
Understanding and Modulating Antibody Fine Specificity: Lessons from Combinatorial Biology

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Relevance of Functional Epitope Mapping for Immunologists and Antibody Engineers
3. Screening Reactivity of Antibodies against Antigen Fragments: The Simplest Way to Locate Epitopes
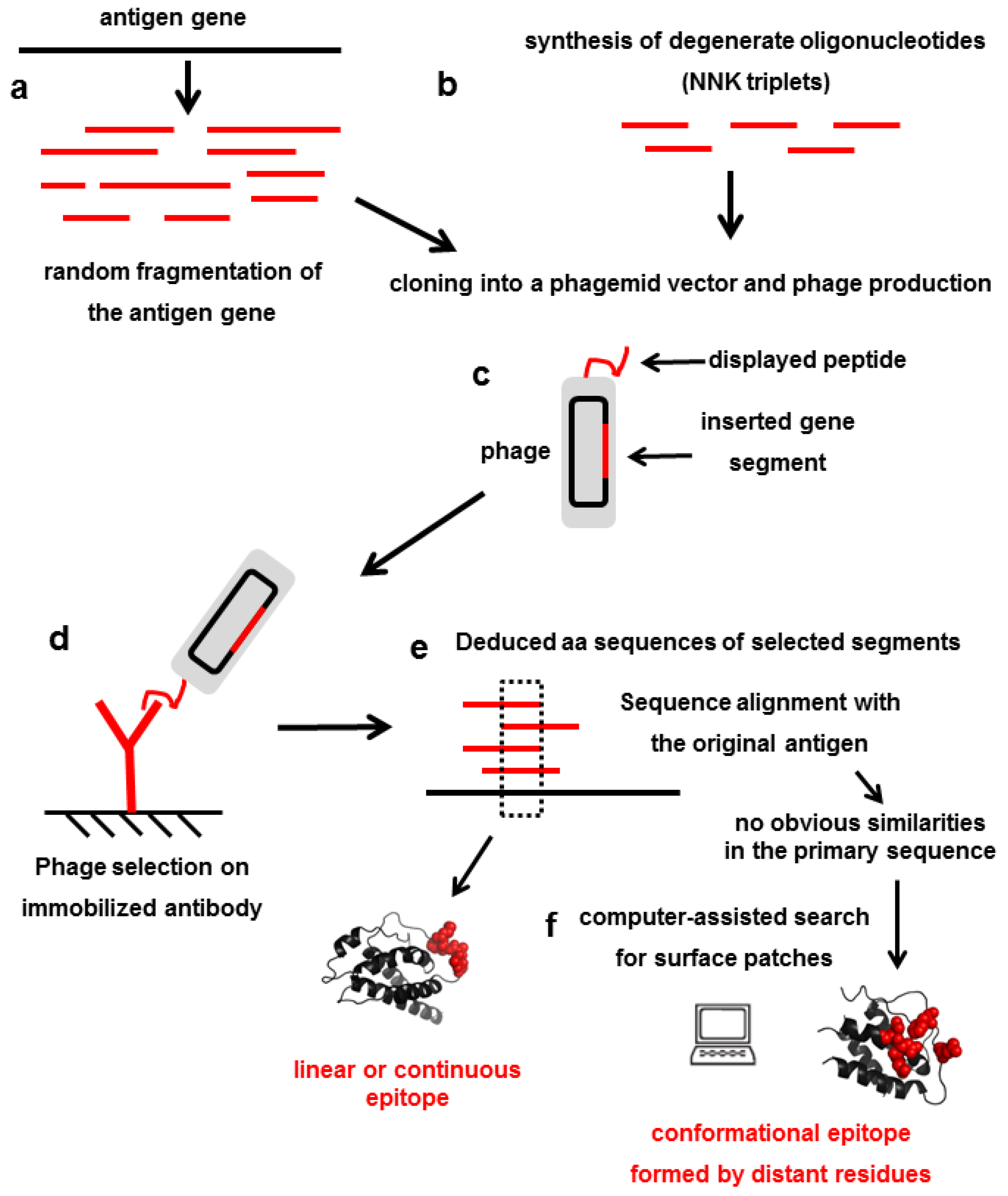
4. Mimicking Epitopes with Short Random peptides: An Indirect Approach for Epitope Mapping
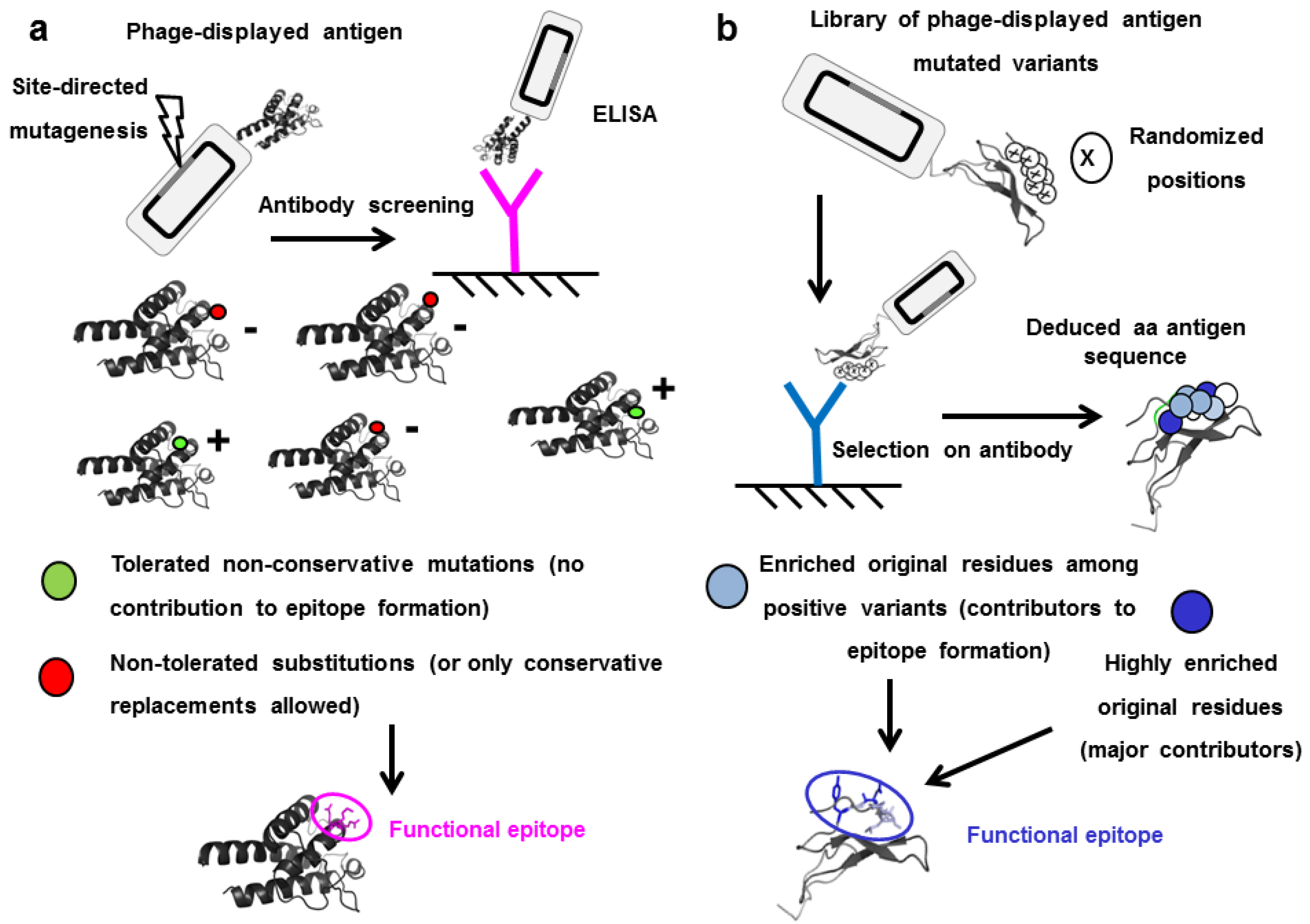
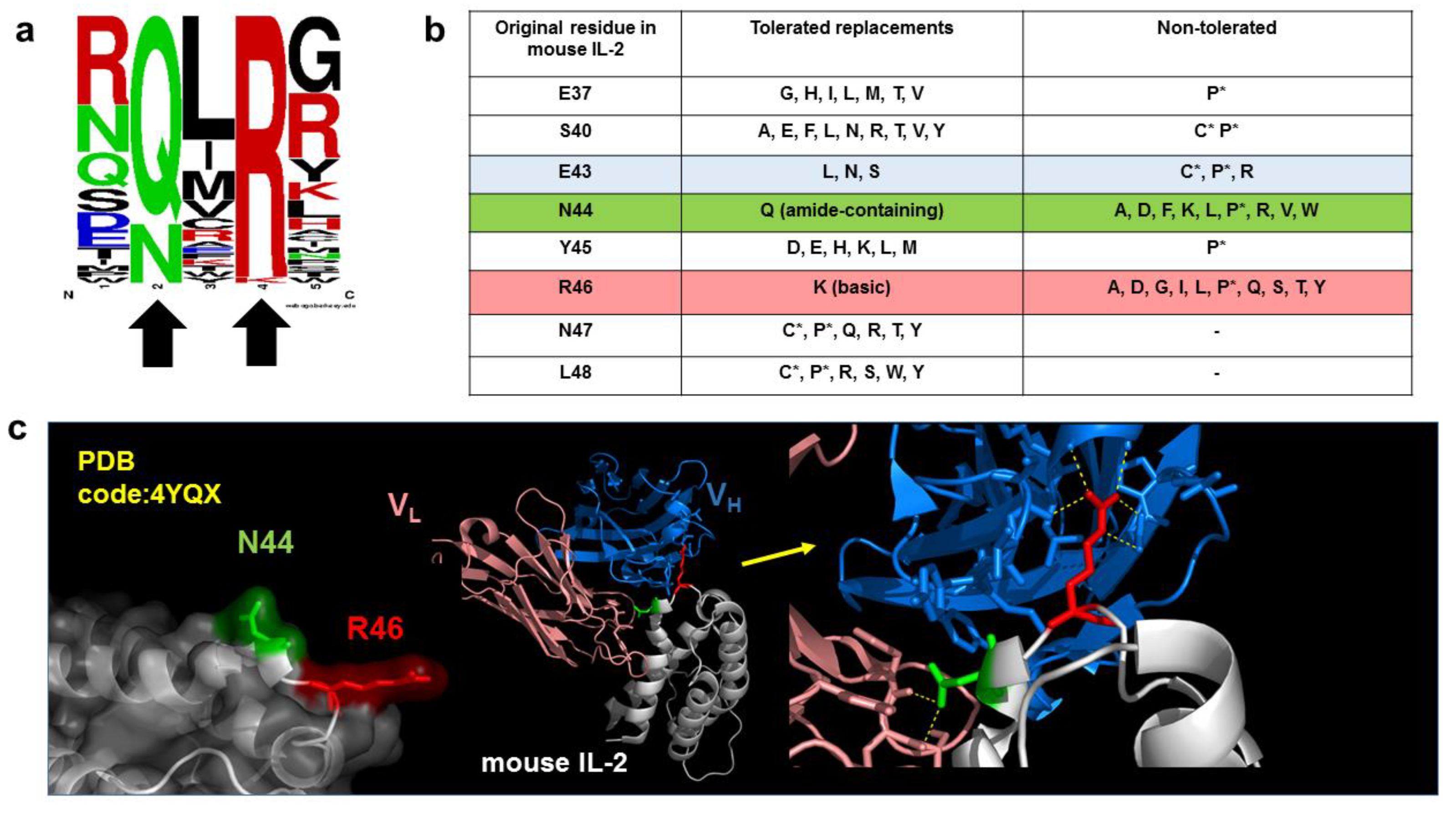
5. Functional Epitope Mapping by Comprehensive Mutagenesis Scanning of Antigen Surface
6. Combinatorial Biology Methods Reveal Singularities in the Chemistry of Epitopes
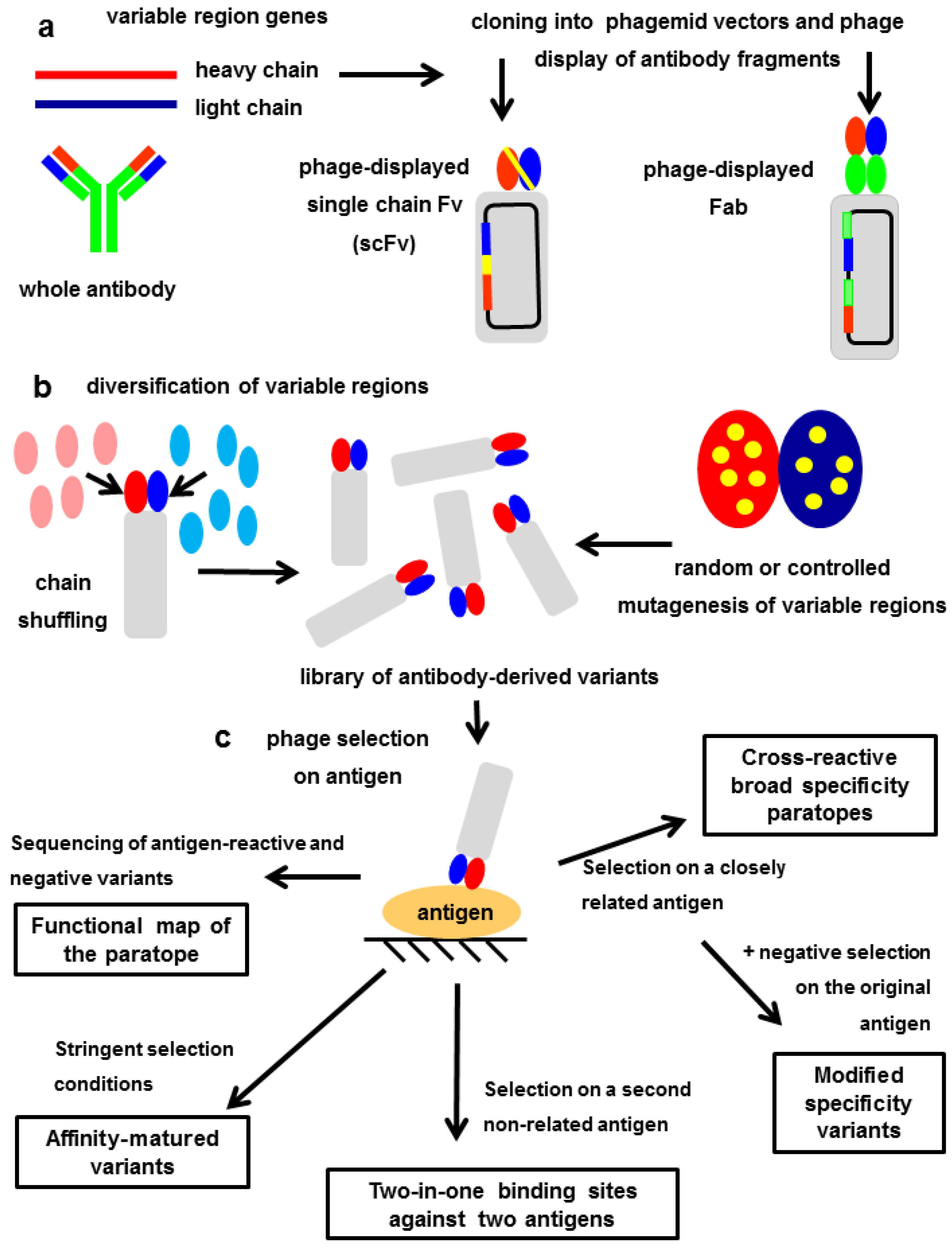
7. Exploring the Other Side of the Interaction: Functional Paratope Mapping
8. In Vitro Affinity Maturation: The Challenge of Increasing the Interaction Strength without Losing Fine Specificity
9. Modulating the Original Antibody Specificity towards Closely Related Antigens
10. Totally Divergent Specificities in A Single Binding Site: Two-in-One Paratopes
11. General Remarks and Future Prospects
Funding
Data Availability Statement
Conflicts of Interest
References
- Klinmant, N.R.; Press, J.L. The characterization fo the B-cell repertoire specific for the 2,4-dinitrophenyl and 2,4,6-trinitrophenyl determinants in neonatal BALB/c mice. J. Exp. Med. 1975, 141, 1133–1146. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G.M.; Berek, C.; Kaartinen, M.; Milstein, C. Somatic mutation and the maturation of immune response to 2-phenyl oxazolone. Nature 1984, 312, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Kolar, G.R.; Capra, J.D. Immunoglobulins: Structure and function. In Fundamental Immunology, 5th ed.; Paul, W.E., Ed.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2003; pp. 47–68. [Google Scholar]
- Ehrlich, P. Die Werbemessung des Diphterieheilserums und Deren Theoretische Grundlagen; 1897. Translated in Collected papers of Paul Ehrlich; Himmelweit, Marquardt, Dale, Eds.; Pergamon: London, UK, 1957; Volume 2, pp. 107–125. [Google Scholar]
- Davies, D.R.; Cohen, G.H. Interactions of protein antigens with antibodies. Proc. Natl. Acad. Sci. USA 1996, 93, 7–12. [Google Scholar] [CrossRef]
- Cunningham, B.C.; Wells, J.A. Comparison of a structural and a functional epitope. J. Mol. Biol. 1993, 234, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P.; Petrenko, V.A. Phage display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef] [PubMed]
- Gai, S.A.; Wittrup, K.D. Yeast surface display for protein engineering and characterization. Curr. Opin. Struct. Biol. 2007, 17, 467–473. [Google Scholar] [CrossRef]
- Boyman, O.; Kovar, M.; Rubinstein, M.P.; Surh, C.; Sprent, J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 2006, 311, 1924–1927. [Google Scholar] [CrossRef]
- Rojas, G.; Pupo, A.; Leon, K.; Avellanet, J.; Carmenate, T.; Sidhu, S. Deciphering the molecular bases of the biological effects of antibodies against Interleukin-2: A versatile platform for fine epitope mapping. Immunobiology 2013, 218, 105–113. [Google Scholar] [CrossRef]
- Rojas, G.; Infante, Y.C.; Pupo, A.; Carmenate, T. Fine specificity of antibodies against Interleukin-2 explains their paradoxical immunomodulatory effects. mAbs 2014, 6, 273–285. [Google Scholar] [CrossRef][Green Version]
- Klein, C.; Lammens, A.; Schäfer, W.; Georges, G.; Schwaiger, M.; Ekkehard, M.; Hopfner, K.-P.; Umaña, P.; Niederfellner, G. Epitope interactions of monoclonal antibodies targeting CD20 and their relationships to functional properties. mAbs 2013, 5, 22–33. [Google Scholar] [CrossRef]
- Edwards, B.M.; Barash, S.C.; Main, S.H.; Choi, G.H.; Minter, R.; Ullrich, S.; Williams, E.; Du Fou, L.; Wilton, J.; Albert, V.R.; et al. The remarkable flexibility of the human antibody repertoire; isolation of over one thousand different antibodies to a single protein, BLyS. J. Mol. Biol. 2003, 334, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Schofield, D.J.; Pope, A.R.; Clementel, V.; Buckell, J.; Chapple, S.D.J.; Clarke, K.F.; Conquer, J.S.; Crofts, A.S.; Crowther, S.R.E.; Dyson, M.R.; et al. Application of phage display to high throughput antibody generation and characterization. Genome Biol. 2007, 8, R254. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Storz, U.; Doranz, B. Enhancing antibody patent protection using epitope mapping information. mAbs 2018, 10, 204–209. [Google Scholar] [CrossRef]
- Montagut, C.; Dalmases, A.; Belosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Voigt, M.; Braig, F.; Göthel, M.; Schulte, A.; Lamszus, K.; Bokemeyer, C.; Binder, M. Functional dissection of the Epidermal Growth Factor receptor epitopes targeted by panitumumab and cetuximab. Neoplasia 2012, 14, 1023–1031. [Google Scholar] [CrossRef]
- Arena, S.; Bellosillo, B.; Siravegna, G.; Martínez, G.A.; Cañadas, I.; Lazzari, L.; Ferruz, N.; Russo, M.; Misale, S.; González, I.; et al. Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin. Cancer Res. 2015, 21, 2157–2166. [Google Scholar] [CrossRef]
- Bertotti, A.; Papp, E.; Jones, S.; Adleff, V.; Anagnostou, V.; Lupo, B.; Sausen, M.; Phallen, J.; Hruban, C.A.; Tokheim, C.; et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015, 526, 263–267. [Google Scholar] [CrossRef]
- Braig, F.; Marz, M.; Schieferdecker, A.; Schulte, A.; Voigt, M.; Stein, A.; Grob, T.; Alawi, M.; Indenbirken, D.; Kriegs, M.; et al. Epidermal growth factor receptor mutation mediates cross-resistance to panitumumab and cetuximab in gastrointestinal cancer. Oncotarget 2015, 6, 12035–12047. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe 2021, 29, 44–57. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayl, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef]
- Reineke, U. Antibody epitope mapping using arrays of synthetic peptides. Methods Mol. Biol. 2004, 248, 443–463. [Google Scholar]
- Midoro-Horiuti, T.; Goldblum, R.M. Epitope mapping with membrane-bound synthetic overlapping peptides. Methods Mol. Biol. 2014, 1131, 421–426. [Google Scholar] [PubMed]
- Smith, G.P. Phage Display: Simple Evolution in a Petri Dish (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2019, 58, 14428–14437. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Filamentous phusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, F.; Salvi, R.; Garulli, C.; Kalogris, C.; Arima, S.; Tardella, L.; Monaci, P.; Pupa, S.M.; Tagliabue, T.; Montani, M.; et al. Identification of relevant conformational epitopes on the HER2 oncoprotein by using large fragment phage display (LFPD). PLoS ONE 2013, 8, e58358. [Google Scholar] [CrossRef]
- Moreira, G.M.S.G.; Fühner, V.; Hust, M. Epitope mapping by phage display. Methods Mol. Biol. 2018, 1701, 497–518. [Google Scholar]
- Fühner, V.; Heine, P.A.; Zilkens, K.J.C.; Meier, D.; Roth, K.D.R.; Moreira, G.M.S.G.; Hust, M.; Russo, G. Epitope Mapping via Phage Display from Single-Gene Libraries. Methods Mol. Biol. 2019, 1904, 353–375. [Google Scholar]
- Cariccio, V.L.; Domina, M.; Benfatto, S.; Venza, M.; Venza, I.; Faleri, A.; Bruttini, M.; Bartolini, E.; Giuliani, M.M.; Santini, L.; et al. Phage display revisited: Epitope mapping of a monoclonal antibody against Neisseria meningitides adhesion A using the PROFILER technology. mAbs 2016, 8, 741–750. [Google Scholar] [CrossRef]
- Volk, A.-L.; Hu, F.J.; Rockberg, J. Epitope mapping of monoclonal and polyclonal antibodies using bacterial cell surface display of gene fragment libraries. Methods Mol. Biol. 2014, 1131, 485–500. [Google Scholar]
- Volk, A.-L.; Hu, F.J.; Rockberg, J. Epitope mapping of antibodies using bacterial cell surface display of gene fragment libraries. Methods Mol. Biol. 2018, 1785, 141–157. [Google Scholar]
- Felici, F.; Castagnoli, L.; Musacchio, A.; Japelli, R.; Cesareni, G. Selection of antibody ligands from a large library of oligopeptides expressed on a multivalent exposition vector. J. Mol. Biol. 1991, 222, 301–310. [Google Scholar] [CrossRef]
- Midoro-Horiuti, T.; Goldblum, R.M. Epitope mapping with random phage display library. Methods Mol. Biol. 2014, 1131, 477–484. [Google Scholar] [PubMed]
- Bonnycastle, L.L.; Mehroke, J.S.; Rashed, M.; Gong, X.; Smith, J.K. Probing the basis of antibody reactivity with a panel of constrained peptide libraries displayed by filamentous phage. J. Mol. Biol. 1996, 258, 747–762. [Google Scholar] [CrossRef]
- Rojas, G.; Pupo, A.; Aleman, M.R.; Vispo, N.S. Preferential selection of Cys-constrained peptides from a random phage-displayed library by anti-glucitollysine antibodies. J. Pept. Sci. 2008, 14, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Rodriguez, Y.; Gazarian, T.; Rowley, M.; Majluf-Cruz, A.; Gazarian, K.J. Collection of phage-peptide probes for HIV-1 immunodominant loop-epitopes. J. Microbiol. Methods 2007, 68, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.L.; Craig, L.; Mehroke, J.S.; Rashed, M.; Zwick, M.B.; Kenar, K.; Toone, E.J.; Greenspan, N.; Auzanneau, F.I.; Marino-Albernas, J.-R.; et al. Exploring the basis of peptide-carbohydrate crossreactivity: Evidence for discrimination by peptides between closely related anti-carbohydrate antibodies. Proc. Natl. Acad. Sci. USA 1997, 94, 2454–2459. [Google Scholar] [CrossRef]
- Newton-Northup, J.R. Contending with target unrelated peptides from phage display. J. Mol. Imaging Dinam. 2012, 2, e101. [Google Scholar] [CrossRef]
- Chin, C.F.; Lai, J.Y.; Choong, Y.S.; Anthony, A.A.; Ismail, A.; Lim, T.S. Delineation of B-cell epitopes of Salmonella enterica serovar Typhi hemolysin E: Potential antibody therapeutic target. Sci. Rep. 2017, 7, 2176. [Google Scholar] [CrossRef]
- Huang, J.; Ru, B.; Li, S.; Lin, H.; Guo, F.-B. SAROTUP: Scanner and reporter of target-unrelated peptides. J. Biomed. Biotechnol. 2010, 2010, 101932. [Google Scholar] [CrossRef]
- Vispo, N.S.; Araña, M.J.; Chinea, G.; Ojalvo, A.G.; Cesareni, G. Characterization of epitopes on human Interleukin-2 using phage-displayed peptide libraries: Insights into antibody-peptide interactions. Hybridoma 1999, 18, 251–255. [Google Scholar] [CrossRef]
- Saphire, E.O.; Montero, M.; Menendez, A.; van Houten, N.E.; Irving, M.E.; Pantophlet, R.; Zwick, M.B.; Parren, P.W.H.I.; Burton, D.R.; Scott, J.K.; et al. Structure of a high affinity “mimotope” peptide bound to HIV-1 neutralizing b12 explains its inability to elicit gp120 cross-reactive antibodies. J. Mol. Biol. 2007, 369, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Bumbaca, D.; Wong, A.; Drake, E.; Reyes, A.E.; Lin, B.C.; Stephan, J.P.; Desnoyers, L.; Shen, B.-Q.; Dennis, M.S. Highly specific off-target binding identified and eliminated during the humanization of an antibody against FGF receptor 4. mAbs 2011, 3, 376–386. [Google Scholar] [CrossRef]
- Bublil, E.M.; Tarnovitski Freund, N.; Mayrose, I.; Penn, O.; Roitburd-Berman, A.; Rubinstein, N.D.; Pupko, T.; Gershoni, J.M. Stepwise prediction of conformational discontinuous B-cell epitopes using the mapitope algorithm. Proteins 2007, 68, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Gutteridge, A.; Honda, W.; Kanehisa, M. MIMOX: A web tool for phage display based epitope mapping. BMC Bioinform. 2006, 7, 56. [Google Scholar] [CrossRef]
- Moreau, V.; Granier, C.; Villard, S.; Laune, D.; Molina, F. Discontinuous epitope prediction based on mimotope analysis. Struct. Bioinform. 2006, 22, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Mayrose, I.; Shlomi, T.; Rubinstein, N.D.; Gershoni, J.M.; Ruppin, E.; Sharan, R.; Pupko, T. Epitope mapping using combinatorial phage-display libraries: A graph-based algorithm. Nucleic. Acids Res. 2007, 35, 69–78. [Google Scholar] [CrossRef]
- Negi, S.-S.; Braun, W. Automated detection of conformational epitopes using phage display peptide sequences. Bioinform. Biol. Insights 2009, 3, 71–81. [Google Scholar]
- Pacios, L.F.; Tordesillas, L.; Palacin, A.; Sanchez-Monge, R.; Salcedo, G.; Diaz-Perales, A. LocaPep: Localization of epitope protein surfaces using peptides from phage display libraries. J. Chem. Inf. Modeling 2011, 51, 1465–1473. [Google Scholar] [CrossRef]
- Chen, W.H.; Sun, P.P.; Lu, Y.; Guo, W.W.; Huang, Y.X.; Ma, Z.Q. MimoPro: A more efficient web-based tool for epitope prediction using phage display libraries. BMC Bioinform. 2011, 12, 199. [Google Scholar] [CrossRef]
- Halperin, R.F.; Stafford, P.; Emery, J.S.; Navalkar, K.A.; Johnston, S.A. GuiTope: An application for mapping randon-sequence peptides to protein sequences. BMC Bioinform. 2012, 13, 1. [Google Scholar] [CrossRef]
- Huang, Y.X.; Bao, Y.L.; Guo, S.Y.; Wang, Y.; Zhou, C.G. Pep-3D-Search: A method for B cell epitope prediction based on mimotope analysis. BMC Bioinform. 2008, 9, 538. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.; Humbert, M.; Benz, A.; Dietrich, U. 3D-Epitope-Explorer (3DEX): Localization of conformational epitopes within three-dimensional structures of proteins. J. Comput. Chem. 2005, 26, 879–887. [Google Scholar] [CrossRef]
- Riemer, A.B.; Förster-Waldl, E.; Bramswig, K.H.; Pollak, A.; Zielinski, C.C.; Pehamberger, H.; Lode, H.N.; Scheiner, O.; Jensen-Jarolim, E. Induction of IgG antibodies against the GD2 carbohydrate tumor antigen by vaccination with peptide mimotopes. Eur. J. Immunol. 2006, 36, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Knittelfelder, R.; Riemer, A.B.; Jensen-Jarolim, E. Mimotope vaccination-from allergy to cancer. Expert Opin. Biol. Ther. 2009, 9, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.C.; Perdue, S.S. Site-directed mutagenesis in epitope mapping. Methods 1996, 9, 508–515. [Google Scholar] [CrossRef]
- Rojas, G.; Tundidor, Y.; Infante, Y.C. High throughput functional epitope mapping: Revisiting phage display platform to scan target antigen surface. mAbs 2014, 6, 1368–1376. [Google Scholar] [CrossRef]
- Rojas, G. Fine epitope mapping based on phage display and extensive mutagenesis of the target antigen. Methods Mol. Biol. 2014, 1131, 447–476. [Google Scholar]
- Lamdan, H.; Gavilondo, J.V.; Munoz, Y.; Pupo, A.; Huerta, V.; Musacchio, A.; Perez, L.; Ayala, M.; Rojas, G.; Balint, R.F.; et al. Affinity maturation and fine functional mapping of an antibody fragment against a novel neutralizing epitope on human vascular endothelial growth factor. Mol. Biosyst. 2013, 9, 2097–2106. [Google Scholar] [CrossRef]
- Tundidor, Y.; Garcia-Hernandez, C.P.; Pupo, A.; Infante, Y.C.; Rojas, G. Delineating the functional map of the interaction between nimotuzumab and the epidermal growth factor receptor. mAbs 2014, 6, 1013–1025. [Google Scholar] [CrossRef]
- Tundidor, Y.; Ponce, L.F.; Chao, L.; Solozábal, J.; Hust, M.; Dübel, S.; Rojas, G. Affinity-matured variants derived from nimotuzumab keep the original fine specificity and exhibit superior biological activity. Sci. Rep. 2020, 10, 1194. [Google Scholar] [CrossRef]
- Infante, Y.C.; Pupo, A.; Rojas, G. A combinatorial mutagenesis approach for functional epitope mapping on phage-displayed target antigen: Application to antibodies against epidermal growth factor. mAbs 2014, 6, 637–648. [Google Scholar] [CrossRef]
- Chao, G.; Cochran, R.; Wittrup, K.D. Fine epitope mapping of anti-epidermal growth factor receptor antibodies through random mutagenesis and yeast surface display. J. Mol. Biol. 2004, 342, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Gaiotto, T.; Hufton, S. Cross-neutralising nanobodies bind to a conserved pocket in the hemagglutinin stem region identified using yeast display and deep mutational scanning. PLoS ONE 2016, 11, e0164296. [Google Scholar] [CrossRef] [PubMed]
- van Blarcom, T.; Rossi, A.; Foletti, D.; Sundar, P.; Pitts, S.; Melton, Z.; Telman, D.; Zhao, L.; Cheung, W.L.; Berka, J.; et al. Epitope mapping using yeast display and next generation sequencing. Methods Mol. Biol. 2018, 785, 89–118. [Google Scholar]
- van Blarcom, T.; Rossi, A.; Foletti, D.; Sundar, P.; Bee, C.; Witt, J.M.; Melton, Z.; Hasa-Moreno, A.; Shaughnessy, L.; Telman, D.; et al. Precise and efficient epitope determination through library design, yeast display and next generation sequencing. J. Mol. Biol. 2014, 427, 1513–1534. [Google Scholar] [CrossRef] [PubMed]
- Wrenbeck, E.E.; Klesmith, J.R.; Stapleton, J.A.; Adeniran, A.; Tyo, K.E.J.; Whitehead, T.A. Plasmid-based one-pot saturation mutagenesis. Nat. Methods 2016, 13, 928. [Google Scholar]
- Kowalsky, C.A.; Faber, M.S.; Nath, A.; Dann, H.E.; Kelly, V.W.; Liu, L.; Shanker, P.; Wagner, E.K.; Maynerd, J.A.; Chan, C.; et al. Rapid fine conformational epitope mapping using comprehensive mutagenesis and deep sequencing. J. Biol. Chem. 2015, 290, 26457–26470. [Google Scholar] [CrossRef] [PubMed]
- Acquaye-Seedah, E.; Reczek, E.E.; Russell, H.H.; DiVenere, A.M.; Sandman, S.O.; Collins, J.H.; Stein, C.A.; Whitehead, T.A.; Maynard, J.A. Characterization of individual human antibodies that bind pertussis toxin stimulated by acellular immunization. Infect. Immun. 2018, 86, e0004-18. [Google Scholar] [CrossRef] [PubMed]
- Medina-Cucurella, A.V.; Zhu, Y.; Bowen, S.J.; Bergeron, L.M.; Whitehead, T.A. Pro region engineering of nerve growth factor by deep mutational scanning enables a yeast platform for conformational epitope mapping of anti-NGF monoclonal antibodies. Biocatal. Prot. Eng. Nanobiotechnol. 2018, 115, 1925–1937. [Google Scholar] [CrossRef]
- Francino-Urdaniz, I.M.; Steiner, P.J.; Kirby, M.B.; Zhao, F.; Haas, C.M.; Barman, S.; Rhodes, E.R.; Leonard, A.C.; Peng, L.; Sprenger, K.G.; et al. One-shot identification of SARS-CoV-2 S RBD escape mutants using yeast screening. Cell Rep. 2021, 36, 109627. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Dingens, A.S.; Bloom, J.D. Complete map of SARS-CoV-2 RBD mutations that escape the monoclonal antibody LY-CoV555 and its cocktail with LY-CoV016. Cell Rep. Med. 2021, 2, 100255. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.D.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 2021, 29, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Francino-Urdaniz, I.M.; Whitehead, T.A. An overview of methods for the structural and functional mapping of epitopes recognized by anti-SARS-CoV-2 antibodies. RSC Chem. Biol. 2021, 2, 1580–1589. [Google Scholar] [CrossRef]
- Najar, T.A.; Khare, S.; Pandey, R.; Gupta, S.K.; Varadarajan, R. Mapping protein binding sites and conformational epitopes using cysteine labeling and yeast surface display. Structure 2017, 25, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Spangler, J.B.; Tomala, J.; Luca, V.C.; Jude, K.M.; Dong, S.; Ring, A.M.; Votavova, P.; Pepper, M.; Kovar, M.; Garcia, K.C. Antibodies to Interleukin-2 elicit selective T cell subset potentiation through distinct conformational mechanisms. Immunity 2015, 42, 815–825. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R.; Hasegawa, M. Models of amino acid substitution and applications to mitochondrial protein evolution. Mol. Biol. Evol. 1998, 15, 1600–1611. [Google Scholar] [CrossRef]
- Jonson, P.H.; Petersen, S.B. A critical view of conservative mutations. Prot. Eng. 2001, 14, 397–402. [Google Scholar] [CrossRef]
- Yampolski, L.Y.; Stoltzfus, A. The exchangeability of amino acids in proteins. Genetics 2005, 170, 1459–1472. [Google Scholar] [CrossRef]
- Kang, A.S.; Jones, T.M.; Burton, D.R. Antibody redesign by chain shuffling from random combinatorial immunoglobulin libraries. Proc. Natl. Acad. Sci. USA 1991, 88, 1120–11123. [Google Scholar] [CrossRef]
- Rojas, G.; Talavera, A.; Munoz, Y.; Rengifo, E.; Krengel, U.; Angstrom, J.; Gavilondo, J.V.; Moreno, E. Light chain shuffling results in successful phage display selection of functional prokaryotic-expressed antibody fragments to N-glycolyl GM3 ganglioside. J. Immunol. Methods 2004, 293, 71–83. [Google Scholar] [CrossRef]
- Gerstner, R.B.; Carter, P.; Lowman, H.B. Sequence plasticity in the antigen-binding site of a therapeutic anti-HER2 antibody. J. Mol. Biol. 2002, 321, 851–862. [Google Scholar] [CrossRef]
- Vajdos, F.F.; Adams, C.W.; Breece, T.N.; Presta, L.G.; de Vos, A.M.; Sidhu, S.S. Comprehensive functional maps of the antigen-binding site of an anti-ErbB2 antibody obtained with shotgun scanning mutagenesis. J. Mol. Biol. 2002, 320, 415–428. [Google Scholar] [CrossRef]
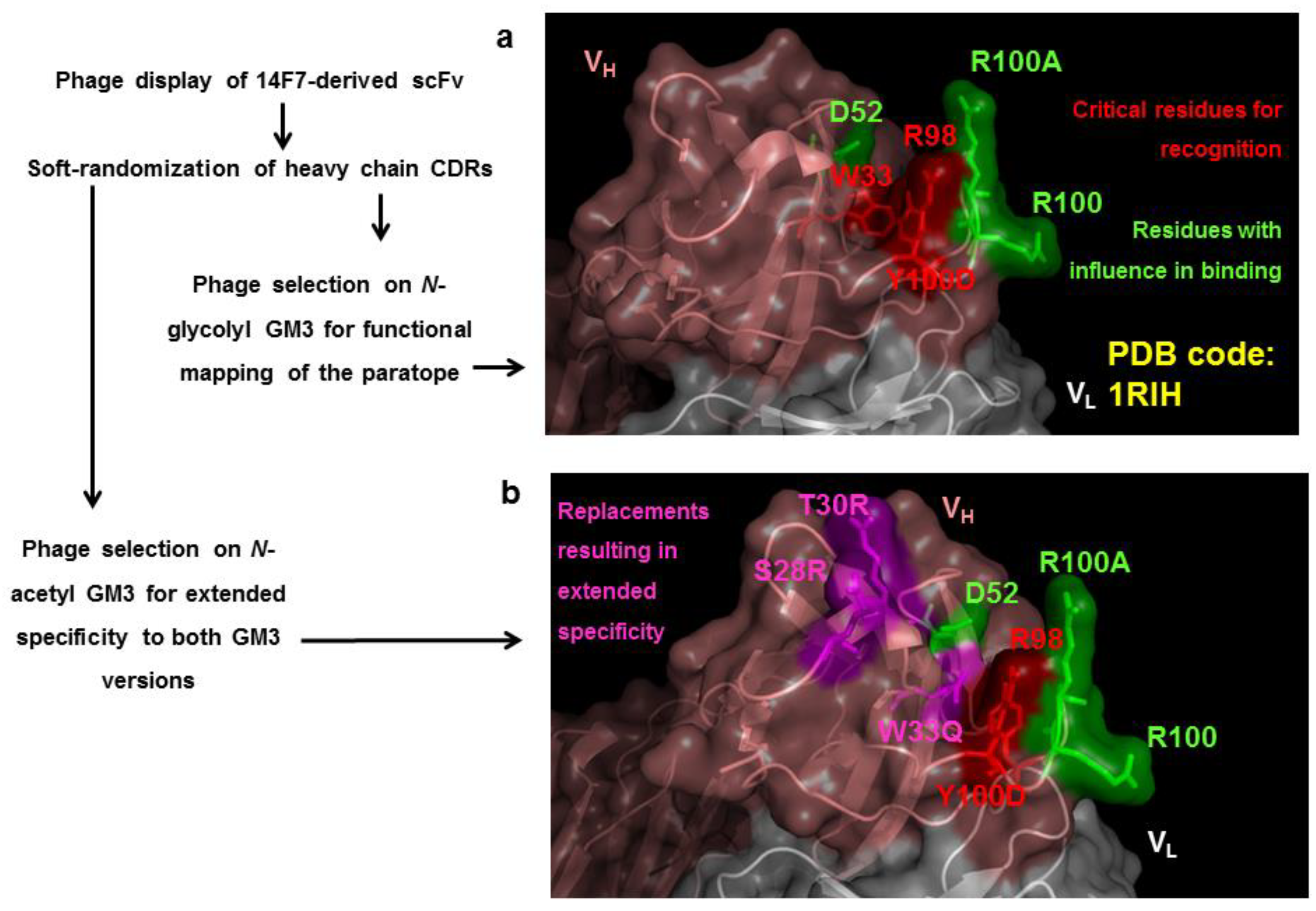
- Rojas, G.; Pupo, A.; Gomez, S.; Krengel, U.; Moreno, E. Engineering the binding site of an antibody against N-glycolyl GM3: From functional mapping to novel anti-ganglioside specificities. ACS Chem. Biol. 2013, 8, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Chames, P.; Willemsen, R.A.; Rojas, G.; Dieckmann, D.; Rem, L.; Schuler, G.; Bolhuis, R.L.; Hoogenboom, H.R. TCR-like human antibodies expressed on human CTLs mediate antibody affinity-dependent cytolytic activity. J. Immunol. 2002, 169, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Thie, H.; Voedisch, B.; Dübel, S.; Hust, M.; Schirrmann, T. Affinity maturation by phage display. Methods Mol. Biol. 2009, 525, 309–322. [Google Scholar] [PubMed]
- Van Deventer, J.A.; Wittrup, K.D. Yeast surface display for antibody isolation: Library construction, library screening, and affinity maturation. Methods Mol. Biol. 2014, 1131, 151–181. [Google Scholar]
- Lou, J.; Geren, I.; Garcia-Rodriguez, C.; Forsyth, C.M.; Wen, W.; Knopp, K.; Brown, J.; Smith, T.; Smith, L.A.; Marks, J.D. Affinity maturation of human botulinum neurotoxin antibodies by light chain shuffling via yeast mating. Protein Eng. Des. Sel. 2010, 23, 311–319. [Google Scholar] [CrossRef]
- Hur, B.U.; Choi, H.J.; Song, S.Y.; Yoon, J.B.; Liu, L.K.; Cha, S.H. Development of the dual-vector system-III (DVS-III), which facilitates affinity maturation of a Fab antibody via light chain shuffling. Immunol. Lett. 2010, 132, 24–30. [Google Scholar] [CrossRef]
- Tiller, K.E.; Li, L.; Kumar, S.; Julian, M.C.; Garde, S.; Tessier, P.M. Arginine mutations in antibody complementarity-determining regions display context-dependent affinity/specificity trade-offs. J. Biol. Chem. 2017, 292, 16638–16652. [Google Scholar] [CrossRef]
- Wu, H.; Beuerlein, G.; Nie, Y.; Smith, H.; Lee, B.A.; Hensler, M.; Huse, W.D.; Watkins, J.D. Stepwise in vitro affinity maturation of Vitaxin, an avb3-specific humanized mAb. Proc. Natl. Acad. Sci. USA 1998, 95, 6037–6042. [Google Scholar] [CrossRef]
- Shi, L.; Wheeler, J.C.; Sweet, R.W.; Lu, J.; Luo, J.; Tornetta, M.; Whitaker, B.; Reddy, R.; Brittingham, R.; Borozdina, L.; et al. De novo selection of high-affinity antibodies from synthetic Fab libraries displayed on phage as pIX fusion proteins. J. Mol. Biol. 2010, 397, 385–396. [Google Scholar] [CrossRef]
- Yang, W.P.; Green, K.; Pinz-Sweeney, S.; Briones, A.T.; Burton, D.R.; Barbas, C.F. CDR walking mutagenesis for the affinity maturation of a potent human anti-HIV-1 antibody into the picomolar range. J. Mol. Biol. 1995, 254, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Steidl, S.; Ratsch, O.; Brocks, B.; Dürr, M.; Thomassen-Wolf, E. In vitro affinity maturation of human GM-CSF antibodies by targeted CDR diversification. Mol. Immunol. 2008, 46, 135–144. [Google Scholar] [CrossRef]
- Fiedler, M.; Horn, C.; Bandtlow, C.; Schwab, M.E.; Skerra, A. An engineered IN-1 Fab fragment with improved affinity for theNogo-A axonal growth inhibitor permits immunochemical detection and shows enhanced neutralizing activity. Prot. Eng. 2002, 15, 931–941. [Google Scholar] [CrossRef][Green Version]
- Crombet, T.; Osorio, M.; Cruz, T.; Roca, C.; del Castillo, R.; Mon, R.; Iznaga-Escobar, N.; Figueredo, R.; Koropatnick, J.; Renginfo, E.; et al. Use of the humanized anti-Epidermal Growth Factor receptor monoclonal antibody h-R3 in combination with radiotherapy in the treatment of locally advanced head and neck cancer patients. J. Clin. Oncol. 2004, 22, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.P.; Schier, R.; McCall, A.M.; Simmons, H.H.; Horak, E.M.; Alpaugh, R.K.; Marks, J.D.; Weiner, L.M. High affinity restricts the localization and tumor penetration of single chain Fv antibody molecules. Cancer Res. 2001, 61, 4750–4755. [Google Scholar]
- Rudnick, S.I.; Lou, J.; Shaller, C.C.; Tang, Y.; Klein-Szanto, A.J.; Weiner, L.M.; Marks, J.D.; Adams, G.P. Influence of affinity and antigen internalization on the uptake and penetration of anti-HER2 antibodies in solid tumors. Cancer Res. 2011, 71, 2250–2259. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yan, H. Skin toxicity with anti-EGFR monoclonal antibody in cancer patients: A meta-analysis of 65 randomized controlled trials. Cancer Chemother. Pharmacol. 2018, 82, 571–583. [Google Scholar] [CrossRef]
- Juweid, M.; Neumann, R.; Paik, C.; Perez-Bacete, M.J.; Sato, J.; van Osdol, W.; Weinstein, J.N. Micropharmacology of monoclonal antibodies in solid tumors: Direct experimental evidence for a binding site barrier. Cancer Res. 1992, 252, 5144–5153. [Google Scholar]
- Iba, Y.; Hayashi, N.; Sawada, J.; Titani, K.; Kurosawa, Y. Changes in the specificity of antibodies against steroid antigens by introduction of mutations into complementarity-determining regions of the V(H) domain. Protein Eng. 1998, 11, 361–370. [Google Scholar] [CrossRef][Green Version]
- Korpimäki, T.; Rosenberg, J.; Virtanen, P.; Lamminmäki, U.; Tuomola, M.; Saviranta, P. Further improvement of broad specificity hapten recognition with protein engineering. Protein Eng. 2003, 16, 37–46. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Riaño-Umbarila, L.; Contreras-Ferrat, G.; Olamendi-Portugal, T.; Morelos-Juárez, C.; Corzo, G.; Possani, L.D.; Becerril, B. Exploiting cross-reactivity to neutralize two different scorpion venoms with one single chain antibody fragment. J. Biol. Chem. 2011, 286, 6143–6151. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Garcia-Rodriguez, C.; Lou, J.; Wen, W.; Conrad, F.; Zhai, W.; Smith, T.J.; Smith, L.A.; Marks, J.D. A three monoclonal antibody combination potently neutralizes multiple botulinum neurotoxin serotype F subtypes. PLoS ONE 2017, 12, e0174187. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rodriguez, C.; Razai, A.; Geren, I.N.; Lou, J.; Conrad, F.; Wen, W.-H.; Farr-Jones, S.; Smith, T.J.; Brown, J.L.; Skerry, J.C.; et al. A three monoclonal antibody combination potently neutralizes multiple botulinum neurotoxin serotype E subtypes. Toxins 2018, 10, 105. [Google Scholar] [CrossRef]
- Kwok, H.F.; Botkjaer, K.A.; Tape, C.J.; Huang, Y.; McCafferty, J.; Murphy, G. Development of a ‘mouse and human cross-reactive’ affinity-matured exosite inhibitory human antibody specific to TACE (ADAM17) for cancer immunotherapy. Protein Eng. Des. Sel. 2014, 27, 179–190. [Google Scholar] [CrossRef][Green Version]
- Mehta, N.; Maddineni, S.; Kelly, R.L.; Lee, R.B.; Hunter, S.A.; Silberstein, J.L.; Sperberg, R.A.P.; Miller, C.L.; Rabe, A.; Labanieh, L.; et al. An engineered antibody binds a distinct epitope and is a potent inhibitor of murine and human VISTA. Sci. Rep. 2020, 10, 15171. [Google Scholar] [CrossRef]
- Bostrom, J.; Yu, S.F.; Kan, D.; Appleton, B.A.; Lee, C.V.; Billeci, K.; Man, W.; Peale, F.; Ross, S.; Wiesmann, C.; et al. Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science 2009, 323, 1610–1614. [Google Scholar] [CrossRef]
- Bostrom, J.; Haber, L.; Koenig, P.; Kelley, R.F.; Fuh, G. High affinity antigen recognition of the dual specific variants of Herceptin is entropy-driven in spite of structural plasticity. PLoS ONE 2011, 6, e17887. [Google Scholar] [CrossRef]
- Schaefer, G.; Haber, L.; Crocker, L.M.; Shia, S.; Shao, L.; Dowbenko, D.; Totpal, K.; Wong, A.; Lee, C.V.; Stawicki, S.; et al. A Two-in-One antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell 2011, 20, 472–486. [Google Scholar] [CrossRef]
- Lee, C.V.; Koenig, P.; Fuh, G. Two-in-One antibody engineered from a humanized interleukin 4 antibody through mutation in heavy chain complementarity-determining regions. mAbs 2014, 6, 622–627. [Google Scholar] [CrossRef]
- Koenig, P.; Lee, C.V.; Sanowar, S.; Wu, P.; Stinson, J.; Harris, S.F.; Fuh, G. Deep sequencing-guided design of a high affinity dual specificity antibody to target two angiogenic factors in neovascular age-related macular degeneration. J. Biol. Chem. 2015, 290, 21773–21786. [Google Scholar] [CrossRef]
- Koenig, P.; Sanowar, S.; Lee, C.V.; Fuh, G. Tuning the specificity of a Two-in-One Fab against three angiogenic antigens by fully utilizing the information of deep mutational scanning. mAbs 2017, 9, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, R.; Jensen, K.; Fenn, S.; Speck, J.; Krause, K.; Meier, A.; Röth, M.; Fauser, S.; Kimbung, R.; Logan, D.T.; et al. DutaFabs are engineered therapeutic Fab fragments that can bind two targets simultaneously. Nat. Commun. 2021, 12, 708. [Google Scholar] [CrossRef] [PubMed]
- Rouet, R.; Jackson, K.J.L.; Langley, D.B.; Christ, D. Next-generation sequencing of antibody display repertoires. Front. Immunol. 2018, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Dyson, M.R.; Masters, E.; Pazeraitis, D.; Perera, R.L.; Syrjanen, J.L.; Surade, S.; Thorsteinson, N.; Parthiban, K.; Jones, P.C.; Sattar, M.; et al. Beyond affinity: Selection of antibody variants with optimal biophysical properties and reduced immunogenicity from mammalian display libraries. mAbs 2020, 12, 1829335. [Google Scholar] [CrossRef] [PubMed]
- Koerber, J.T.; Thomsen, N.D.; Hannigan, B.T.; Degrado, W.F.; Wells, J.A. Nature-inspired design of motif-specific antibody scaffolds. Nat. Biotechnol. 2013, 31, 916–921. [Google Scholar] [CrossRef]
- Stoevesandt, O.; Taussig, M.J. Phospho-specific antibodies by design. Nat. Biotechnol. 2013, 31, 889–891. [Google Scholar] [CrossRef]
- Pantazes, R.J.; Maranas, C.D. OptCDR: A general computational method for the design of antibodies complementarity determining regions for targeted epitope binding. Protein Eng. Des. Sel. 2010, 23, 849–858. [Google Scholar] [CrossRef]
- Entzminger, K.C.; Hyun, J.-M.; Pantazes, R.J.; Patterson-Orazem, A.C.; Querquez, A.N.; Frye, Z.P.; Hughes, R.A.; Ellington, A.D.; Lieberman, R.L.; Maranas, C.D.; et al. De novo design of antibody complementarity determining regions binding a FLAG tetra-peptide. Sci. Rep. 2017, 7, 10295. [Google Scholar] [CrossRef]
- Li, T.; Pantazes, R.J.; Maranas, C.D. OptMAVEn—A new framework for the de novo design of antibody variable region models targeting specific antigen epitopes. PLoS ONE 2014, 9, e105954. [Google Scholar] [CrossRef]
- Poosarla, V.G.; Li, T.; Goh, B.C.; Schulten, K.; Wood, T.K.; Maranas, C.D. Computational de novo design of antibodies binding to a peptide with high affinity. Biotechnol. Bioeng. 2017, 114, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Sormanni, P.; Aprile, F.A.; Vendruscolo, M. Rational design of antibodies targeting specific epitopes within intrinsically disordered proteins. Proc. Natl, Acad. Sci. USA 2015, 112, 9902–9907. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Taylor, R.D.; Griffin, L.; Coker, S.-F.; Adams, R.; Ceska, T.; Shi, J.; Lawson, A.D.G.; Baker, T. Computational design of an epitope-specific Keap1 binding antibody using hotspots residues grafting and CDR loop swapping. Sci. Rep. 2017, 7, 41306. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wang, Y.; Hull, M.; Schultz, P.G.; Wang, F. Rational design of CXCR4 specific antibodies with elongated CDRs. J. Am. Chem. Soc. 2014, 136, 10557–10560. [Google Scholar] [CrossRef]
- Liu, T.; Fu, G.; Luo, X.; Liu, Y.; Wang, Y.; Wang, R.E.; Schultz, P.G.; Wang, F. Rational design of antibody protease inhibitors. J. Am. Chem. Soc. 2015, 137, 4042–4045. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas, G. Understanding and Modulating Antibody Fine Specificity: Lessons from Combinatorial Biology. Antibodies 2022, 11, 48. https://doi.org/10.3390/antib11030048
Rojas G. Understanding and Modulating Antibody Fine Specificity: Lessons from Combinatorial Biology. Antibodies. 2022; 11(3):48. https://doi.org/10.3390/antib11030048
Chicago/Turabian StyleRojas, Gertrudis. 2022. "Understanding and Modulating Antibody Fine Specificity: Lessons from Combinatorial Biology" Antibodies 11, no. 3: 48. https://doi.org/10.3390/antib11030048
APA StyleRojas, G. (2022). Understanding and Modulating Antibody Fine Specificity: Lessons from Combinatorial Biology. Antibodies, 11(3), 48. https://doi.org/10.3390/antib11030048