
Rational Design and In Vivo Characterization of mRNA-Encoded Broadly Neutralizing Antibody Combinations against HIV-1

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Recombinant IgG and scFv-Fc Expression, Purification, and Characterization
2.2. mRNA-LNP Production and Formulation
2.3. Animal Experiments
2.4. Quantification of IgG in Animal Sera
2.5. TZM-bl In Vitro Neutralization Assay
2.6. Pharmacometric Analysis
3. Results
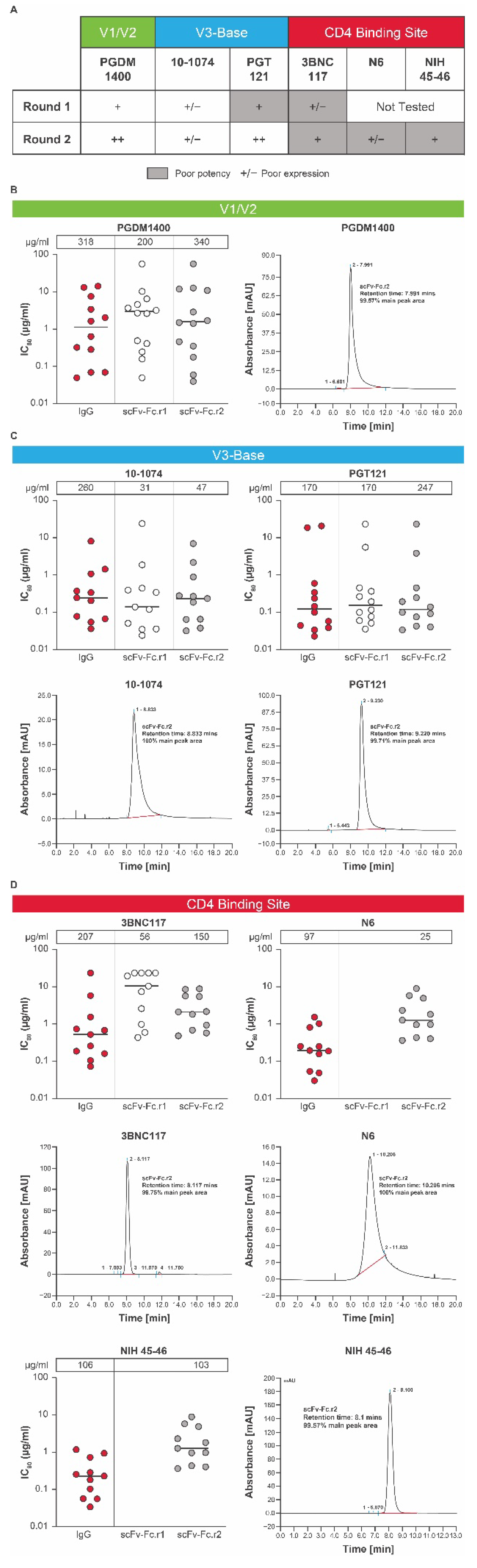
3.1. Design of mRNA-Encoded Antibody Combinations Using Single-Chain Conversion
3.2. Expression of scFv-Fc Antibodies
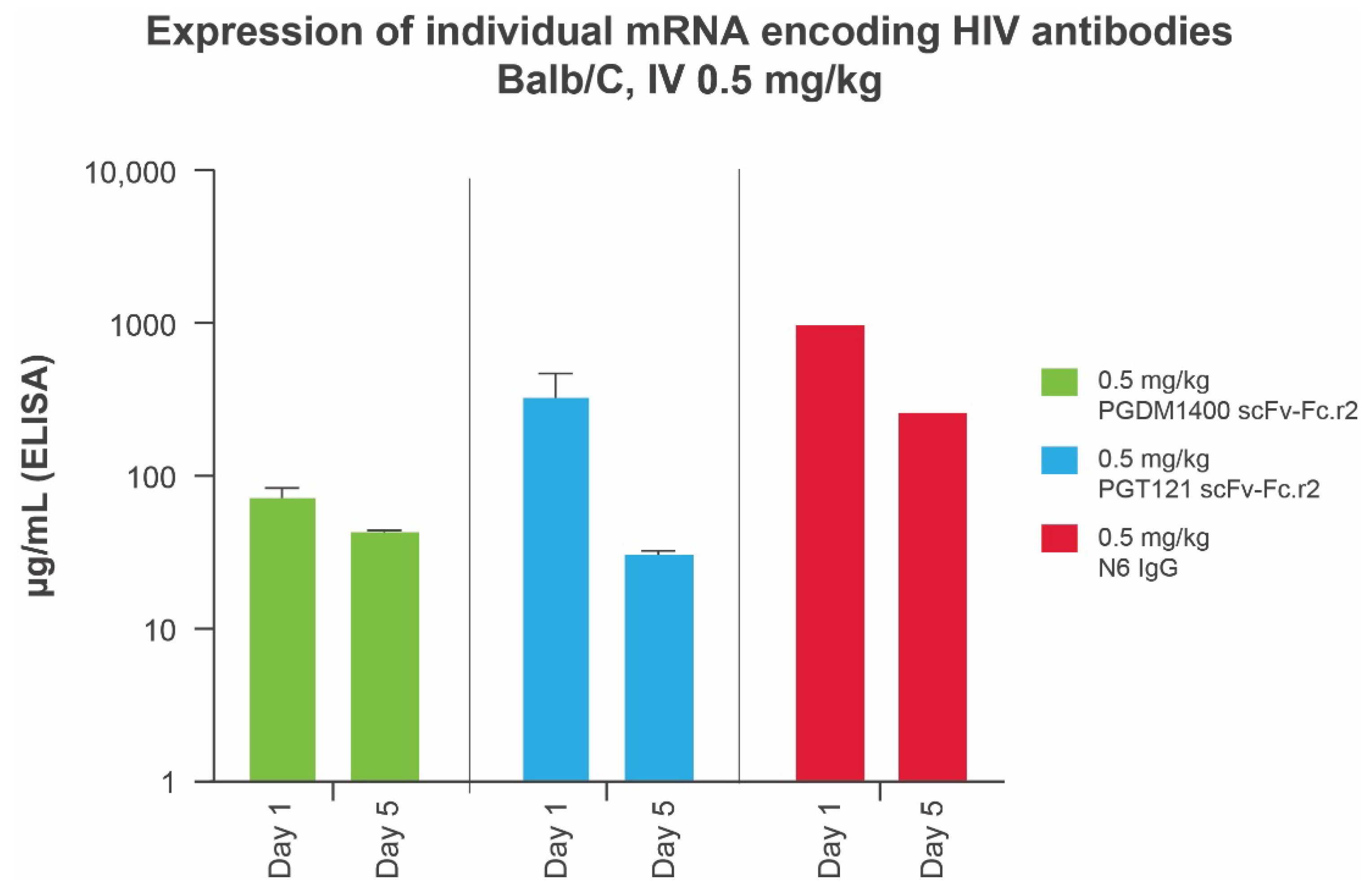
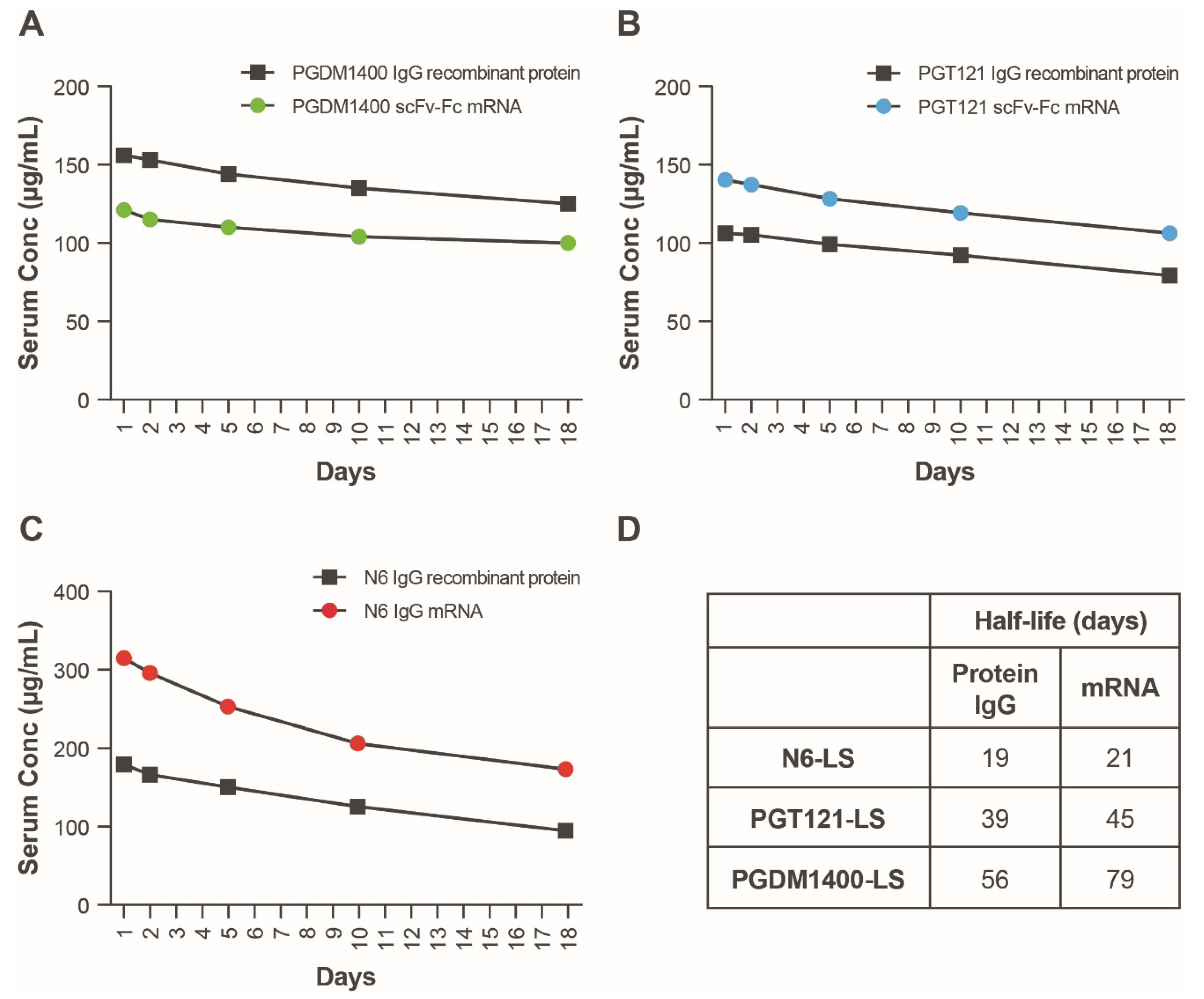
3.3. In Vivo Expression of Individual Components of Candidate HIV mRNA-LNP bnAbs
3.4. Selection of a Lead HIV bnAb mRNA-LNP Combination
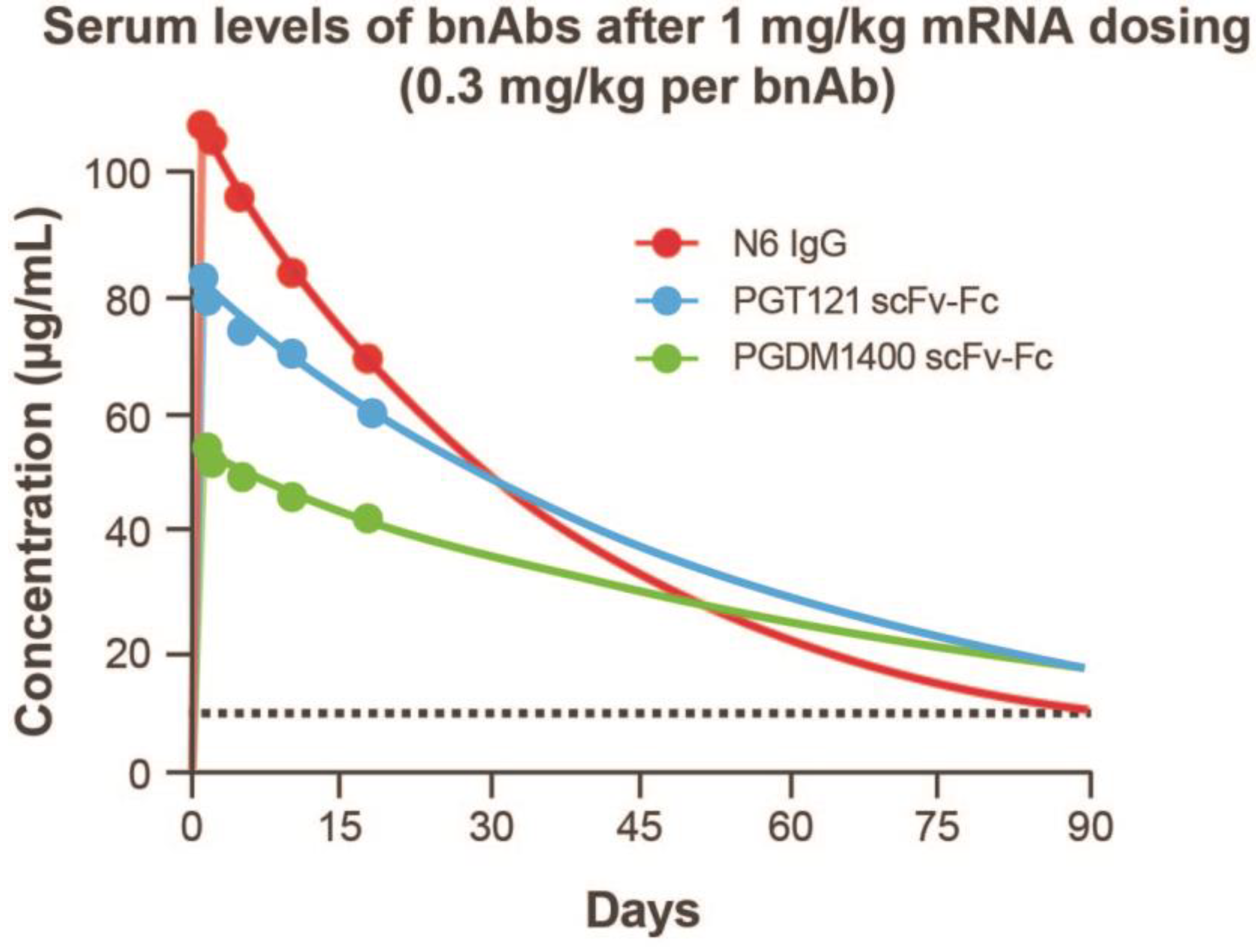
3.5. In Vivo Expression of an mRNA-LNP Encoded HIV bnAb Combination
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pierpont, T.M.; Limper, C.; Richards, K.L. Past, present, and future of rituximab—The world’s first oncology monoclonal antibody therapy. Front. Oncol. 2018, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Olivo, M.A.; Urruela, M.A.; McGahan, L.; Pollono, E.N.; Suarez-Almazor, M.E. Rituximab for rheumatoid arthritis. Cochrane Database Syst. Rev. 2015, 1, CD007356. [Google Scholar] [CrossRef] [PubMed]
- Connor, E.M. Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics 1998, 102, 531–537. [Google Scholar] [CrossRef]
- Lavoie, P.M.; Solimano, A.; Taylor, R.; Kwan, E.; Claydon, J.; Turvey, S.E.; Marr, N. Outcomes of respiratory syncytial virus immunoprophylaxis in infants using an abbreviated dosing regimen of palivizumab. JAMA Pediatr. 2016, 170, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Chandele, A.; Sharma, A. Current status of therapeutic monoclonal antibodies against SARS-CoV-2. PLoS Pathog. 2021, 17, e1009885. [Google Scholar] [CrossRef]
- Nowak, M.A. Variability of HIV infections. J. Theor. Biol. 1992, 155, 1–20. [Google Scholar] [CrossRef]
- Shapiro, M.B.; Cheever, T.; Malherbe, D.C.; Pandey, S.; Reed, J.; Yang, E.S.; Wang, K.; Pegu, A.; Chen, X.; Siess, D.; et al. Single-dose bNAb cocktail or abbreviated ART post-exposure regimens achieve tight SHIV control without adaptive immunity. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Mendoza, P.; Grüll, H.; Nogueira, L.; Pai, J.A.; Butler, A.L.; Millard, K.; Lehmann, C.; Suárez, I.; Oliveira, T.; Lorenzi, J.C.C.; et al. Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature 2018, 561, 479–484. [Google Scholar] [CrossRef]
- Stephenson, K.E.; Julg, B.; Tan, C.S.; Zash, R.; Walsh, S.R.; Rolle, C.-P.; Monczor, A.N.; Lupo, S.; Gelderblom, H.C.; Ansel, J.L.; et al. Safety, pharmacokinetics and antiviral activity of PGT121, a broadly neutralizing monoclonal antibody against HIV-1: A randomized, placebo-controlled, phase 1 clinical trial. Nat. Med. 2021, 27, 1718–1724. [Google Scholar] [CrossRef]
- Mosa, A.I. Antigenic variability. Front. Immunol. 2020, 11, 2057. [Google Scholar] [CrossRef]
- Gottlieb, R.L.; Nirula, A.; Chen, P.; Boscia, J.; Heller, B.; Morris, J.; Huhn, G.; Cardona, J.; Mocherla, B.; Stosor, V.; et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: A randomized clinical trial. JAMA 2021, 325, 632–644. [Google Scholar] [CrossRef]
- Mahomed, S.; Garrett, N.; Karim, Q.A.; Zuma, N.Y.; Capparelli, E.; Baxter, C.; Gengiah, T.; Archary, D.; Samsunder, N.; Rose, N.D.; et al. Assessing the safety and pharmacokinetics of the anti-HIV monoclonal antibody CAP256V2LS alone and in combination with VRC07-523LS and PGT121 in South African women: Study protocol for the first-in-human CAPRISA 012B phase I clinical trial. BMJ Open 2020, 10, e042247. [Google Scholar]
- Millham, L.R.; Scott, J.A.; Sax, P.E.; Shebl, F.M.; Reddy, K.P.; Losina, E.; Walensky, R.P.; Freedberg, K.A. Clinical and economic impact of ibalizumab for people with multidrug-resistant HIV in the United States. JAIDS J. Acquir. Immune Defic. Syndr. 2020, 83, 148–156. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Thran, M.; Mukherjee, J.; Pönisch, M.; Fiedler, K.; Thess, A.; Mui, B.L.; Hope, M.J.; Tam, Y.K.; Horscroft, N.; Heidenreich, R.; et al. mRNA mediates passive vaccination against infectious agents, toxins, and tumors. EMBO Mol. Med. 2017, 9, 1434–1447. [Google Scholar] [CrossRef]
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. mRNA vaccines for infectious diseases: Principles, delivery and clinical translation. Nat. Rev. Drug Discov. 2021, 20, 817–838. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Study to Evaluate the Efficacy, Safety, and Immunogenicity of mRNA-1647 Cytomegalovirus (CMV) Vaccine in Healthy Participants 16 to 40 Years of Age. Available online: https://clinicaltrials.gov/ct2/show/NCT05085366 (accessed on 23 May 2022).
- ClinicalTrials.gov. Safety and Immunogenicity of mRNA-1653, a Combined Human Metapneumovirus (hMPV) and Parain-Fluenza Virus Type 3 (PIV3) Vaccine, in Healthy Adults, and Children 12 to 59 Months of Age with Serologic Evidence of Prior Exposure. Available online: https://clinicaltrials.gov/ct2/show/NCT04144348 (accessed on 23 May 2022).
- Patel, M.R.; Bauer, T.M.; Jimeno, A.; Wang, D.; Lorusso, P.; Do, K.T.; Stemmer, S.M.; Maurice-Dror, C.; Geva, R.; Zacharek, S.; et al. A phase I study of mRNA-2752, a lipid nanoparticle encapsulating mRNAs encoding human OX40L, IL-23, and IL-36γ, for intratumoral (iTu) injection alone and in combination with durvalumab. J. Clin. Oncol. 2020, 38, 3092. [Google Scholar] [CrossRef]
- August, A.; Attarwala, H.Z.; Himansu, S.; Kalidindi, S.; Lu, S.; Pajon, R.; Han, S.; Lecerf, J.-M.; Tomassini, J.E.; Hard, M.; et al. A phase 1 trial of lipid-encapsulated mRNA encoding a monoclonal antibody with neutralizing activity against Chikungunya virus. Nat. Med. 2021, 27, 2224–2233. [Google Scholar] [CrossRef]
- Julg, B.; Tartaglia, L.J.; Keele, B.F.; Wagh, K.; Pegu, A.; Sok, D.; Abbink, P.; Schmidt, S.D.; Wang, K.; Chen, X.; et al. Broadly neutralizing antibodies targeting the HIV-1 envelope V2 apex confer protection against a clade C SHIV challenge. Sci. Transl. Med. 2017, 9, eaal1321. [Google Scholar] [CrossRef]
- Mouquet, H.; Scharf, L.; Euler, Z.; Liu, Y.; Eden, C.; Scheid, J.F.; Halper-Stromberg, A.; Gnanapragasam, P.N.P.; Spencer, D.I.R.; Seaman, M.S.; et al. Complex-type N -glycan recognition by potent broadly neutralizing HIV antibodies. Proc. Natl. Acad. Sci. USA 2012, 109, E3268–E3277. [Google Scholar] [CrossRef]
- Huang, J.; Kang, B.H.; Ishida, E.; Zhou, T.; Griesman, T.; Sheng, Z.; Wu, F.; Doria-Rose, N.A.; Zhang, B.; McKee, K.; et al. Identification of a CD4-binding-site antibody to HIV that evolved near-pan neutralization breadth. Immunity 2016, 45, 1108–1121. [Google Scholar] [CrossRef] [PubMed]
- King, A.C.; Woods, M.; Liu, W.; Lu, Z.; Gill, D.; Krebs, M.R.H. High-throughput measurement, correlation analysis, and machine-learning predictions for pH and thermal stabilities of Pfizer-generated antibodies. Protein Sci. 2011, 20, 1546–1557. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.-C.; Aulabaugh, A.; Jin, G.; Cowling, R.; Bard, J.; Malamas, M.; Ellestad, G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 2004, 332, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Sarzotti-Kelsoe, M.; Bailer, R.T.; Turk, E.; Lin, C.-L.; Bilska, M.; Greene, K.M.; Gao, H.; Todd, C.A.; Ozaki, D.A.; Seaman, M.S.; et al. Optimization and validation of the TZM-bl assay for standardized assessments of neutralizing antibodies against HIV-1. J. Immunol. Methods 2013, 409, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Montefiori, D.C. Measuring HIV neutralization in a luciferase reporter gene assay. Methods Mol. Biol. 2009, 485, 395–405. [Google Scholar] [CrossRef]
- Kortemme, T.; Morozov, A.V.; Baker, D. An orientation-dependent hydrogen bonding potential improves prediction of specificity and structure for proteins and protein–protein complexes. J. Mol. Biol. 2003, 326, 1239–1259. [Google Scholar] [CrossRef]
- Lazaridis, T.; Karplus, M. Effective energy function for proteins in solution. Proteins Struct. Funct. Bioinform. 1999, 35, 133–152. [Google Scholar] [CrossRef]
- Van Hoecke, L.; Roose, K. How mRNA therapeutics are entering the monoclonal antibody field. J. Transl. Med. 2019, 17, 1–14. [Google Scholar] [CrossRef]
- Zalevsky, J.; Chamberlain, A.K.; Horton, H.M.; Karki, S.; Leung, I.W.L.; Sproule, T.J.; Lazar, G.A.; Roopenian, D.C.; Desjarlais, J.R. Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 2010, 28, 157–159. [Google Scholar] [CrossRef]
- Decamp, A.; Hraber, P.; Bailer, R.T.; Seaman, M.S.; Ochsenbauer, C.; Kappes, J.; Gottardo, R.; Edlefsen, P.; Self, S.; Tang, H.; et al. Global panel of HIV-1 env reference strains for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 2014, 88, 2489–2507. [Google Scholar] [CrossRef]
- Klein, J.S.; Gnanapragasam, P.N.P.; Galimidi, R.P.; Foglesong, C.P.; West, A.P., Jr.; Bjorkman, P.J. Examination of the contributions of size and avidity to the neutralization mechanisms of the anti-HIV antibodies b12 and 4E10. Proc. Natl. Acad. Sci. USA 2009, 106, 7385–7390. [Google Scholar] [CrossRef]
- Kose, N.; Fox, J.M.; Sapparapu, G.; Bombardi, R.; Tennekoon, R.N.; de Silva, A.D.; Elbashir, S.M.; Theisen, M.A.; Humphris-Narayanan, E.; Ciaramella, G.; et al. A lipid-encapsulated mRNA encoding a potently neutralizing human monoclonal antibody protects against chikungunya infection. Sci. Immunol. 2019, 4, eaaw6647. [Google Scholar] [CrossRef]
- Avery, L.B.; Wang, M.; Kavosi, M.S.; Joyce, A.; Kurz, J.C.; Fan, Y.-Y.; Dowty, M.; Zhang, M.; Zhang, Y.; Cheng, A.; et al. Utility of a human FcRn transgenic mouse model in drug discovery for early assessment and prediction of human pharmacokinetics of monoclonal antibodies. mAbs 2016, 8, 1064–1078. [Google Scholar] [CrossRef]
- Mayer, K.H.; Seaton, K.E.; Huang, Y.; Grunenberg, N.; Isaacs, A.; Allen, M.; Ledgerwood, J.E.; Frank, I.; Sobieszczyk, M.E.; Baden, L.R.; et al. Safety, pharmacokinetics, and immunological activities of multiple intravenous or subcutaneous doses of an anti-HIV monoclonal antibody, VRC01, administered to HIV-uninfected adults: Results of a phase 1 randomized trial. PLoS Med. 2017, 14, e1002435. [Google Scholar] [CrossRef]
- Scheid, J.F.; Horwitz, J.A.; Bar-On, Y.; Kreider, E.F.; Lu, C.-L.; Lorenzi, J.C.C.; Feldmann, A.; Braunschweig, M.; Nogueira, L.; Oliveira, T.; et al. HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature 2016, 535, 556–560. [Google Scholar] [CrossRef]
- Caskey, M.; Schoofs, T.; Grüll, H.; Settler, A.; Karagounis, T.; Kreider, E.F.; Murrell, B.; Pfeifer, N.; Nogueira, L.; Oliveira, T.; et al. Antibody 10-1074 suppresses viremia in HIV-1-infected individuals. Nat. Med. 2017, 23, 185–191. [Google Scholar] [CrossRef]
- Julg, B.; Stephenson, K.E.; Wagh, K.; Tan, S.C.; Zash, R.; Walsh, S.; Ansel, J.; Kanjilal, D.; Nkolola, J.; Walker-Sperling, V.E.K.; et al. Safety and antiviral activity of triple combination broadly neutralizing monoclonal antibody therapy against HIV-1: A phase 1 clinical trial. Nat. Med. 2022, 28, 1288–1296. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, W.; Sun, M.; Li, T. Broadly neutralizing antibodies for HIV-1: Efficacies, challenges and opportunities. Emerg. Microbes Infect. 2020, 9, 194–206. [Google Scholar] [CrossRef]
- Wagh, K.; Bhattacharya, T.; Williamson, C.; Robles, A.; Bayne, M.; Garrity, J.; Rist, M.; Rademeyer, C.; Yoon, H.; Lapedes, A.; et al. Optimal combinations of broadly neutralizing antibodies for prevention and treatment of HIV-1 clade C infection. PLoS Pathog. 2016, 12, e1005520. [Google Scholar] [CrossRef]
- Kong, R.; Louder, M.K.; Wagh, K.; Bailer, R.T.; Decamp, A.; Greene, K.; Gao, H.; Taft, J.D.; Gazumyan, A.; Liu, C.; et al. Improving neutralization potency and breadth by combining broadly reactive HIV-1 antibodies targeting major neutralization epitopes. J. Virol. 2015, 89, 2659–2671. [Google Scholar] [CrossRef]
- Walsh, S.R.; Seaman, M.S. Broadly neutralizing antibodies for HIV-1 prevention. Front. Immunol. 2021, 12, 712122. [Google Scholar] [CrossRef] [PubMed]
- Corey, L.; Gilbert, P.B.; Juraska, M.; Montefiori, D.C.; Morris, L.; Karuna, S.T.; Edupuganti, S.; Mgodi, N.M.; Decamp, A.C.; Rudnicki, E.; et al. Two randomized trials of neutralizing antibodies to prevent HIV-1 acquisition. N. Engl. J. Med. 2021, 384, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Playford, E.G.; Munro, T.; Mahler, S.M.; Elliott, S.; Gerometta, M.; Hoger, K.L.; Jones, M.L.; Griffin, P.; Lynch, K.D.; Carroll, H.; et al. Safety, tolerability, pharmacokinetics, and immunogenicity of a human monoclonal antibody targeting the G glycoprotein of henipaviruses in healthy adults: A first-in-human, randomised, controlled, phase 1 study. Lancet Infect. Dis. 2020, 20, 445–454. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Narayanan, E.; Falcone, S.; Elbashir, S.M.; Attarwala, H.; Hassett, K.; Seaman, M.S.; Carfi, A.; Himansu, S. Rational Design and In Vivo Characterization of mRNA-Encoded Broadly Neutralizing Antibody Combinations against HIV-1. Antibodies 2022, 11, 67. https://doi.org/10.3390/antib11040067
Narayanan E, Falcone S, Elbashir SM, Attarwala H, Hassett K, Seaman MS, Carfi A, Himansu S. Rational Design and In Vivo Characterization of mRNA-Encoded Broadly Neutralizing Antibody Combinations against HIV-1. Antibodies. 2022; 11(4):67. https://doi.org/10.3390/antib11040067
Chicago/Turabian StyleNarayanan, Elisabeth, Samantha Falcone, Sayda M. Elbashir, Husain Attarwala, Kimberly Hassett, Michael S. Seaman, Andrea Carfi, and Sunny Himansu. 2022. "Rational Design and In Vivo Characterization of mRNA-Encoded Broadly Neutralizing Antibody Combinations against HIV-1" Antibodies 11, no. 4: 67. https://doi.org/10.3390/antib11040067
APA StyleNarayanan, E., Falcone, S., Elbashir, S. M., Attarwala, H., Hassett, K., Seaman, M. S., Carfi, A., & Himansu, S. (2022). Rational Design and In Vivo Characterization of mRNA-Encoded Broadly Neutralizing Antibody Combinations against HIV-1. Antibodies, 11(4), 67. https://doi.org/10.3390/antib11040067