granulomatosis with polyangiitis: An overview. Front. Immunol. 2014, 5, 549. [Google Scholar] [CrossRef]
- Yazici, H.; Tascilar, K.; Yazici, Y. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology classification criteria sets for three types of antineutrophilic cytoplasmic antibody-associated vasculitis. Curr. Opin. Rheumatol. 2023, 35, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lyons, P.A.; Rayner, T.F.; Trivedi, S.; Holle, J.U.; Watts, R.A.; Jayne, D.R.; Baslund, B.; Brenchley, P.; Bruchfeld, A.; Chaudhry, A.N.; et al. Genetically distinct subsets within ANCA-associated vasculitis. N. Engl. J. Med. 2012, 367, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Roshandel, D.; Sherva, R.; Monach, P.A.; Lu, E.Y.; Kung, T.; Carrington, K.; Zhang, S.S.; Pulit, S.L.; Ripke, S.; et al. Association of granulomatosis with polyangiitis (Wegener’s) with HLA-DPB1*04 and SEMA6A gene variants: Evidence from genome-wide analysis. Arthritis Rheum. 2013, 65, 2457–2468. [Google Scholar] [CrossRef]
- Rahmattulla, C.; Mooyaart, A.L.; van Hooven, D.; Schoones, J.W.; Bruijn, J.A.; Dekkers, O.M.; European Vasculitis Genetics, C.; Bajema, I.M. Genetic variants in ANCA-associated vasculitis: A meta-analysis. Ann. Rheum. Dis. 2016, 75, 1687–1692. [Google Scholar] [CrossRef]
- Fujimoto, S.; Watts, R.A.; Kobayashi, S.; Suzuki, K.; Jayne, D.R.; Scott, D.G.; Hashimoto, H.; Nunoi, H. Comparison of the epidemiology of anti-neutrophil cytoplasmic antibody-associated vasculitis between Japan and the U.K. Rheumatology 2011, 50, 1916–1920. [Google Scholar] [CrossRef]
- Cao, Y.; Yang, J.; Colby, K.; Hogan, S.L.; Hu, Y.; Jennette, C.E.; Berg, E.A.; Zhang, Y.; Jennette, J.C.; Falk, R.J.; et al. High basal activity of the PTPN22 gain-of-function variant blunts leukocyte responsiveness negatively affecting IL-10 production in ANCA vasculitis. PLoS ONE 2012, 7, e42783. [Google Scholar] [CrossRef]
- Relle, M.; Fohr, B.; Fasola, F.; Schwarting, A. Genetics and pathophysiology of granulomatosis with polyangiitis (GPA) and its main autoantigen proteinase 3. Mol. Cell. Probes 2016, 30, 366–373. [Google Scholar] [CrossRef]
- Ciavatta, D.J.; Yang, J.; Preston, G.A.; Badhwar, A.K.; Xiao, H.; Hewins, P.; Nester, C.M.; Pendergraft, W.F., III; Magnuson, T.R.; Jennette, J.C.; et al. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J. Clin. Investig. 2010, 120, 3209–3219. [Google Scholar] [CrossRef]
- Jones, B.E.; Yang, J.; Muthigi, A.; Hogan, S.L.; Hu, Y.; Starmer, J.; Henderson, C.D.; Poulton, C.J.; Brant, E.J.; Pendergraft, W.F., III; et al. Gene-Specific DNA Methylation Changes Predict Remission in Patients with ANCA-Associated Vasculitis. J. Am. Soc. Nephrol. 2017, 28, 1175–1187. [Google Scholar] [CrossRef]
- Xiao, H.; Heeringa, P.; Hu, P.; Liu, Z.; Zhao, M.; Aratani, Y.; Maeda, N.; Falk, R.J.; Jennette, J.C. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J. Clin. Investig. 2002, 110, 955–963. [Google Scholar] [CrossRef]
- Little, M.A.; Smyth, L.; Salama, A.D.; Mukherjee, S.; Smith, J.; Haskard, D.; Nourshargh, S.; Cook, H.T.; Pusey, C.D. Experimental autoimmune vasculitis: An animal model of anti-neutrophil cytoplasmic autoantibody-associated systemic vasculitis. Am. J. Pathol. 2009, 174, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Hellmich, B.; Csernok, E.; Trabandt, A.; Gross, W.L.; Ernst, M. Granulocyte-macrophage colony-stimulating factor (GM-CSF) but not granulocyte colony-stimulating factor (G-CSF) induces plasma membrane expression of proteinase 3 (PR3) on neutrophils in vitro. Clin. Exp. Immunol. 2000, 120, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, A.P.; Limburg, P.C.; Kallenberg, C.G. Activation, apoptosis, and clearance of neutrophils in Wegener’s granulomatosis. Ann. N. Y. Acad. Sci. 2005, 1051, 1–11. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ma, T.T.; Huang, Y.M.; Wang, C.; Zhao, M.H.; Chen, M. Coagulation and fibrinolysis index profile in patients with ANCA-associated vasculitis. PLoS ONE 2014, 9, e97843. [Google Scholar] [CrossRef]
- Claudel, S.E.; Tucker, B.M.; Kleven, D.T.; Pirkle, J.L., Jr.; Murea, M. Narrative Review of Hypercoagulability in Small-Vessel Vasculitis. Kidney Int. Rep. 2020, 5, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Kallenberg, C.G. Pathogenesis of ANCA-associated vasculitides. Ann. Rheum. Dis. 2011, 70 (Suppl. S1), i59–i63. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Hattanda, F.; Nakazawa, D.; Watanabe-Kusunoki, K.; Kusunoki, Y.; Shida, H.; Masuda, S.; Nishio, S.; Tomaru, U.; Atsumi, T.; Ishizu, A. The presence of anti-neutrophil extracellular trap antibody in patients with microscopic polyangiitis. Rheumatology 2019, 58, 1293–1298. [Google Scholar] [CrossRef]
- Wang, H.; Wang, C.; Zhao, M.H.; Chen, M. Neutrophil extracellular traps can activate alternative complement pathways. Clin. Exp. Immunol. 2015, 181, 518–527. [Google Scholar] [CrossRef]
- Nakazawa, D.; Masuda, S.; Tomaru, U.; Ishizu, A. Pathogenesis and therapeutic interventions for ANCA-associated vasculitis. Nat. Rev. Rheumatol. 2019, 15, 91–101. [Google Scholar] [CrossRef]
- Wilde, B.; Thewissen, M.; Damoiseaux, J.; van Paassen, P.; Witzke, O.; Tervaert, J.W. T cells in ANCA-associated vasculitis: What can we learn from lesional versus circulating T cells? Arthritis Res. Ther. 2010, 12, 204. [Google Scholar] [CrossRef] [PubMed]
- Hutton, H.L.; Holdsworth, S.R.; Kitching, A.R. ANCA-Associated Vasculitis: Pathogenesis, Models, and Preclinical Testing. Semin. Nephrol. 2017, 37, 418–435. [Google Scholar] [CrossRef] [PubMed]
- Hilhorst, M.; Shirai, T.; Berry, G.; Goronzy, J.J.; Weyand, C.M. T cell-macrophage interactions and granuloma formation in vasculitis. Front. Immunol. 2014, 5, 432. [Google Scholar] [CrossRef] [PubMed]
- Dolff, S.; Witzke, O.; Wilde, B. Th17 cells in renal inflammation and autoimmunity. Autoimmun. Rev. 2019, 18, 129–136. [Google Scholar] [CrossRef]
- Martinez Valenzuela, L.; Bordignon Draibe, J.; Fulladosa Oliveras, X.; Bestard Matamoros, O.; Cruzado Garrit, J.M.; Torras Ambros, J. T-lymphocyte in ANCA-associated vasculitis: What do we know? A pathophysiological and therapeutic approach. Clin. Kidney J. 2019, 12, 503–511. [Google Scholar] [CrossRef]
- Perez, C.; Prajapati, K.; Burke, B.; Plaza-Rojas, L.; Zeleznik-Le, N.J.; Guevara-Patino, J.A. NKG2D signaling certifies effector CD8 T cells for memory formation. J. Immunother. Cancer 2019, 7, 48. [Google Scholar] [CrossRef]
- Vaglio, A.; Buzio, C.; Zwerina, J. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): State of the art. Allergy 2013, 68, 261–273. [Google Scholar] [CrossRef]
- Sanders, J.S.; Stegeman, C.A.; Kallenberg, C.G. The Th1 and Th2 paradigm in ANCA-associated vasculitis. Kidney Blood Press. Res. 2003, 26, 215–220. [Google Scholar] [CrossRef]
- Reumaux, D.; Duthilleul, P.; Roos, D. Pathogenesis of diseases associated with antineutrophil cytoplasm autoantibodies. Hum. Immunol. 2004, 65, 1–12. [Google Scholar] [CrossRef]
- Popa, E.R.; Stegeman, C.A.; Kallenberg, C.G.; Tervaert, J.W. Staphylococcus aureus and Wegener’s granulomatosis. Arthritis Res. Ther. 2002, 4, 77–79. [Google Scholar] [CrossRef]
- Paroli, M.; Caccavale, R.; Fiorillo, M.T.; Spadea, L.; Gumina, S.; Candela, V.; Paroli, M.P. The Double Game Played by Th17 Cells in Infection: Host Defense and Immunopathology. Pathogens 2022, 11, 1547. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.J.; Jennette, J.C. ANCA are pathogenic--oh yes they are! J. Am. Soc. Nephrol. 2002, 13, 1977–1979. [Google Scholar] [CrossRef] [PubMed]
- Shochet, L.; Holdsworth, S.; Kitching, A.R. Animal Models of ANCA Associated Vasculitis. Front. Immunol. 2020, 11, 525. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.J.; Terrell, R.S.; Charles, L.A.; Jennette, J.C. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc. Natl. Acad. Sci. USA 1990, 87, 4115–4119. [Google Scholar] [CrossRef]
- Keogan, M.T.; Esnault, V.L.; Green, A.J.; Lockwood, C.M.; Brown, D.L. Activation of normal neutrophils by anti-neutrophil cytoplasm antibodies. Clin. Exp. Immunol. 1992, 90, 228–234. [Google Scholar] [CrossRef]
- Radford, D.J.; Savage, C.O.; Nash, G.B. Treatment of rolling neutrophils with antineutrophil cytoplasmic antibodies causes conversion to firm integrin-mediated adhesion. Arthritis Rheum. 2000, 43, 1337–1345. [Google Scholar] [CrossRef]
- Taekema-Roelvink, M.E.J.; Kooten, C.V.; Kooij, S.V.; Heemskerk, E.; Daha, M.R. Proteinase 3 enhances endothelial monocyte chemoattractant protein-1 production and induces increased adhesion of neutrophils to endothelial cells by upregulating intercellular cell adhesion molecule-1. J. Am. Soc. Nephrol. 2001, 12, 932–940. [Google Scholar] [CrossRef]
- Heeringa, P.; Brouwer, E.; Klok, P.A.; Huitema, M.G.; van den Born, J.; Weening, J.J.; Kallenberg, C.G. Autoantibodies to myeloperoxidase aggravate mild anti-glomerular-basement-membrane-mediated glomerular injury in the rat. Am. J. Pathol. 1996, 149, 1695–1706. [Google Scholar]
- Kobayashi, K.; Shibata, T.; Sugisaki, T. Aggravation of rat nephrotoxic serum nephritis by anti-myeloperoxidase antibodies. Kidney Int. 1995, 47, 454–463. [Google Scholar] [CrossRef]
- Brouwer, E.; Huitema, M.G.; Klok, P.A.; de Weerd, H.; Tervaert, J.W.; Weening, J.J.; Kallenberg, C.G. Antimyeloperoxidase-associated proliferative glomerulonephritis: An animal model. J. Exp. Med. 1993, 177, 905–914. [Google Scholar] [CrossRef]
- Tomasson, G.; Grayson, P.C.; Mahr, A.D.; Lavalley, M.; Merkel, P.A. Value of ANCA measurements during remission to predict a relapse of ANCA-associated vasculitis--a meta-analysis. Rheumatology 2012, 51, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, M.; Ye, H.; Yu, F.; Guo, X.H.; Zhao, M.H. Long-term outcomes of patients with propylthiouracil-induced anti-neutrophil cytoplasmic auto-antibody-associated vasculitis. Rheumatology 2008, 47, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Gao, Y.; Guo, X.H.; Zhao, M.H. Propylthiouracil-induced antineutrophil cytoplasmic antibody-associated vasculitis. Nat. Rev. Nephrol. 2012, 8, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Bansal, P.J.; Tobin, M.C. Neonatal microscopic polyangiitis secondary to transfer of maternal myeloperoxidase-antineutrophil cytoplasmic antibody resulting in neonatal pulmonary hemorrhage and renal involvement. Ann. Allergy Asthma Immunol. 2004, 93, 398–401. [Google Scholar] [CrossRef]
- Schlieben, D.J.; Korbet, S.M.; Kimura, R.E.; Schwartz, M.M.; Lewis, E.J. Pulmonary-renal syndrome in a newborn with placental transmission of ANCAs. Am. J. Kidney Dis. 2005, 45, 758–761. [Google Scholar] [CrossRef]
- Rovin, B.H.; Adler, S.G.; Barratt, J.; Bridoux, F.; Burdge, K.A.; Chan, T.M.; Cook, H.T.; Fervenza, F.C.; Gibson, K.L.; Glassock, R.J.; et al. Executive summary of the KDIGO 2021 Guideline for the Management of Glomerular Diseases. Kidney Int. 2021, 100, 753–779. [Google Scholar] [CrossRef]
- Stone, J.H.; Merkel, P.A.; Spiera, R.; Seo, P.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.; St Clair, E.W.; Turkiewicz, A.; Tchao, N.K.; et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N. Engl. J. Med. 2010, 363, 221–232. [Google Scholar] [CrossRef]
- Yates, M.; Watts, R.A.; Bajema, I.M.; Cid, M.C.; Crestani, B.; Hauser, T.; Hellmich, B.; Holle, J.U.; Laudien, M.; Little, M.A.; et al. EULAR/ERA-EDTA recommendations for the management of ANCA-associated vasculitis. Ann. Rheum. Dis. 2016, 75, 1583–1594. [Google Scholar] [CrossRef]
- Ruth, A.J.; Kitching, A.R.; Kwan, R.Y.; Odobasic, D.; Ooi, J.D.; Timoshanko, J.R.; Hickey, M.J.; Holdsworth, S.R. Anti-neutrophil cytoplasmic antibodies and effector CD4+ cells play nonredundant roles in anti-myeloperoxidase crescentic glomerulonephritis. J. Am. Soc. Nephrol. 2006, 17, 1940–1949. [Google Scholar] [CrossRef]
- Jennette, J.C.; Falk, R.J. B cell-mediated pathogenesis of ANCA-mediated vasculitis. Semin. Immunopathol. 2014, 36, 327–338. [Google Scholar] [CrossRef]
- Dudreuilh, C.; Fakhouri, F.; Vigneau, C.; Augusto, J.F.; Machet, M.C.; Rabot, N.; Chapal, M.; Charpy, V.; Barbet, C.; Buchler, M.; et al. The Presence of Renal IgG Deposits in Necrotizing Crescentic Glomerulonephritis Associated with ANCA Is Not Related to Worse Renal Clinical Outcomes. Kidney Dis. 2020, 6, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Fauci, A.S.; Wolff, S.M. Wegener’s granulomatosis: Studies in eighteen patients and a review of the literature. 1973. Medicine 1994, 73, 315–324. [Google Scholar] [CrossRef]
- Van Paassen, P.; Tervaert, J.W.; Heeringa, P. Mechanisms of vasculitis: How pauci-immune is ANCA-associated renal vasculitis? Nephron Exp. Nephrol. 2007, 105, e10–e16. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Schreiber, A.; Heeringa, P.; Falk, R.J.; Jennette, J.C. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am. J. Pathol. 2007, 170, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Dairaghi, D.J.; Powers, J.P.; Ertl, L.S.; Baumgart, T.; Wang, Y.; Seitz, L.C.; Penfold, M.E.; Gan, L.; Hu, P.; et al. C5a receptor (CD88) blockade protects against MPO-ANCA GN. J. Am. Soc. Nephrol. 2014, 25, 225–231. [Google Scholar] [CrossRef]
- Schreiber, A.; Xiao, H.; Jennette, J.C.; Schneider, W.; Luft, F.C.; Kettritz, R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J. Am. Soc. Nephrol. 2009, 20, 289–298. [Google Scholar] [CrossRef]
- Hao, J.; Meng, L.Q.; Xu, P.C.; Chen, M.; Zhao, M.H. p38MAPK, ERK and PI3K signaling pathways are involved in C5a-primed neutrophils for ANCA-mediated activation. PLoS ONE 2012, 7, e38317. [Google Scholar] [CrossRef]
- Wu, E.Y.; McInnis, E.A.; Boyer-Suavet, S.; Mendoza, C.E.; Aybar, L.T.; Kennedy, K.B.; Poulton, C.J.; Henderson, C.D.; Hu, Y.; Hogan, S.L.; et al. Measuring Circulating Complement Activation Products in Myeloperoxidase- and Proteinase 3-Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Rheumatol. 2019, 71, 1894–1903. [Google Scholar] [CrossRef]
- Ohlsson, S.; Holm, L.; Hansson, C.; Ohlsson, S.M.; Gunnarsson, L.; Pettersson, A.; Skattum, L. Neutrophils from ANCA-associated vasculitis patients show an increased capacity to activate the complement system via the alternative pathway after ANCA stimulation. PLoS ONE 2019, 14, e0218272. [Google Scholar] [CrossRef]
- Brilland, B.; Garnier, A.S.; Chevailler, A.; Jeannin, P.; Subra, J.F.; Augusto, J.F. Complement alternative pathway in ANCA-associated vasculitis: Two decades from bench to bedside. Autoimmun. Rev. 2020, 19, 102424. [Google Scholar] [CrossRef]
- Jayne, D. Complement inhibition in ANCA vasculitis. Nephrol. Ther. 2019, 15, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Manthey, H.D.; Woodruff, T.M.; Taylor, S.M.; Monk, P.N. Complement component 5a (C5a). Int. J. Biochem. Cell. Biol. 2009, 41, 2114–2117. [Google Scholar] [CrossRef] [PubMed]
- Raby, A.C.; Holst, B.; Davies, J.; Colmont, C.; Laumonnier, Y.; Coles, B.; Shah, S.; Hall, J.; Topley, N.; Kohl, J.; et al. TLR activation enhances C5a-induced pro-inflammatory responses by negatively modulating the second C5a receptor, C5L2. Eur. J. Immunol. 2011, 41, 2741–2752. [Google Scholar] [CrossRef]
- Hao, J.; Huang, Y.M.; Zhao, M.H.; Chen, M. The interaction between C5a and sphingosine-1-phosphate in neutrophils for antineutrophil cytoplasmic antibody mediated activation. Arthritis Res. Ther. Ther. 2014, 16, R142. [Google Scholar] [CrossRef] [PubMed]
- Kallenberg, C.G.; Heeringa, P. Complement is crucial in the pathogenesis of ANCA-associated vasculitis. Kidney Int. 2013, 83, 16–18. [Google Scholar] [CrossRef]
- Chen, S.F.; Wang, F.M.; Li, Z.Y.; Yu, F.; Chen, M.; Zhao, M.H. The functional activities of complement factor H are impaired in patients with ANCA-positive vasculitis. Clin. Immunol. 2017, 175, 41–50. [Google Scholar] [CrossRef]
- Chen, S.F.; Wang, F.M.; Li, Z.Y.; Yu, F.; Chen, M.; Zhao, M.H. Complement Factor H Inhibits Anti-Neutrophil Cytoplasmic Autoantibody-Induced Neutrophil Activation by Interacting with Neutrophils. Front. Immunol. 2018, 9, 559. [Google Scholar] [CrossRef]
- Cheng, L.; Gou, S.J.; Qiu, H.Y.; Ma, L.; Fu, P. Complement regulatory proteins in kidneys of patients with anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Clin. Exp. Immunol. 2018, 191, 116–124. [Google Scholar] [CrossRef]
- Choi, H.; Kim, Y.; Jung, S.M.; Song, J.J.; Park, Y.B.; Lee, S.W. Low serum complement 3 level is associated with severe ANCA-associated vasculitis at diagnosis. Clin. Exp. Nephrol. 2019, 23, 223–230. [Google Scholar] [CrossRef]
- Garcia, L.; Pena, C.E.; Maldonado, R.A.; Costi, C.; Mamberti, M.; Martins, E.; Garcia, M.A. Increased renal damage in hypocomplementemic patients with ANCA-associated vasculitis: Retrospective cohort study. Clin. Rheumatol. 2019, 38, 2819–2824. [Google Scholar] [CrossRef]
- Nguyen, Y.; Guillevin, L. Eosinophilic Granulomatosis with Polyangiitis (Churg-Strauss). Semin. Respir. Crit. Care Med. 2018, 39, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Sinico, R.A.; Di Toma, L.; Maggiore, U.; Bottero, P.; Radice, A.; Tosoni, C.; Grasselli, C.; Pavone, L.; Gregorini, G.; Monti, S.; et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis Rheum. 2005, 52, 2926–2935. [Google Scholar] [CrossRef] [PubMed]
- Comarmond, C.; Cacoub, P. Granulomatosis with polyangiitis (Wegener): Clinical aspects and treatment. Autoimmun. Rev. 2014, 13, 1121–1125. [Google Scholar] [CrossRef]
- Moosig, F.; Bremer, J.P.; Hellmich, B.; Holle, J.U.; Holl-Ulrich, K.; Laudien, M.; Matthis, C.; Metzler, C.; Nolle, B.; Richardt, G.; et al. A vasculitis centre based management strategy leads to improved outcome in eosinophilic granulomatosis and polyangiitis (Churg-Strauss, EGPA): Monocentric experiences in 150 patients. Ann. Rheum. Dis. 2013, 72, 1011–1017. [Google Scholar] [CrossRef]
- Valent, P.; Klion, A.D.; Horny, H.P.; Roufosse, F.; Gotlib, J.; Weller, P.F.; Hellmann, A.; Metzgeroth, G.; Leiferman, K.M.; Arock, M.; et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J. Allergy Clin. Immunol. 2012, 130, 607–612.e9. [Google Scholar] [CrossRef]
- Trivioli, G.; Terrier, B.; Vaglio, A. Eosinophilic granulomatosis with polyangiitis: Understanding the disease and its management. Rheumatology 2020, 59 (Suppl. S3), iii84–iii94. [Google Scholar] [CrossRef]
- Kiene, M.; Csernok, E.; Muller, A.; Metzler, C.; Trabandt, A.; Gross, W.L. Elevated interleukin-4 and interleukin-13 production by T cell lines from patients with Churg-Strauss syndrome. Arthritis Rheum. 2001, 44, 469–473. [Google Scholar] [CrossRef]
- Tsurikisawa, N.; Saito, H.; Oshikata, C.; Tsuburai, T.; Akiyama, K. Decreases in the numbers of peripheral blood regulatory T cells, and increases in the levels of memory and activated B cells, in patients with active eosinophilic granulomatosis and polyangiitis. J. Clin. Immunol. 2013, 33, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Emmi, G.; Silvestri, E.; Marconi, R.; Carrai, V.; Fanelli, T.; Zucchini, P.; Marasca, R.; Vannucchi, A.M.; Emmi, L.; Prisco, D.; et al. First report of FIP1L1-PDGFRalpha-positive eosinophilic granulomatosis with polyangiitis. Rheumatolology 2015, 54, 1751–1753. [Google Scholar] [CrossRef]
- Polzer, K.; Karonitsch, T.; Neumann, T.; Eger, G.; Haberler, C.; Soleiman, A.; Hellmich, B.; Csernok, E.; Distler, J.; Manger, B.; et al. Eotaxin-3 is involved in Churg-Strauss syndrome--a serum marker closely correlating with disease activity. Rheumatolology 2008, 47, 804–808. [Google Scholar] [CrossRef]
- Terrier, B.; Bieche, I.; Maisonobe, T.; Laurendeau, I.; Rosenzwajg, M.; Kahn, J.E.; Diemert, M.C.; Musset, L.; Vidaud, M.; Sene, D.; et al. Interleukin-25: A cytokine linking eosinophils and adaptive immunity in Churg-Strauss syndrome. Blood 2010, 116, 4523–4531. [Google Scholar] [CrossRef] [PubMed]
- Jakiela, B.; Sanak, M.; Szczeklik, W.; Sokolowska, B.; Plutecka, H.; Mastalerz, L.; Musial, J.; Szczeklik, A. Both Th2 and Th17 responses are involved in the pathogenesis of Churg-Strauss syndrome. Clin. Exp. Rheumatol. 2011, 29 (Suppl. S64), S23–S34. [Google Scholar] [PubMed]
- Yates, M.; Watts, R. ANCA-associated vasculitis. Clin. Med. (Lond.) 2017, 17, 60–64. [Google Scholar] [CrossRef]
- Rowaiye, O.O.; Kusztal, M.; Klinger, M. The kidneys and ANCA-associated vasculitis: From pathogenesis to diagnosis. Clin. Kidney J. 2015, 8, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Said, M.S. Upper respiratory tract symptoms, renal involvement and vasculitis: A case report and review of wegener granulomatosis. J. Clin. Med. Res. 2010, 2, 189–193. [Google Scholar]
- Parra-Garcia, G.D.; Callejas-Rubio, J.L.; Rios-Fernandez, R.; Sainz-Quevedo, M.; Ortego-Centeno, N. Otolaryngologic manifestations of systemic vasculitis. Acta Otorrinolaringol. Esp. 2012, 63, 303–310. [Google Scholar] [CrossRef]
- Alba, M.A.; Flores-Suarez, L.F.; Henderson, A.G.; Xiao, H.; Hu, P.; Nachman, P.H.; Falk, R.J.; Charles Jennette, J. Interstital lung disease in ANCA vasculitis. Autoimmun. Rev. 2017, 16, 722–729. [Google Scholar] [CrossRef]
- Watkins, A.S.; Kempen, J.H.; Choi, D.; Liesegang, T.L.; Pujari, S.S.; Newcomb, C.; Nussenblatt, R.B.; Rosenbaum, J.T.; Thorne, J.E.; Foster, C.S.; et al. Ocular disease in patients with ANCA-positive vasculitis. J. Ocul. Biol. Dis. Infor. 2009, 3, 12–19. [Google Scholar] [CrossRef]
- Demirkesen, C. Approach to cutaneous vasculitides with special emphasis on small vessel vasculitis: Histopathology and direct immunofluorescence. Curr. Opin. Rheumatol. 2017, 29, 39–44. [Google Scholar] [CrossRef]
- Wludarczyk, A.; Szczeklik, W. Neurological manifestations in ANCA-associated vasculitis—Assessment and treatment. Expert Rev. Neurother. 2016, 16, 861–863. [Google Scholar] [CrossRef]
- Storesund, B.; Gran, J.T.; Koldingsnes, W. Severe intestinal involvement in Wegener’s granulomatosis: Report of two cases and review of the literature. Br. J. Rheumatol. 1998, 37, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Berti, A.; Matteson, E.L.; Crowson, C.S.; Specks, U.; Cornec, D. Risk of Cardiovascular Disease and Venous Thromboembolism among Patients with Incident ANCA-Associated Vasculitis: A 20-Year Population-Based Cohort Study. Mayo Clin. Proc. 2018, 93, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H. Wegener’s Granulomatosis Etanercept Trial Research G: Limited versus severe Wegener’s granulomatosis: Baseline data on patients in the Wegener’s granulomatosis etanercept trial. Arthritis Rheum. 2003, 48, 2299–2309. [Google Scholar]
- Sinico, R.A.; Bottero, P. Churg-Strauss angiitis. Best Pract. Res. Clin. Rheumatol. 2009, 23, 355–366. [Google Scholar] [CrossRef]
- Keogh, K.A.; Specks, U. Churg-Strauss syndrome: Clinical presentation, antineutrophil cytoplasmic antibodies, and leukotriene receptor antagonists. Am. J. Med. 2003, 115, 284–290. [Google Scholar] [CrossRef]
- Mukhtyar, C.; Lee, R.; Brown, D.; Carruthers, D.; Dasgupta, B.; Dubey, S.; Flossmann, O.; Hall, C.; Hollywood, J.; Jayne, D.; et al. Modification and validation of the Birmingham Vasculitis Activity Score (version 3). Ann. Rheum. Dis. 2009, 68, 1827–1832. [Google Scholar] [CrossRef] [PubMed]
- Guillevin, L.; Pagnoux, C.; Seror, R.; Mahr, A.; Mouthon, L.; Toumelin, P.L.; French Vasculitis Study, G. The Five-Factor Score revisited: Assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine 2011, 90, 19–27. [Google Scholar] [CrossRef]
- Exley, A.R.; Bacon, P.A.; Luqmani, R.A.; Kitas, G.D.; Gordon, C.; Savage, C.O.; Adu, D. Development and initial validation of the Vasculitis Damage Index for the standardized clinical assessment of damage in the systemic vasculitides. Arthritis Rheum. 1997, 40, 371–380. [Google Scholar] [CrossRef]
- Chung, S.A.; Langford, C.A.; Maz, M.; Abril, A.; Gorelik, M.; Guyatt, G.; Archer, A.M.; Conn, D.L.; Full, K.A.; Grayson, P.C.; et al. 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the Management of Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Rheumatol. 2021, 73, 1366–1383. [Google Scholar] [CrossRef]
- Cupps, T.R.; Edgar, L.C.; Fauci, A.S. Suppression of human B lymphocyte function by cyclophosphamide. J. Immunol. 1982, 128, 2453–2457. [Google Scholar] [CrossRef]
- Reinhold-Keller, E.; Beuge, N.; Latza, U.; de Groot, K.; Rudert, H.; Nolle, B.; Heller, M.; Gross, W.L. An interdisciplinary approach to the care of patients with Wegener’s granulomatosis: Long-term outcome in 155 patients. Arthritis Rheum. 2000, 43, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, G.S.; Kerr, G.S.; Leavitt, R.Y.; Hallahan, C.W.; Lebovics, R.S.; Travis, W.D.; Rottem, M.; Fauci, A.S. Wegener granulomatosis: An analysis of 158 patients. Ann. Intern. Med. 1992, 116, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Pagnoux, C.; Mahr, A.; Hamidou, M.A.; Boffa, J.J.; Ruivard, M.; Ducroix, J.P.; Kyndt, X.; Lifermann, F.; Papo, T.; Lambert, M.; et al. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N. Engl. J. Med. 2008, 359, 2790–2803. [Google Scholar] [CrossRef] [PubMed]
- Von Borstel, A.; Abdulahad, W.H.; Dekkema, G.; Rutgers, A.; Stegeman, C.A.; Veldman, J.; Heeringa, P.; Sanders, J.S. Mycophenolic acid and 6-mercaptopurine both inhibit B-cell proliferation in granulomatosis with polyangiitis patients, whereas only mycophenolic acid inhibits B-cell IL-6 production. PLoS ONE 2020, 15, e0235743. [Google Scholar] [CrossRef] [PubMed]
- Raffray, L.; Guillevin, L. Rituximab treatment of ANCA-associated vasculitis. Expert Opin. Biol. Ther. 2020, 20, 899–910. [Google Scholar] [CrossRef]
- Hofmann, K.; Clauder, A.K.; Manz, R.A. Targeting B Cells and Plasma Cells in Autoimmune Diseases. Front. Immunol. 2018, 9, 835. [Google Scholar] [CrossRef]
- Jones, R.B.; Tervaert, J.W.; Hauser, T.; Luqmani, R.; Morgan, M.D.; Peh, C.A.; Savage, C.O.; Segelmark, M.; Tesar, V.; van Paassen, P.; et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N. Engl. J. Med. 2010, 363, 211–220. [Google Scholar] [CrossRef]
- Unizony, S.; Villarreal, M.; Miloslavsky, E.M.; Lu, N.; Merkel, P.A.; Spiera, R.; Seo, P.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.M.; et al. Clinical outcomes of treatment of anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis based on ANCA type. Ann. Rheum. Dis. 2016, 75, 1166–1169. [Google Scholar] [CrossRef]
- Rovin, B.H.; Caster, D.J.; Cattran, D.C.; Gibson, K.L.; Hogan, J.J.; Moeller, M.J.; Roccatello, D.; Cheung, M.; Wheeler, D.C.; Winkelmayer, W.C.; et al. Management and treatment of glomerular diseases (part 2): Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2019, 95, 281–295. [Google Scholar] [CrossRef]
- McClure, M.; Gopaluni, S.; Jayne, D.; Jones, R. B cell therapy in ANCA-associated vasculitis: Current and emerging treatment options. Nat. Rev. Rheumatol. 2018, 14, 580–591. [Google Scholar] [CrossRef]
- Roberts, D.M.; Jones, R.B.; Smith, R.M.; Alberici, F.; Kumaratne, D.S.; Burns, S.; Jayne, D.R. Rituximab-associated hypogammaglobulinemia: Incidence, predictors and outcomes in patients with multi-system autoimmune disease. J. Autoimmun. 2015, 57, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Tieu, J.; Smith, R.; Basu, N.; Brogan, P.; D’Cruz, D.; Dhaun, N.; Flossmann, O.; Harper, L.; Jones, R.B.; Lanyon, P.C.; et al. Rituximab for maintenance of remission in ANCA-associated vasculitis: Expert consensus guidelines. Rheumatology 2020, 59, e24–e32. [Google Scholar] [CrossRef] [PubMed]
- Zonozi, R.; Wallace, Z.S.; Laliberte, K.; Huizenga, N.R.; Rosenthal, J.M.; Rhee, E.P.; Cortazar, F.B.; Niles, J.L. Incidence, Clinical Features, and Outcomes of Late-Onset Neutropenia from Rituximab for Autoimmune Disease. Arthritis Rheumatol. 2021, 73, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and cancer: Golden anniversary. Nat. Rev. Clin. Oncol. 2009, 6, 638–647. [Google Scholar] [CrossRef]
- Walsh, M.; Merkel, P.A.; Jayne, D.R.W. Plasma Exchange and Glucocorticoids in Severe ANCA-Associated Vasculitis. Reply. N. Engl. J. Med. 2020, 382, 2169. [Google Scholar] [CrossRef]
- Guillevin, L.; Pagnoux, C.; Karras, A.; Khouatra, C.; Aumaitre, O.; Cohen, P.; Maurier, F.; Decaux, O.; Ninet, J.; Gobert, P.; et al. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N. Engl. J. Med. 2014, 371, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, A.J.; Hot, A.; Arndt, F.; Moosig, F.; Guerry, M.J.; Amudala, N.; Smith, R.; Sivasothy, P.; Guillevin, L.; Merkel, P.A.; et al. Rituximab for the treatment of eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Ann. Rheum. Dis. 2016, 75, 396–401. [Google Scholar] [CrossRef]
- Teixeira, V.; Mohammad, A.J.; Jones, R.B.; Smith, R.; Jayne, D. Efficacy and safety of rituximab in the treatment of eosinophilic granulomatosis with polyangiitis. RMD Open 2019, 5, e000905. [Google Scholar] [CrossRef]
- Emmi, G.; Rossi, G.M.; Urban, M.L.; Silvestri, E.; Prisco, D.; Goldoni, M.; Vaglio, A. Scheduled rituximab maintenance reduces relapse rate in eosinophilic granulomatosis with polyangiitis. Ann. Rheum. Dis. 2018, 77, 952–954. [Google Scholar] [CrossRef]
- Thiel, J.; Troilo, A.; Salzer, U.; Schleyer, T.; Halmschlag, K.; Rizzi, M.; Frede, N.; Venhoff, A.; Voll, R.E.; Venhoff, N. Rituximab as Induction Therapy in Eosinophilic Granulomatosis with Polyangiitis Refractory to Conventional Immunosuppressive Treatment: A 36-Month Follow-Up Analysis. J. Allergy Clin. Immunol. Pract. 2017, 5, 1556–1563. [Google Scholar] [CrossRef]
- Menditto, V.G.; Rossetti, G.; Olivari, D.; Angeletti, A.; Rocchi, M.; Gabrielli, A.; Pomponio, G. Rituximab for eosinophilic granulomatosis with polyangiitis: A systematic review of observational studies. Rheumatology 2021, 60, 1640–1650. [Google Scholar] [CrossRef]
- Akiyama, M.; Kaneko, Y.; Takeuchi, T. Rituximab for the treatment of eosinophilic granulomatosis with polyangiitis: A systematic literature review. Autoimmun. Rev. 2021, 20, 102737. [Google Scholar] [CrossRef]
- Canzian, A.; Venhoff, N.; Urban, M.L.; Sartorelli, S.; Ruppert, A.M.; Groh, M.; Girszyn, N.; Taille, C.; Maurier, F.; Cottin, V.; et al. Use of Biologics to Treat Relapsing and/or Refractory Eosinophilic Granulomatosis with Polyangiitis: Data from a European Collaborative Study. Arthritis Rheumatol. 2021, 73, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Terrier, B.; de Moreuil, C.; Bonnotte, B. Rituximab versus conventional therapeutic strategy for remission induction in eosinophilic granulomatosis with polyangiitis: A double-blind, randomized, controlled trial. Arthritis Rheum. 2021, 73 (Suppl. S10). [Google Scholar]
- Wetsel, R.A. Expression of the complement C5a anaphylatoxin receptor (C5aR) on non-myeloid cells. Immunol. Lett. 1995, 44, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Bekker, P.; Dairaghi, D.; Seitz, L.; Leleti, M.; Wang, Y.; Ertl, L.; Baumgart, T.; Shugarts, S.; Lohr, L.; Dang, T.; et al. Characterization of Pharmacologic and Pharmacokinetic Properties of CCX168, a Potent and Selective Orally Administered Complement 5a Receptor Inhibitor, Based on Preclinical Evaluation and Randomized Phase 1 Clinical Study. PLoS ONE 2016, 11, e0164646. [Google Scholar] [CrossRef]
- Jayne, D.R.W.; Bruchfeld, A.N.; Harper, L.; Schaier, M.; Venning, M.C.; Hamilton, P.; Burst, V.; Grundmann, F.; Jadoul, M.; Szombati, I.; et al. Randomized Trial of C5a Receptor Inhibitor Avacopan in ANCA-Associated Vasculitis. J. Am. Soc. Nephrol. 2017, 28, 2756–2767. [Google Scholar] [CrossRef]
- Merkel, P.A.; Niles, J.; Jimenez, R.; Spiera, R.F.; Rovin, B.H.; Bomback, A.; Pagnoux, C.; Potarca, A.; Schall, T.J.; Bekker, P.; et al. Adjunctive Treatment with Avacopan, an Oral C5a Receptor Inhibitor, in Patients with Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. ACR Open Rheumatol. 2020, 2, 662–671. [Google Scholar] [CrossRef]
- Jayne, D.R.W.; Merkel, P.A.; Bekker, P. Avacopan for the Treatment of ANCA-Associated Vasculitis. Reply. N. Engl. J. Med. 2021, 384, e81. [Google Scholar] [CrossRef]
- Kitamura, F.; Yamaguchi, M.; Nishimura, M.; Katsuno, T.; Ito, M.; Sugiyama, H.; Iwagaitsu, S.; Nobata, H.; Kinashi, H.; Ishimoto, T.; et al. Anti-neutrophil cytoplasmic antibody-associated vasculitis complicated by thrombotic microangiopathy with posterior reversible encephalopathy syndrome successfully treated with eculizumab: A case report. Mod. Rheumatol. Case Rep. 2022, 6, 254–259. [Google Scholar] [CrossRef]
- Manenti, L.; Urban, M.L.; Maritati, F.; Galetti, M.; Vaglio, A. Complement blockade in ANCA-associated vasculitis: An index case, current concepts and future perspectives. Intern. Emerg. Med. 2017, 12, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Ribes, D.; Belliere, J.; Piedrafita, A.; Faguer, S. Glucocorticoid-free induction regimen in severe ANCA-associated vasculitis using a combination of rituximab and eculizumab. Rheumatololgy 2019, 58, 2335–2337. [Google Scholar] [CrossRef] [PubMed]
- Riedl, M.A.; Grivcheva-Panovska, V.; Moldovan, D.; Baker, J.; Yang, W.H.; Giannetti, B.M.; Reshef, A.; Andrejevic, S.; Lockey, R.F.; Hakl, R.; et al. Recombinant human C1 esterase inhibitor for prophylaxis of hereditary angio-oedema: A phase 2, multicentre, randomised, double-blind, placebo-controlled crossover trial. Lancet 2017, 390, 1595–1602. [Google Scholar] [CrossRef] [PubMed]
- Molfino, N.A.; Gossage, D.; Kolbeck, R.; Parker, J.M.; Geba, G.P. Molecular and clinical rationale for therapeutic targeting of interleukin-5 and its receptor. Clin. Exp. Allergy 2012, 42, 712–737. [Google Scholar] [CrossRef]
- Wechsler, M.E.; Akuthota, P.; Jayne, D.; Khoury, P.; Klion, A.; Langford, C.A.; Merkel, P.A.; Moosig, F.; Specks, U.; Cid, M.C.; et al. Mepolizumab or Placebo for Eosinophilic Granulomatosis with Polyangiitis. N. Engl. J. Med. 2017, 376, 1921–1932. [Google Scholar] [CrossRef] [PubMed]
- Tsurikisawa, N.; Oshikata, C.; Watanabe, M.; Fukuda, N.; Yamaguchi, T.; Kiyohara, H.; Kaneko, T. Clinical Features of Patients with Active Eosinophilic Granulomatosis with Polyangiitis Successfully Treated with Mepolizumab. Int. Arch. Allergy Immunol. 2021, 182, 744–756. [Google Scholar] [CrossRef]
- Rios-Garces, R.; Prieto-Gonzalez, S.; Hernandez-Rodriguez, J.; Arismendi, E.; Alobid, I.; Penatti, A.E.; Cid, M.C.; Espigol-Frigole, G. Response to mepolizumab according to disease manifestations in patients with eosinophilic granulomatosis with polyangiitis. Eur. J. Intern. Med. 2022, 95, 61–66. [Google Scholar] [CrossRef]
- Caminati, M.; Crisafulli, E.; Lunardi, C.; Micheletto, C.; Festi, G.; Maule, M.; Giollo, A.; Orsolini, G.; Senna, G. Mepolizumab 100 mg in severe asthmatic patients with EGPA in remission phase. J. Allergy Clin. Immunol. Pract. 2021, 9, 1386–1388. [Google Scholar] [CrossRef]
- Manka, L.A.; Guntur, V.P.; Denson, J.L.; Dunn, R.M.; Dollin, Y.T.; Strand, M.J.; Wechsler, M.E. Efficacy and safety of reslizumab in the treatment of eosinophilic granulomatosis with polyangiitis. Ann. Allergy Asthma Immunol. 2021, 126, 696–701.e1. [Google Scholar] [CrossRef]
- Guntur, V.P.; Manka, L.A.; Denson, J.L.; Dunn, R.M.; Dollin, Y.T.; Gill, M.; Kolakowski, C.; Strand, M.J.; Wechsler, M.E. Benralizumab as a Steroid-Sparing Treatment Option in Eosinophilic Granulomatosis with Polyangiitis. J. Allergy Clin. Immunol. Pract. 2021, 9, 1186–1193.e1. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Target | Drug | Trial | Primary Endpoint Results |
|---|---|---|---|
| B cells | RTX | RAVE | Non inferiority to oral CYC for remission induction, superior for relapsing or PR3-ANCA patients |
| B cells | RTX | RITUXVAS | Non inferiority to CYC in pulses for remission induction |
| B cells | RTX | MAINRISTAN | Superiority to AZA for maintenance of remission |
| B cells | RTX | MAINRITSAN 2 | No difference between standard and customized infusion based on B-cell count for relapse rate |
| B cells | RTX | REOVAS | Non inferiority to conventional therapy for remission (CYC/CS) in EGPA |
| C5aR | AVACOPAN | CLASSIC | Safe and effective at day 85 |
| C5aR | AVACOPAN | ADVOCATE | Non inferiority to CS for remission induction |
| C5aR | AVACOPAN | CLEAR | Non inferiority to CS for remission induction |
| IL-5 | MEPO | MIRRA | Non inferiority to placebo for relapsing or refractory EPGA |
| Drug | AAV | Specifications |
|---|---|---|
| Rituximab | GPA and MPA | In adult and pediatric patients 2 years of age and older in combination with glucocorticoids. |
| Avacopan | GPA and MPA | As an adjunctive treatment of adult patients with severe active GPA and MPA in combination with standard therapy. |
| Mepolizumab | EGPA | Adult patients with EGPA. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paroli, M.; Gioia, C.; Accapezzato, D. New Insights into Pathogenesis and Treatment of ANCA-Associated Vasculitis: Autoantibodies and Beyond. Antibodies 2023, 12, 25. https://doi.org/10.3390/antib12010025
Paroli M, Gioia C, Accapezzato D. New Insights into Pathogenesis and Treatment of ANCA-Associated Vasculitis: Autoantibodies and Beyond. Antibodies. 2023; 12(1):25. https://doi.org/10.3390/antib12010025
Chicago/Turabian StyleParoli, Marino, Chiara Gioia, and Daniele Accapezzato. 2023. "New Insights into Pathogenesis and Treatment of ANCA-Associated Vasculitis: Autoantibodies and Beyond" Antibodies 12, no. 1: 25. https://doi.org/10.3390/antib12010025
APA StyleParoli, M., Gioia, C., & Accapezzato, D. (2023). New Insights into Pathogenesis and Treatment of ANCA-Associated Vasculitis: Autoantibodies and Beyond. Antibodies, 12(1), 25. https://doi.org/10.3390/antib12010025