TCDD Inhibition of IgG1 Production in Experimental Autoimmune Encephalomyelitis (EAE) and In Vitro


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
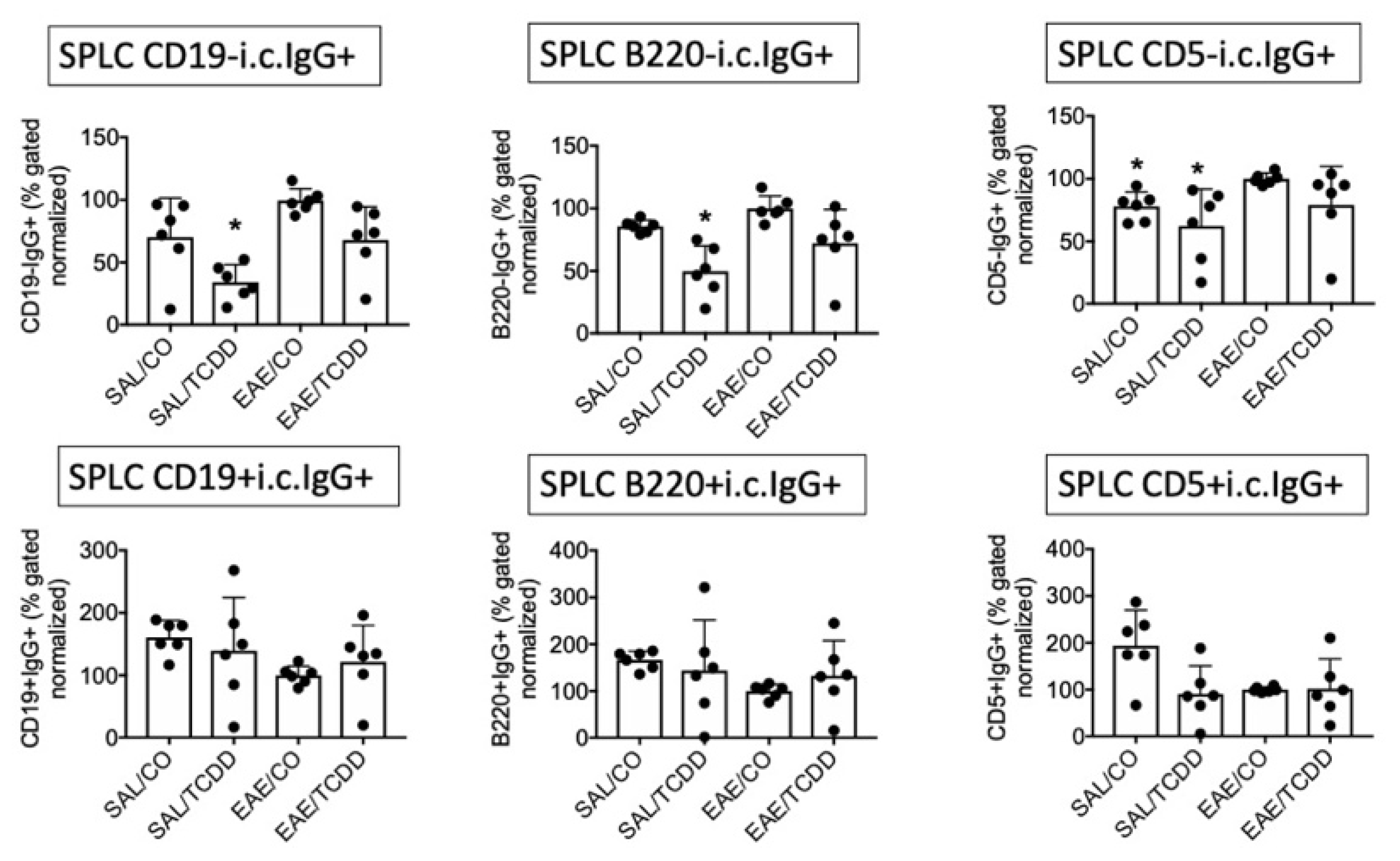
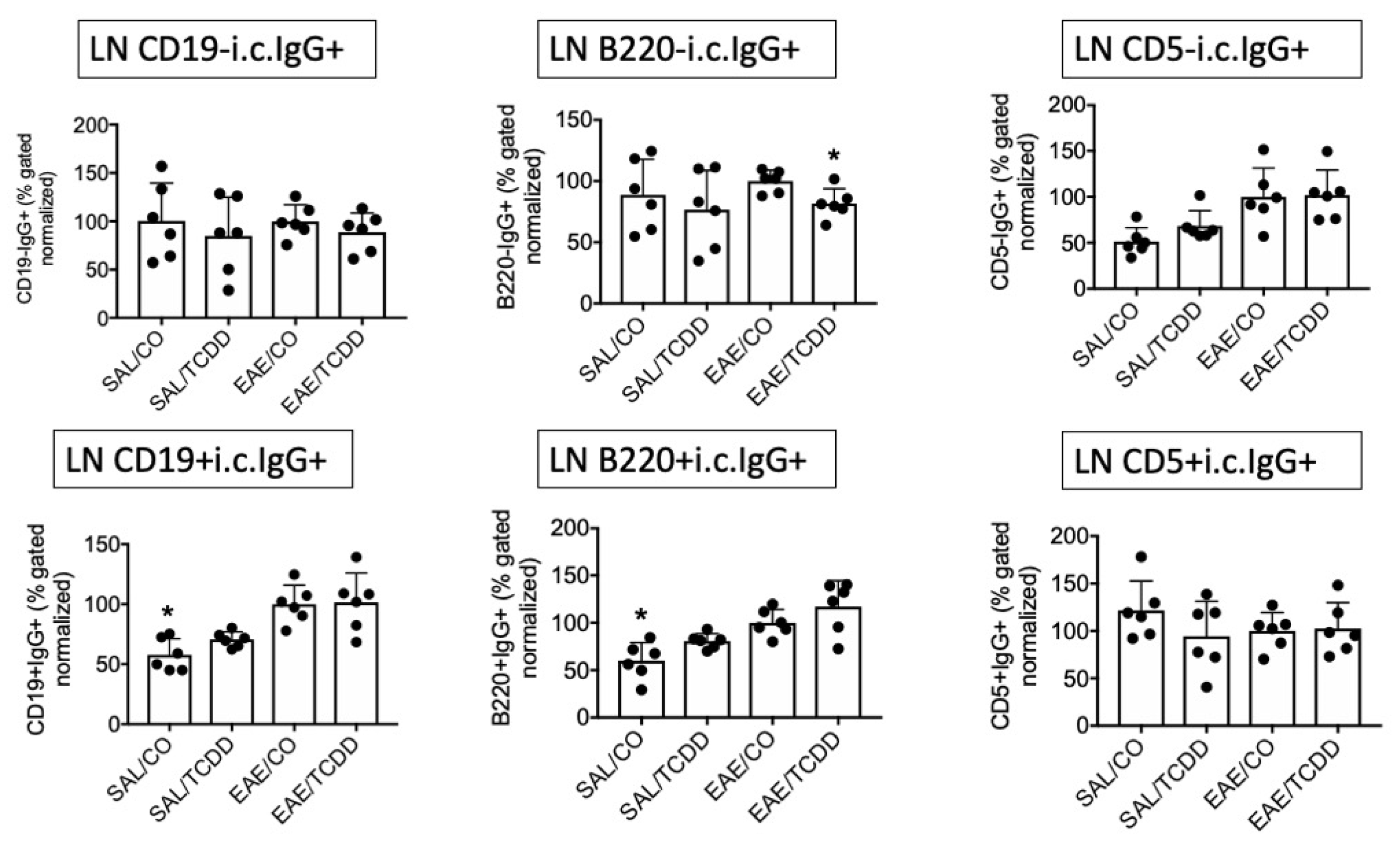
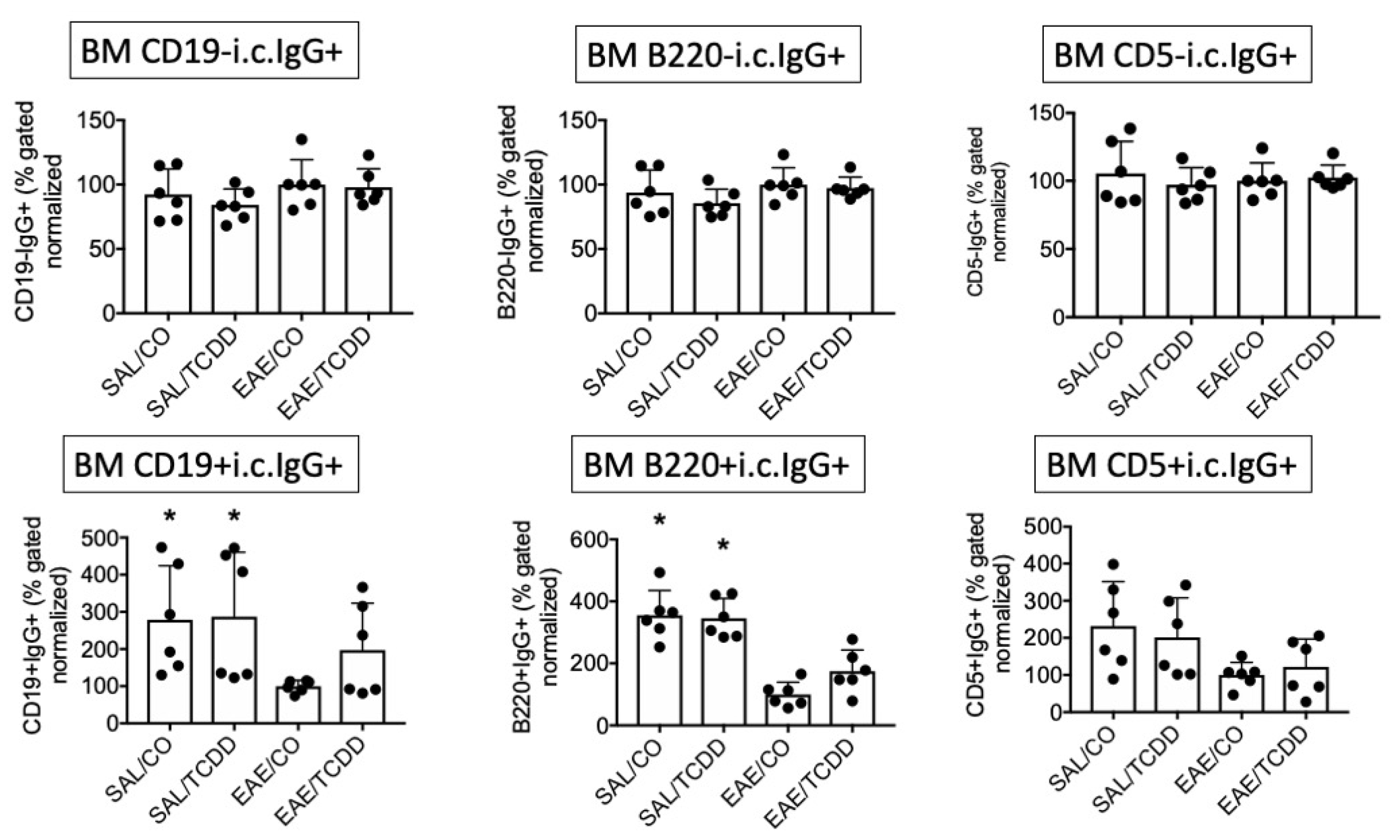
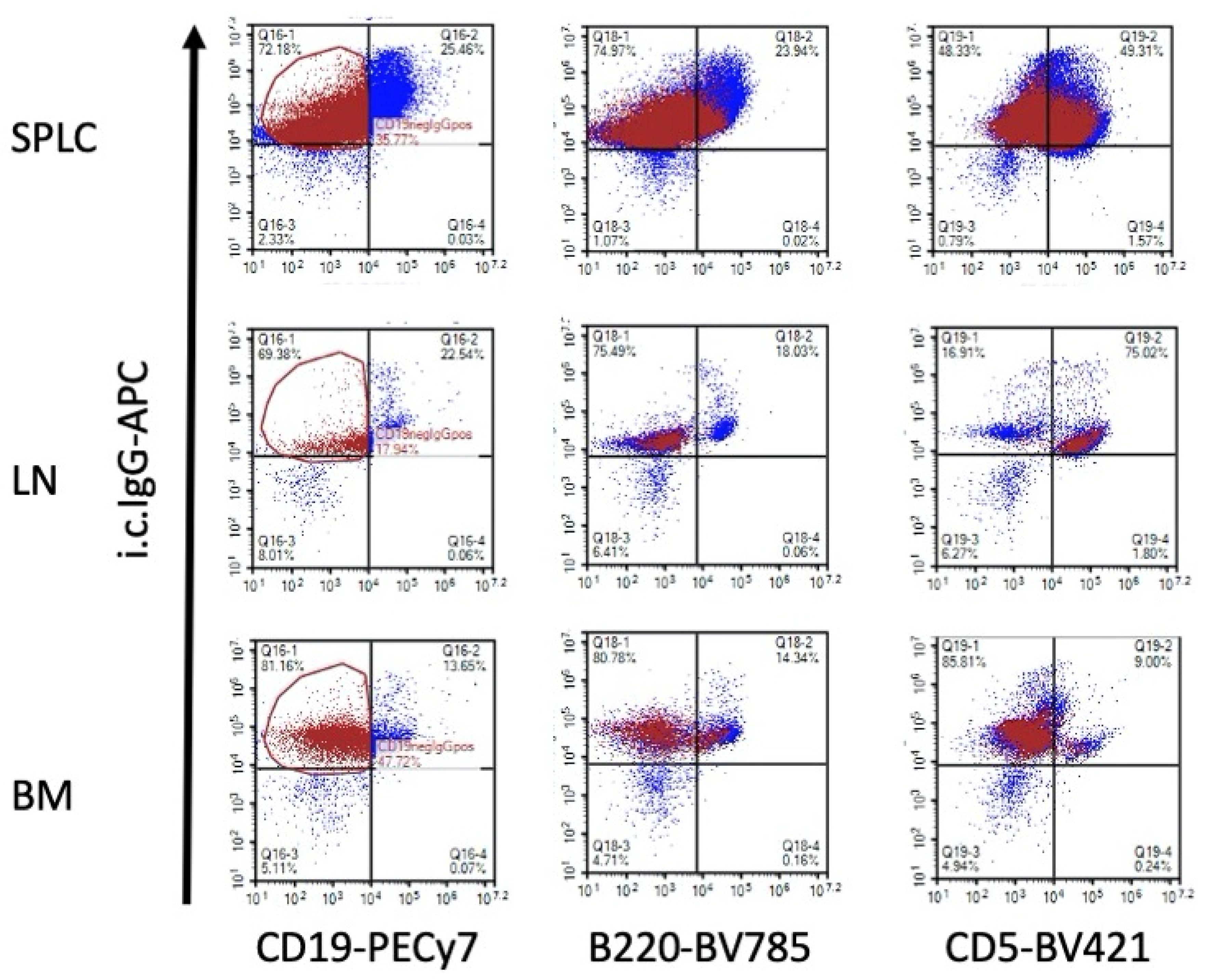
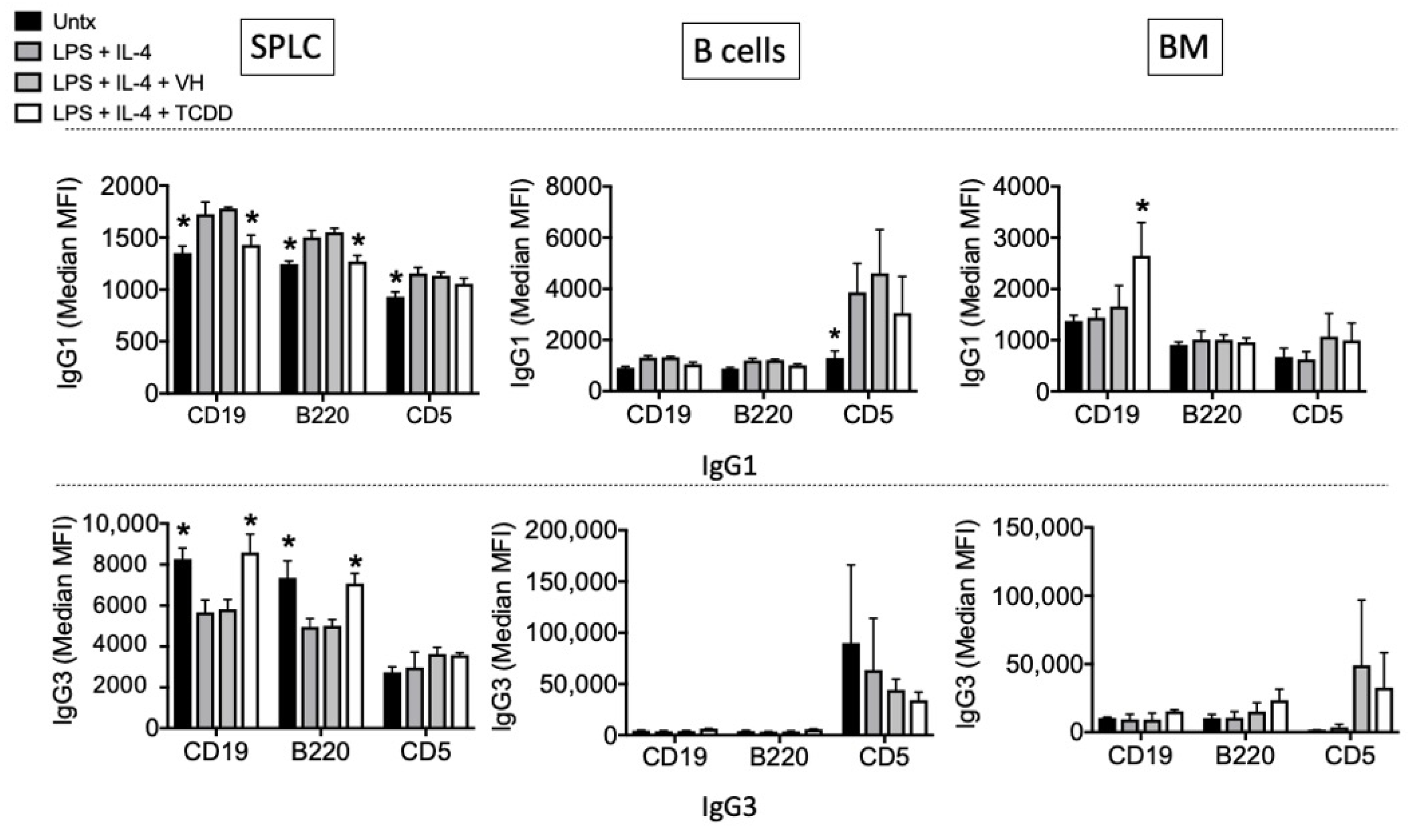
3.1. TCDD Modestly Suppressed the Percent of Intracellular IgG in CD19-, B220- and CD5- Cells in EAE
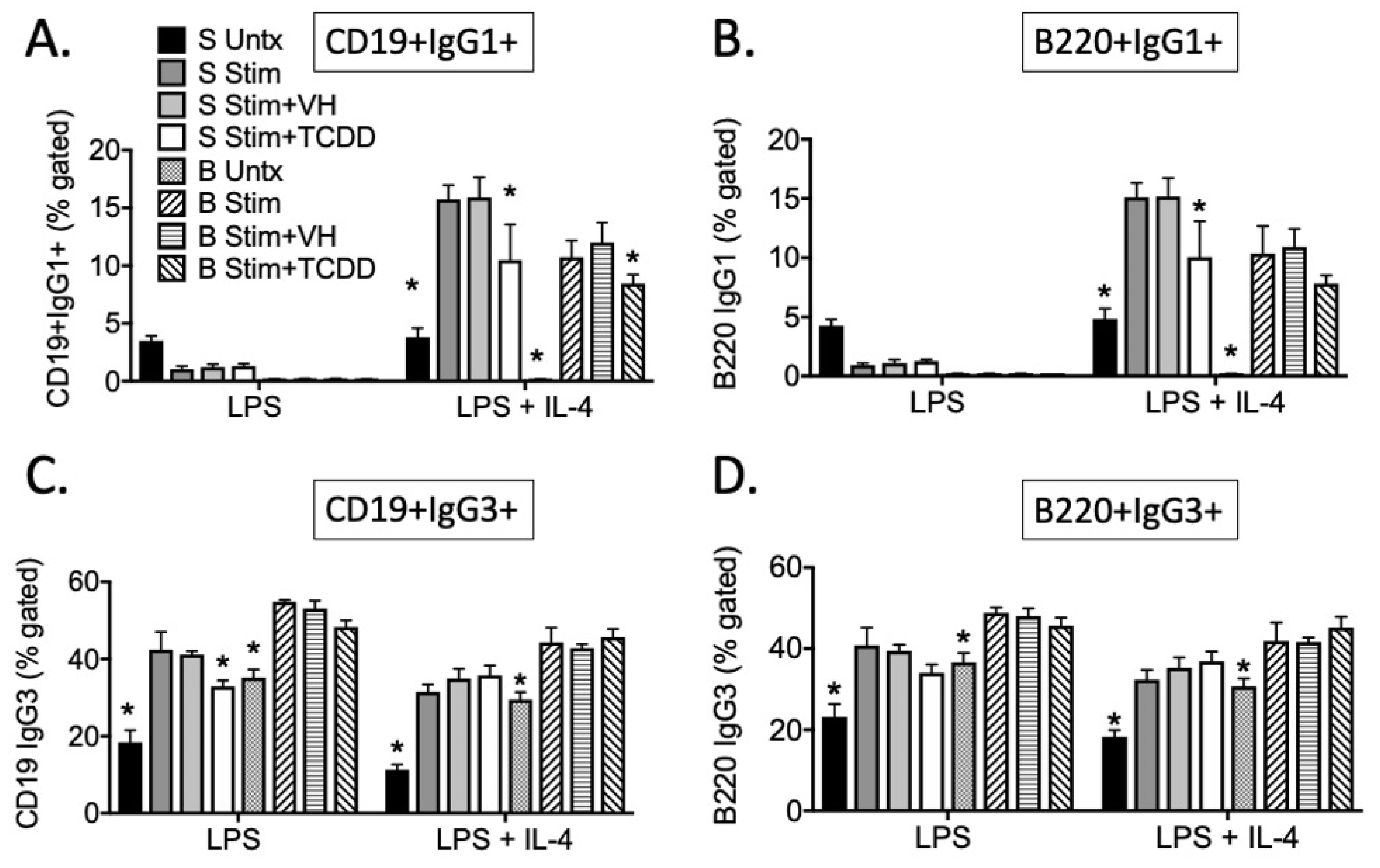
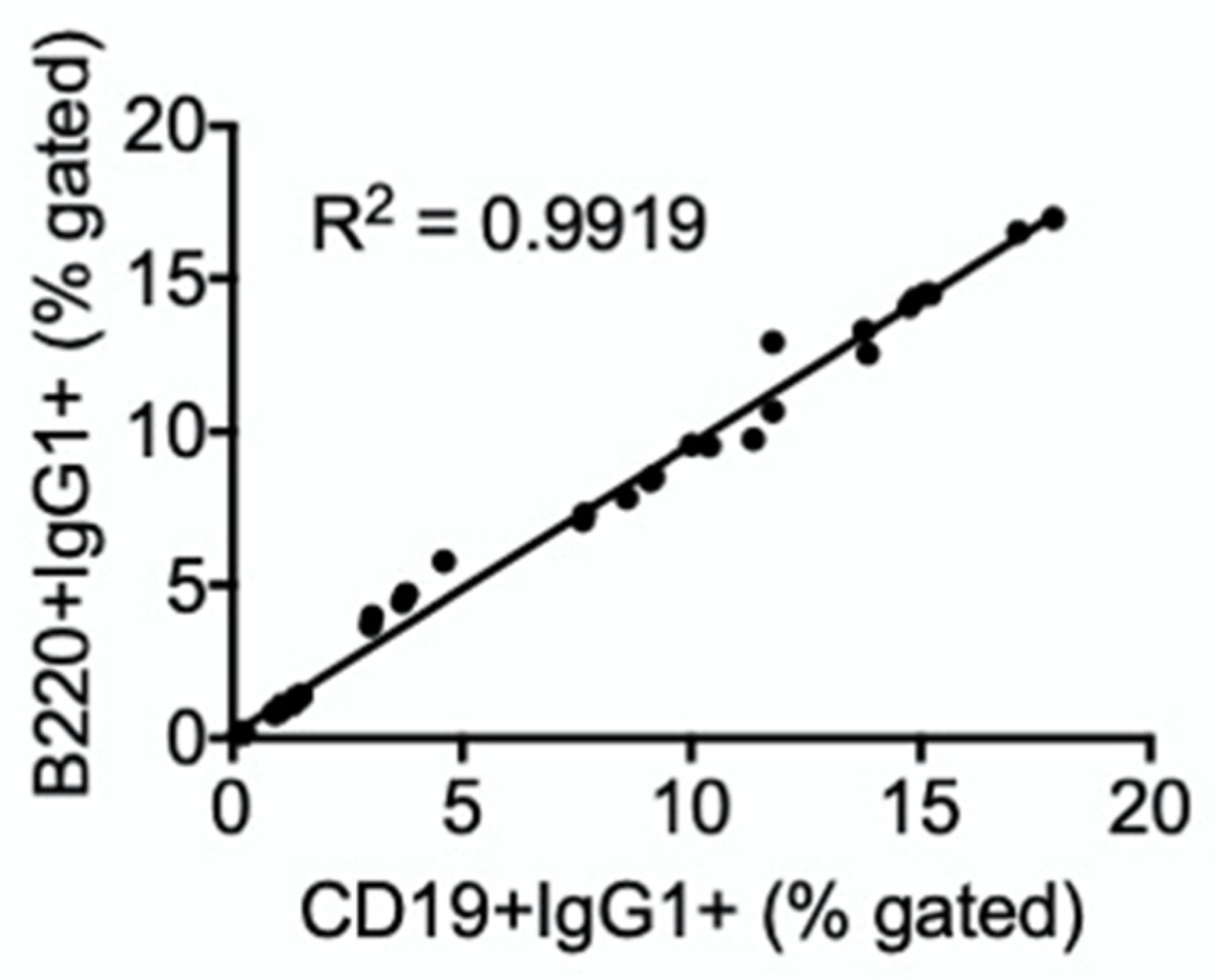
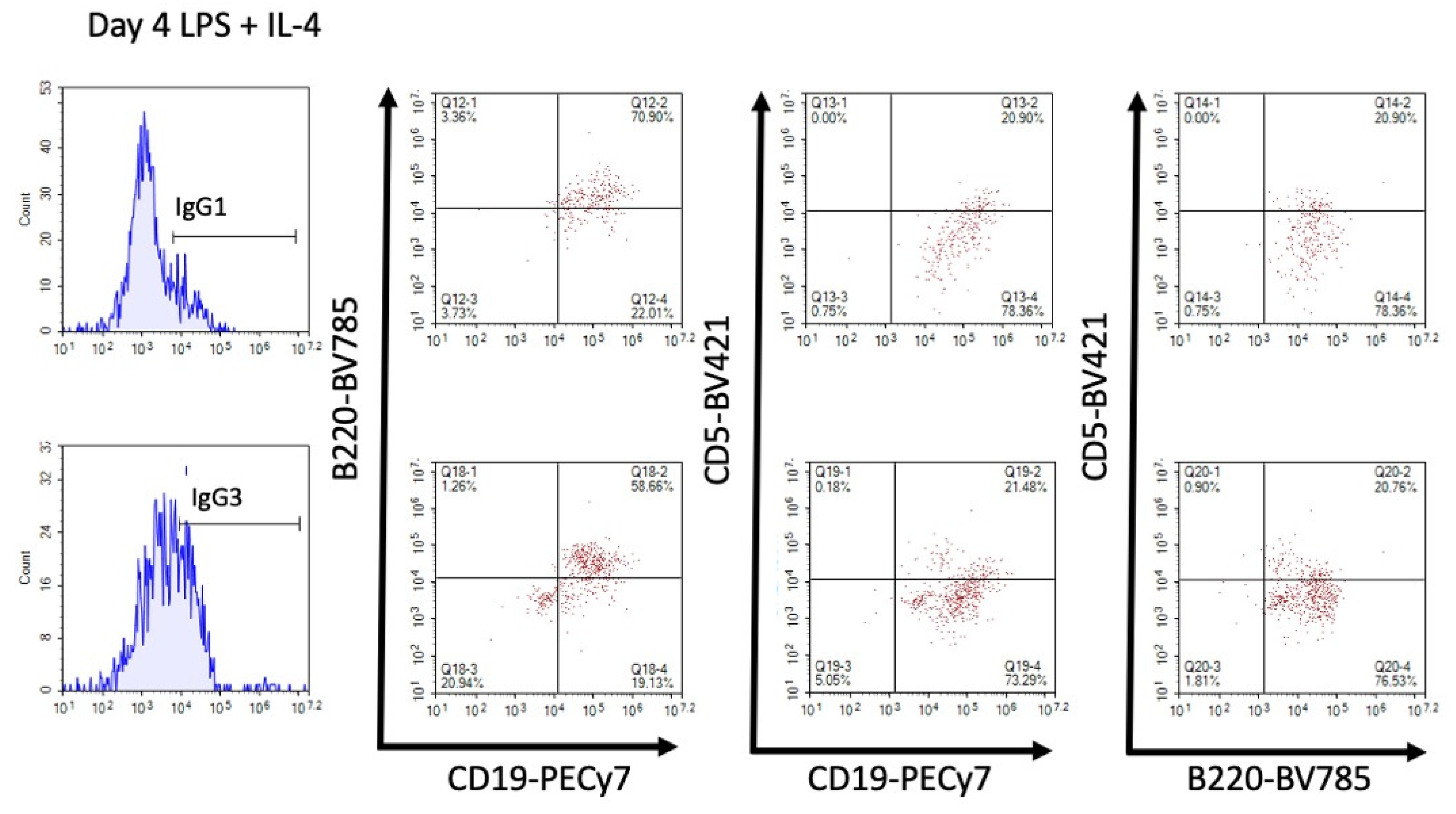
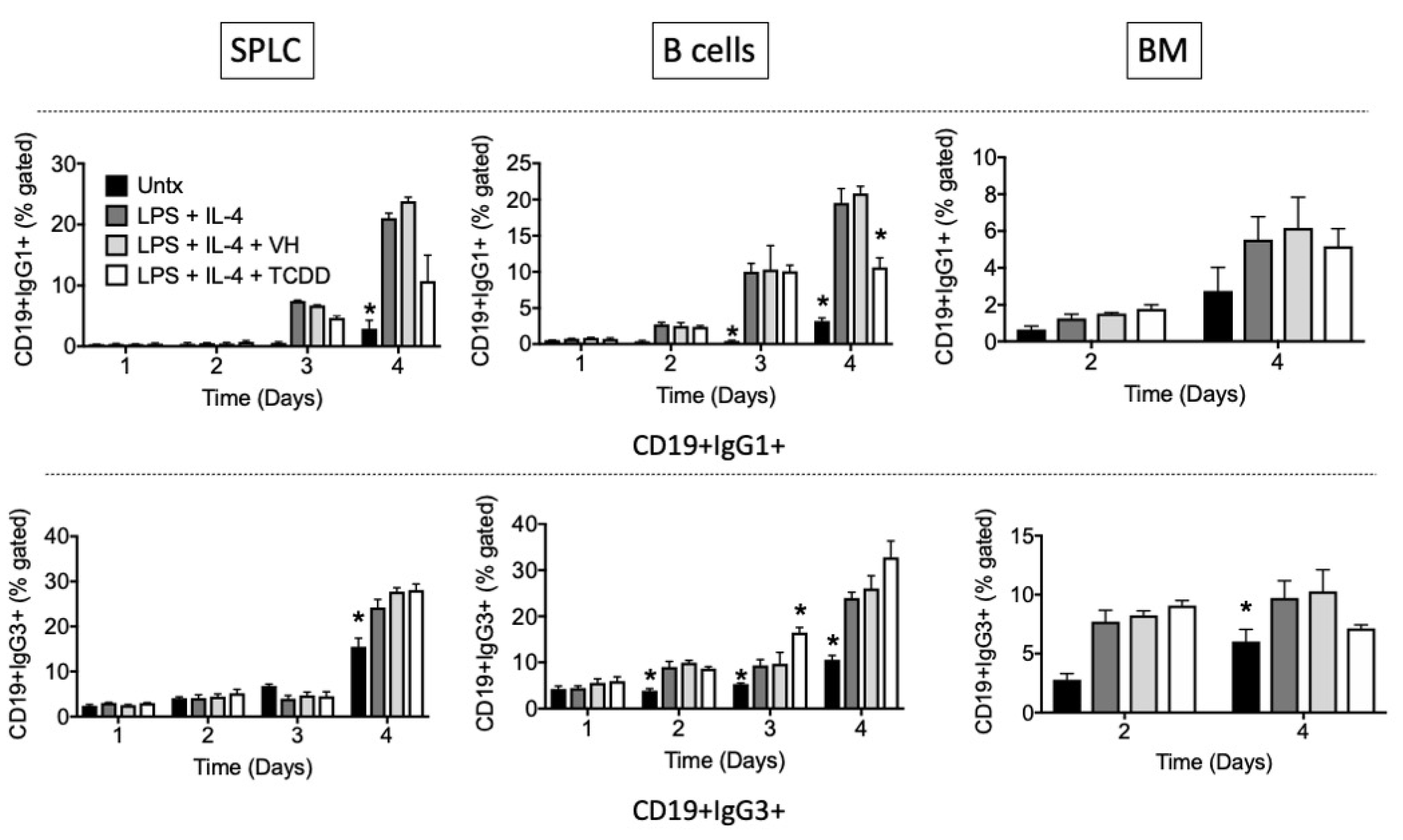
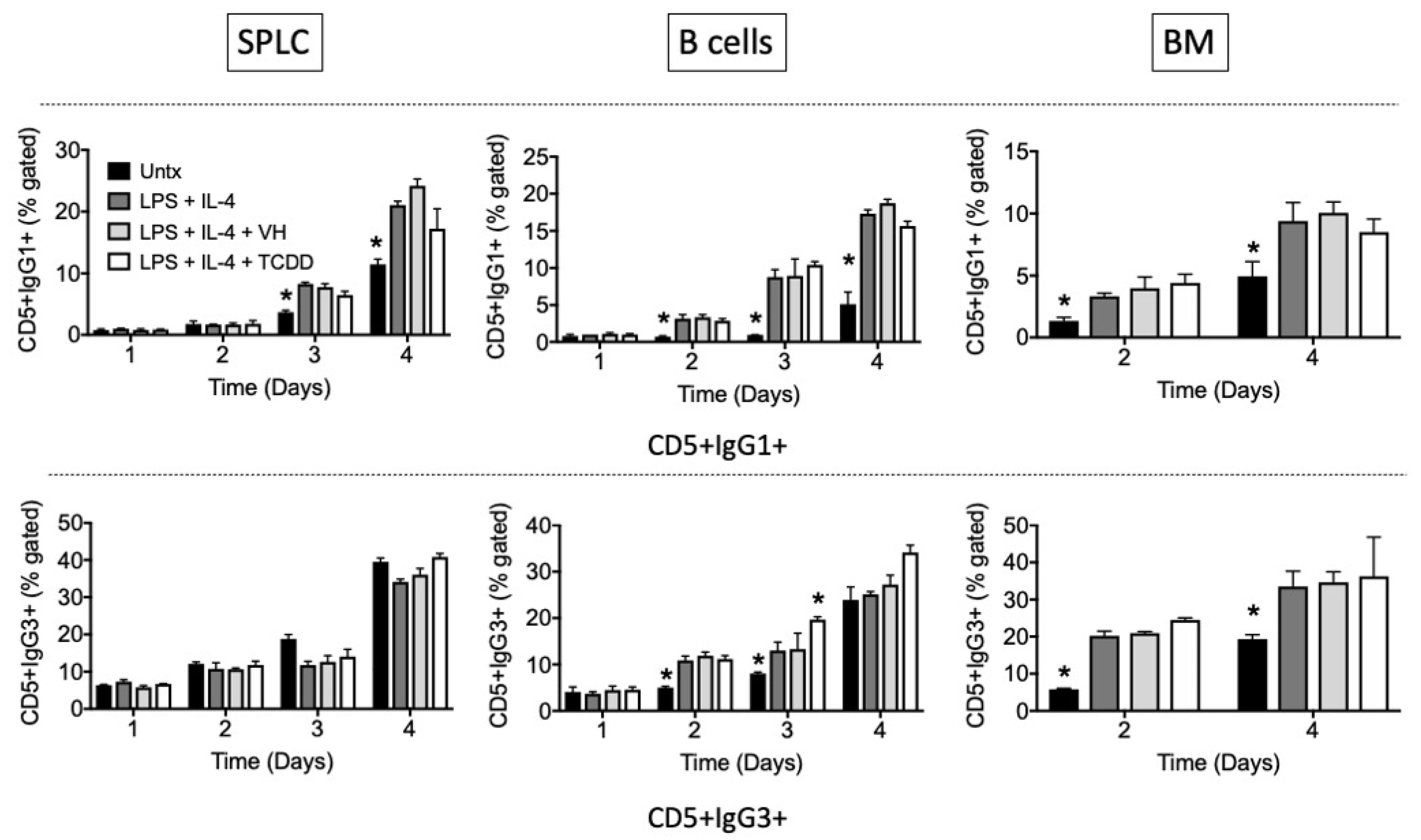
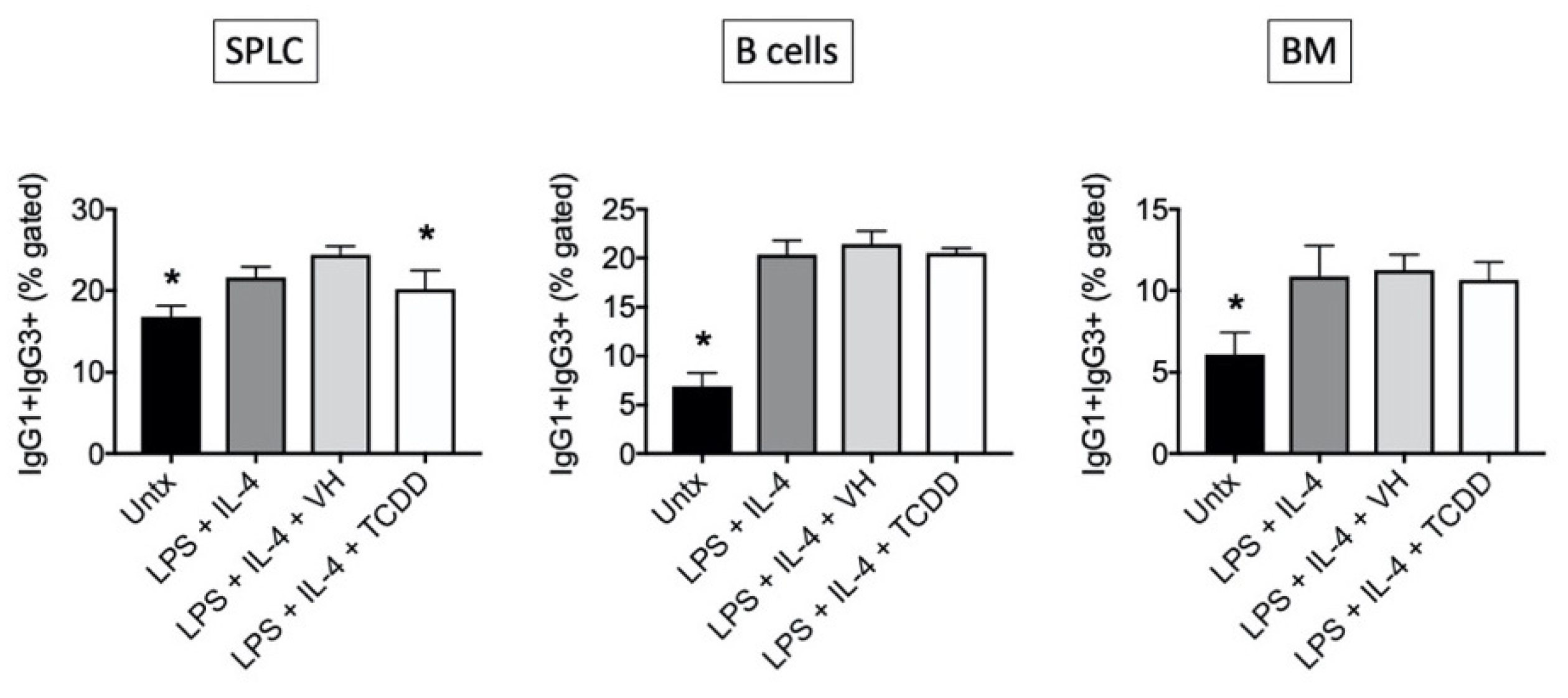
3.2. TCDD Suppressed the Percent of IgG1, but Not IgG3, Expressed on the Cell Surface
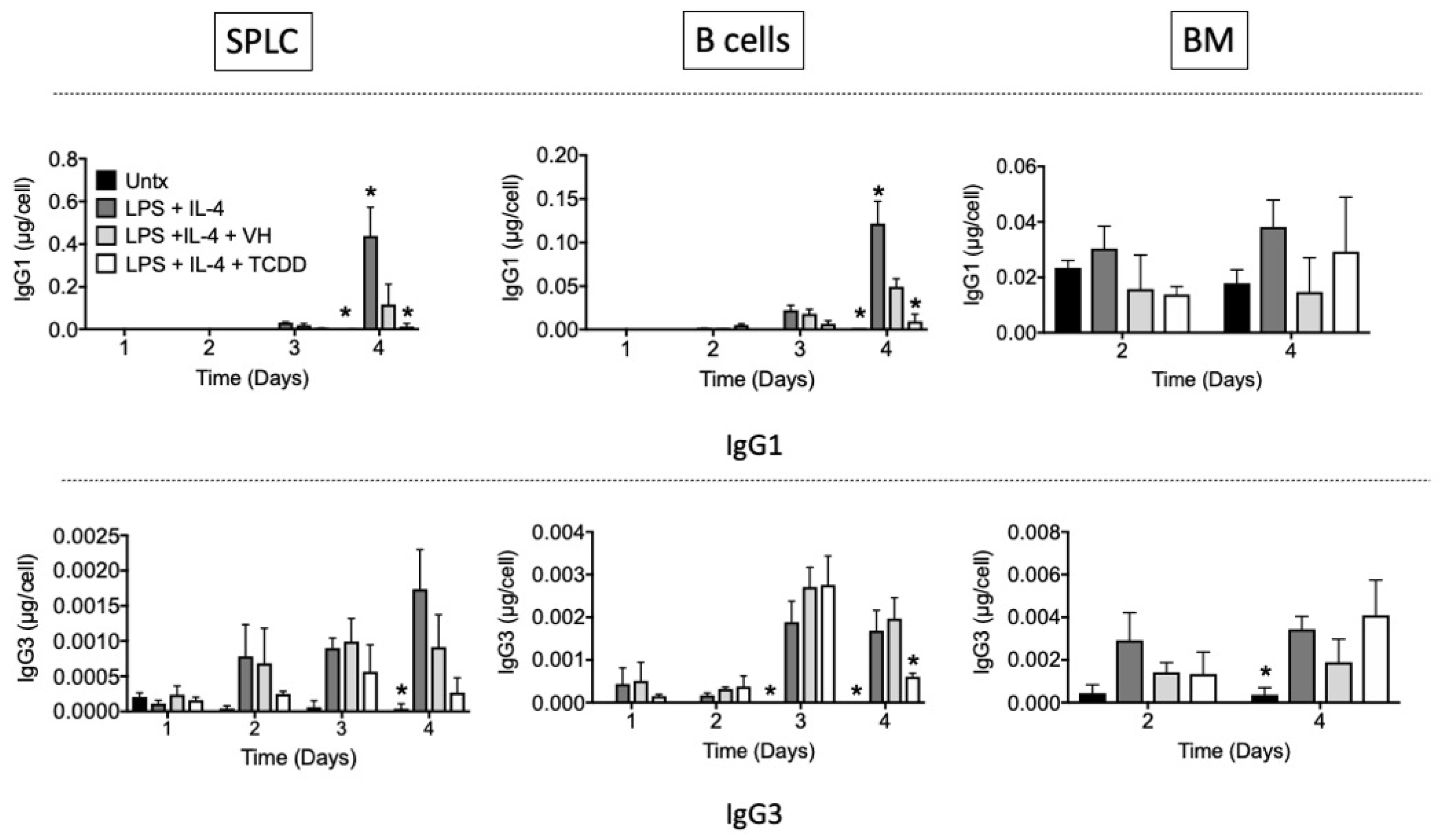
3.3. TCDD Suppressed IgG1 and IgG3 Antibody Secretion
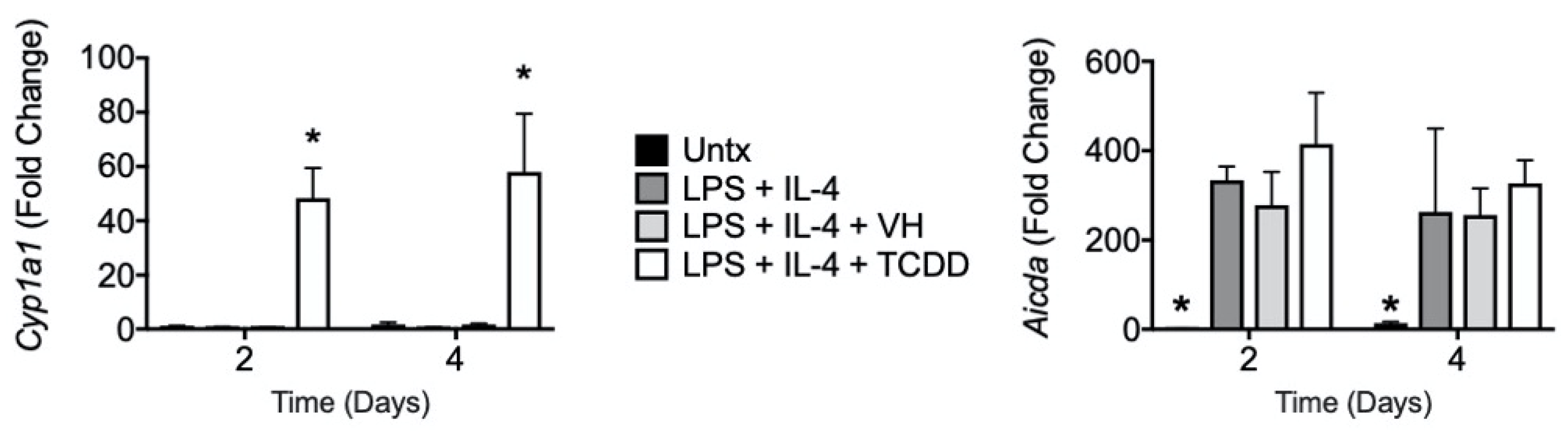
3.4. TCDD Induced Cyp1a1 Gene Expression with No Effect on Aicda
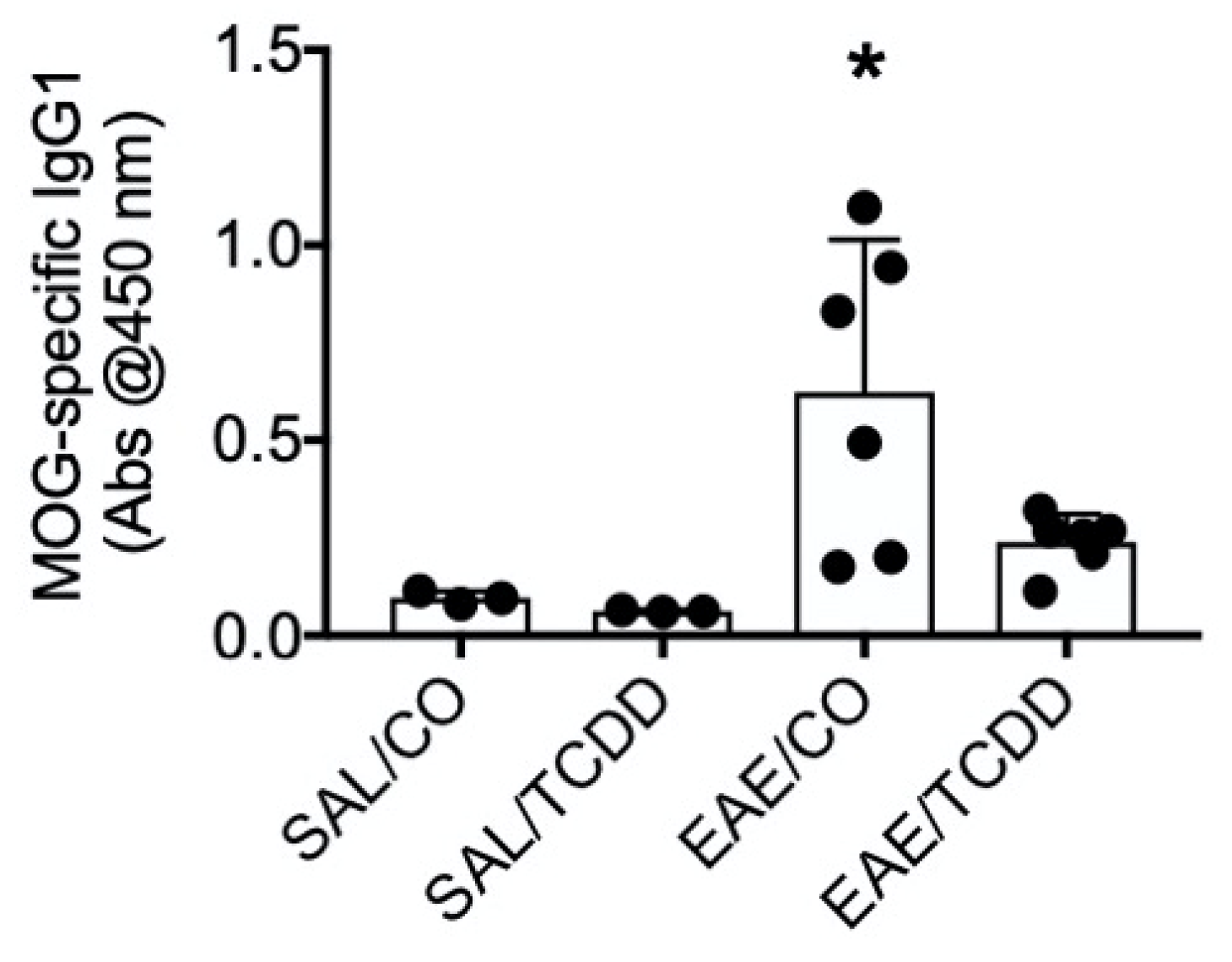
3.5. TCDD Inhibited MOG-Specific IgG1 in EAE Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mandal, P.K. Dioxin: A review of its environmental effects and its aryl hydrocarbon receptor biology. J. Comp. Physiol. B 2005, 175, 221–230. [Google Scholar] [CrossRef]
- Sulentic, C.E.; Kaminski, N.E. The long winding road toward understanding the molecular mechanisms for B-cell suppression by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Sci. 2011, 120 (Suppl 1), S171–S191. [Google Scholar] [CrossRef]
- Sulentic, C.E.; Holsapple, M.P.; Kaminski, N.E. Aryl hydrocarbon receptor-dependent suppression by 2,3,7,8-tetrachlorodibenzo-p-dioxin of IgM secretion in activated B cells. Mol. Pharmacol. 1998, 53, 623–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blevins, L.K.; Zhou, J.; Crawford, R.; Kaminski, N.E. TCDD-mediated suppression of naive human B cell IgM secretion involves aryl hydrocarbon receptor-mediated reduction in STAT3 serine 727 phosphorylation and is restored by interferon-gamma. Cell Signal 2020, 65, 109447. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, R.L.; Sulentic, C.E. The AhR and NF-kappaB/Rel Proteins Mediate the Inhibitory Effect of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin on the 3’ Immunoglobulin Heavy Chain Regulatory Region. Toxicol. Sci. 2015, 148, 443–459. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.; Manzan, M.A.; Crawford, R.B.; Chen, W.; Kaminski, N.E. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated impairment of B cell differentiation involves dysregulation of paired box 5 (Pax5) isoform, Pax5a. J. Pharmacol. Exp. Ther. 2008, 326, 463–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boule, L.A.; Burke, C.G.; Jin, G.B.; Lawrence, B.P. Aryl hydrocarbon receptor signaling modulates antiviral immune responses: Ligand metabolism rather than chemical source is the stronger predictor of outcome. Sci. Rep. 2018, 8, 1826. [Google Scholar] [CrossRef] [PubMed]
- Inouye, K.; Ito, T.; Fujimaki, H.; Takahashi, Y.; Takemori, T.; Pan, X.; Tohyama, C.; Nohara, K. Suppressive effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the high-affinity antibody response in C57BL/6 mice. Toxicol. Sci. 2003, 74, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Karras, J.G.; Conrad, D.H.; Holsapple, M.P. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on interleukin-4-mediated mechanisms of immunity. Toxicol. Lett. 1995, 75, 225–233. [Google Scholar] [CrossRef]
- Kerkvliet, N.I.; Baecher-Steppan, L.; Shepherd, D.M.; Oughton, J.A.; Vorderstrasse, B.A.; DeKrey, G.K. Inhibition of TC-1 cytokine production, effector cytotoxic T lymphocyte development and alloantibody production by 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Immunol. 1996, 157, 2310–2319. [Google Scholar]
- Kummari, E.; Rushing, E.; Nicaise, A.; McDonald, A.; Kaplan, B.L.F. TCDD attenuates EAE through induction of FasL on B cells and inhibition of IgG production. Toxicology 2021, 448, 152646. [Google Scholar] [CrossRef]
- Nohara, K.; Fujimaki, H.; Tsukumo, S.; Inouye, K.; Sone, H.; Tohyama, C. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on T cell-derived cytokine production in ovalbumin (OVA)-immunized C57Bl/6 mice. Toxicology 2002, 172, 49–58. [Google Scholar] [CrossRef]
- Vaidyanathan, B.; Chaudhry, A.; Yewdell, W.T.; Angeletti, D.; Yen, W.F.; Wheatley, A.K.; Bradfield, C.A.; McDermott, A.B.; Yewdell, J.W.; Rudensky, A.Y.; et al. The aryl hydrocarbon receptor controls cell-fate decisions in B cells. J. Exp. Med. 2017, 214, 197–208. [Google Scholar] [CrossRef]
- Baccarelli, A.; Mocarelli, P.; Patterson, D.G., Jr.; Bonzini, M.; Pesatori, A.C.; Caporaso, N.; Landi, M.T. Immunologic effects of dioxin: New results from Seveso and comparison with other studies. Environ. Health Perspect. 2002, 110, 1169–1173. [Google Scholar] [CrossRef] [Green Version]
- Neubert, R.; Maskow, L.; Triebig, G.; Broding, H.C.; Jacob-Muller, U.; Helge, H.; Neubert, D. Chlorinated dibenzo-p-dioxins and dibenzofurans and the human immune system: 3. Plasma immunoglobulins and cytokines of workers with quantified moderately-increased body burdens. Life Sci. 2000, 66, 2123–2142. [Google Scholar] [CrossRef]
- Kim, H.A.; Kim, E.M.; Park, Y.C.; Yu, J.Y.; Hong, S.K.; Jeon, S.H.; Park, K.L.; Hur, S.J.; Heo, Y. Immunotoxicological effects of Agent Orange exposure to the Vietnam War Korean veterans. Ind. Health 2003, 41, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Mannetje, A.; Eng, A.; Walls, C.; Dryson, E.; Douwes, J.; Bertazzi, P.; Ryder-Lewis, S.; Scott, D.; Brooks, C.; McLean, D.; et al. Morbidity in New Zealand pesticide producers exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Environ. Int. 2018, 110, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Thomson, C. IgG Structure and Function. Ref. Mod. Biomed. Sci: Encycl. Immunobiol. 2016, 2, 15–22. [Google Scholar]
- Hoftberger, R.; Guo, Y.; Flanagan, E.P.; Lopez-Chiriboga, A.S.; Endmayr, V.; Hochmeister, S.; Joldic, D.; Pittock, S.J.; Tillema, J.M.; Gorman, M.; et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. 2020, 139, 875–892. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.J.; Stokes, J.V.; Kummari, E.; Eells, J.; Kaplan, B.L. Immunomodulation By Subchronic Low Dose 2,3,7,8-Tetrachlorodibenzo-p-Dioxin in Experimental Autoimmune Encephalomyelitis in the Absence of Pertussis Toxin. Toxicol. Sci. 2016, 151, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lightman, S.M.; Utley, A.; Lee, K.P. Survival of Long-Lived Plasma Cells (LLPC): Piecing Together the Puzzle. Front. Immunol. 2019, 10, 965. [Google Scholar] [CrossRef]
- Crowe, J.E.; Weitkamp, J.-H.; Williams, J.V. B-cell development. In Fetal and Neonatal Physiology; W. B. Saunders Company: Philadelphia, PA, USA, 2004; Volume 2, pp. 1518–1522. [Google Scholar]
- Li, G.; Pone, E.J.; Tran, D.C.; Patel, P.J.; Dao, L.; Xu, Z.; Casali, P. Iron inhibits activation-induced cytidine deaminase enzymatic activity and modulates immunoglobulin class switch DNA recombination. J. Biol. Chem. 2012, 287, 21520–21529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Katsuya, K.; Oka, T.; Koizumi, S.; Wakita, D.; Kitamura, H.; Nishimura, T. Effects of AhR ligands on the production of immunoglobulins in purified mouse B cells. Biomed. Res. 2012, 33, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Wourms, M.J.; Sulentic, C.E. The aryl hydrocarbon receptor regulates an essential transcriptional element in the immunoglobulin heavy chain gene. Cell Immunol. 2015, 295, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Pracht, K.; Meinzinger, J.; Daum, P.; Schulz, S.R.; Reimer, D.; Hauke, M.; Roth, E.; Mielenz, D.; Berek, C.; Corte-Real, J.; et al. A new staining protocol for detection of murine antibody-secreting plasma cell subsets by flow cytometry. Eur. J. Immunol. 2017, 47, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Dasgupta, S.; Bandyopadhyay, M. Regulatory B cells in infection, inflammation, and autoimmunity. Cell Immunol. 2020, 352, 104076. [Google Scholar] [CrossRef]
- van de Veen, W.; Stanic, B.; Wirz, O.F.; Jansen, K.; Globinska, A.; Akdis, M. Role of regulatory B cells in immune tolerance to allergens and beyond. J. Allergy Clin. Immunol. 2016, 138, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Blevins, L.K.; Zhou, J.; Crawford, R.B.; Kaminski, N.E. Identification of a Sensitive Human Immunological Target of Aryl Hydrocarbon Receptor Activation: CD5(+) Innate-Like B Cells. Front. Immunol. 2021, 12, 635748. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Zhang, Q.; Yan, H.; Yang, Y.; Wang, P.; Zhang, Y.; Deng, Z.; Yu, M.; Zhou, W.; Wang, Q.; et al. More than one antibody of individual B cells revealed by single-cell immune profiling. Cell Discov. 2019, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Piddlesden, S.J.; Lassmann, H.; Zimprich, F.; Morgan, B.P.; Linington, C. The demyelinating potential of antibodies to myelin oligodendrocyte glycoprotein is related to their ability to fix complement. Am. J. Pathol. 1993, 143, 555–564. [Google Scholar] [CrossRef]
- Calida, D.M.; Constantinescu, C.; Purev, E.; Zhang, G.X.; Ventura, E.S.; Lavi, E.; Rostami, A. Cutting edge: C3, a key component of complement activation, is not required for the development of myelin oligodendrocyte glycoprotein peptide-induced experimental autoimmune encephalomyelitis in mice. J. Immunol. 2001, 166, 723–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piddlesden, S.; Lassmann, H.; Laffafian, I.; Morgan, B.P.; Linington, C. Antibody-mediated demyelination in experimental allergic encephalomyelitis is independent of complement membrane attack complex formation. Clin. Exp. Immunol. 1991, 83, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Kinzel, S.; Lehmann-Horn, K.; Torke, S.; Hausler, D.; Winkler, A.; Stadelmann, C.; Payne, N.; Feldmann, L.; Saiz, A.; Reindl, M.; et al. Myelin-reactive antibodies initiate T cell-mediated CNS autoimmune disease by opsonization of endogenous antigen. Acta Neuropathol. 2016, 132, 43–58. [Google Scholar] [CrossRef] [Green Version]
- Kerkvliet, N.I.; Steppan, L.B.; Vorachek, W.; Oda, S.; Farrer, D.; Wong, C.P.; Pham, D.; Mourich, D.V. Activation of aryl hydrocarbon receptor by TCDD prevents diabetes in NOD mice and increases Foxp3+ T cells in pancreatic lymph nodes. Immunotherapy 2009, 1, 539–547. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, J.; Takeuchi, M.; Usui, Y.; Hattori, T.; Okunuki, Y.; Yamakawa, N.; Kezuka, T.; Kuroda, M.; Goto, H. Suppression of experimental autoimmune uveoretinitis by inducing differentiation of regulatory T cells via activation of aryl hydrocarbon receptor. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; McMurray, R.W. Effects of chronic exposure to DDT and TCDD on disease activity in murine systemic lupus erythematosus. Lupus 2009, 18, 941–949. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicaise, A.J.; McDonald, A.; Sears, E.R.; Sturgis, T.; Kaplan, B.L.F. TCDD Inhibition of IgG1 Production in Experimental Autoimmune Encephalomyelitis (EAE) and In Vitro. Antibodies 2022, 11, 4. https://doi.org/10.3390/antib11010004
Nicaise AJ, McDonald A, Sears ER, Sturgis T, Kaplan BLF. TCDD Inhibition of IgG1 Production in Experimental Autoimmune Encephalomyelitis (EAE) and In Vitro. Antibodies. 2022; 11(1):4. https://doi.org/10.3390/antib11010004
Chicago/Turabian StyleNicaise, Ashleigh J., Amye McDonald, Erin Rushing Sears, Trell Sturgis, and Barbara L. F. Kaplan. 2022. "TCDD Inhibition of IgG1 Production in Experimental Autoimmune Encephalomyelitis (EAE) and In Vitro" Antibodies 11, no. 1: 4. https://doi.org/10.3390/antib11010004
APA StyleNicaise, A. J., McDonald, A., Sears, E. R., Sturgis, T., & Kaplan, B. L. F. (2022). TCDD Inhibition of IgG1 Production in Experimental Autoimmune Encephalomyelitis (EAE) and In Vitro. Antibodies, 11(1), 4. https://doi.org/10.3390/antib11010004