
Glycoengineering of Therapeutic Antibodies with Small Molecule Inhibitors

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Immunoglobulins and Their Glycans
2.1. Immunoglobulins and Their Functions
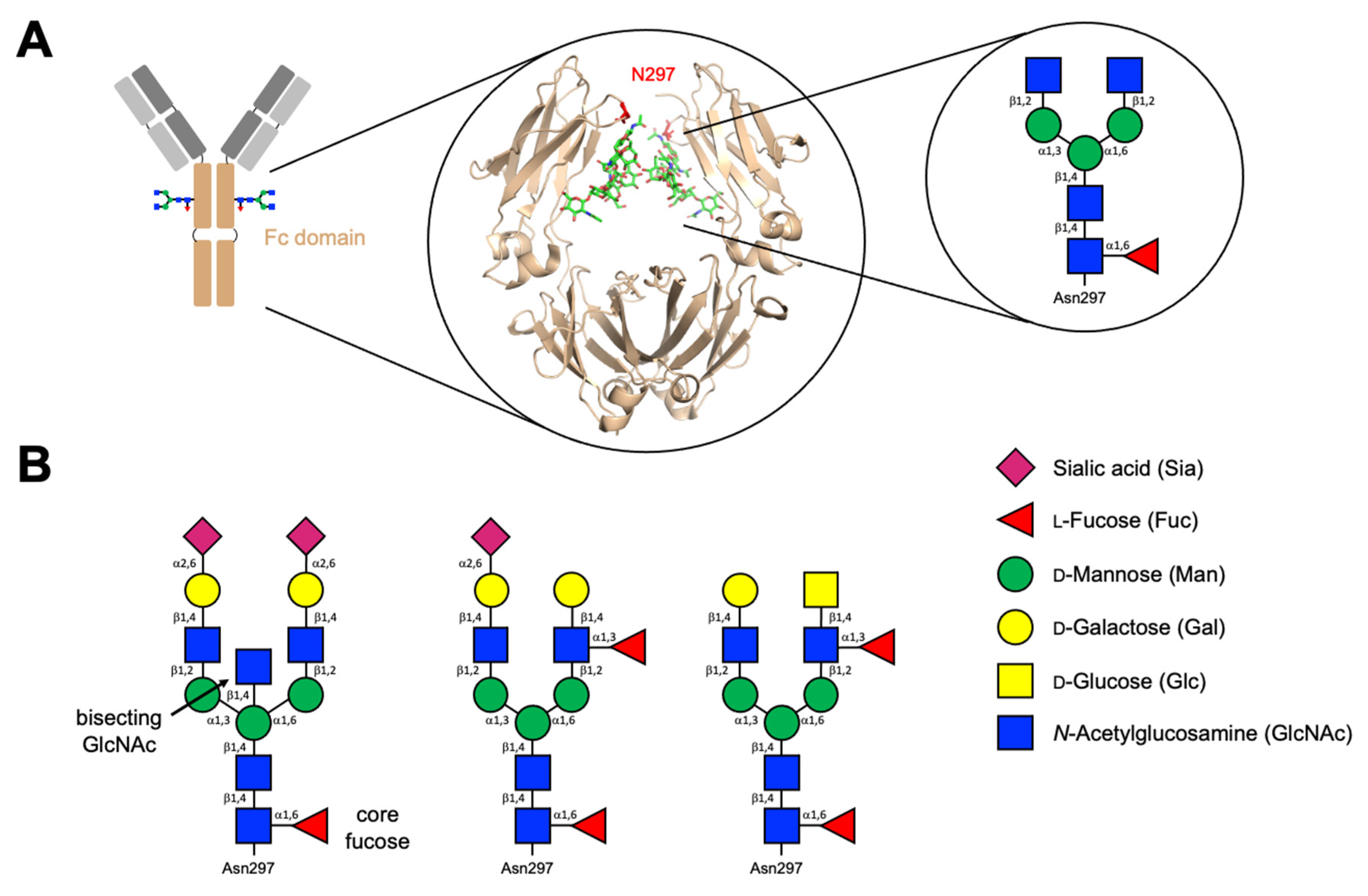
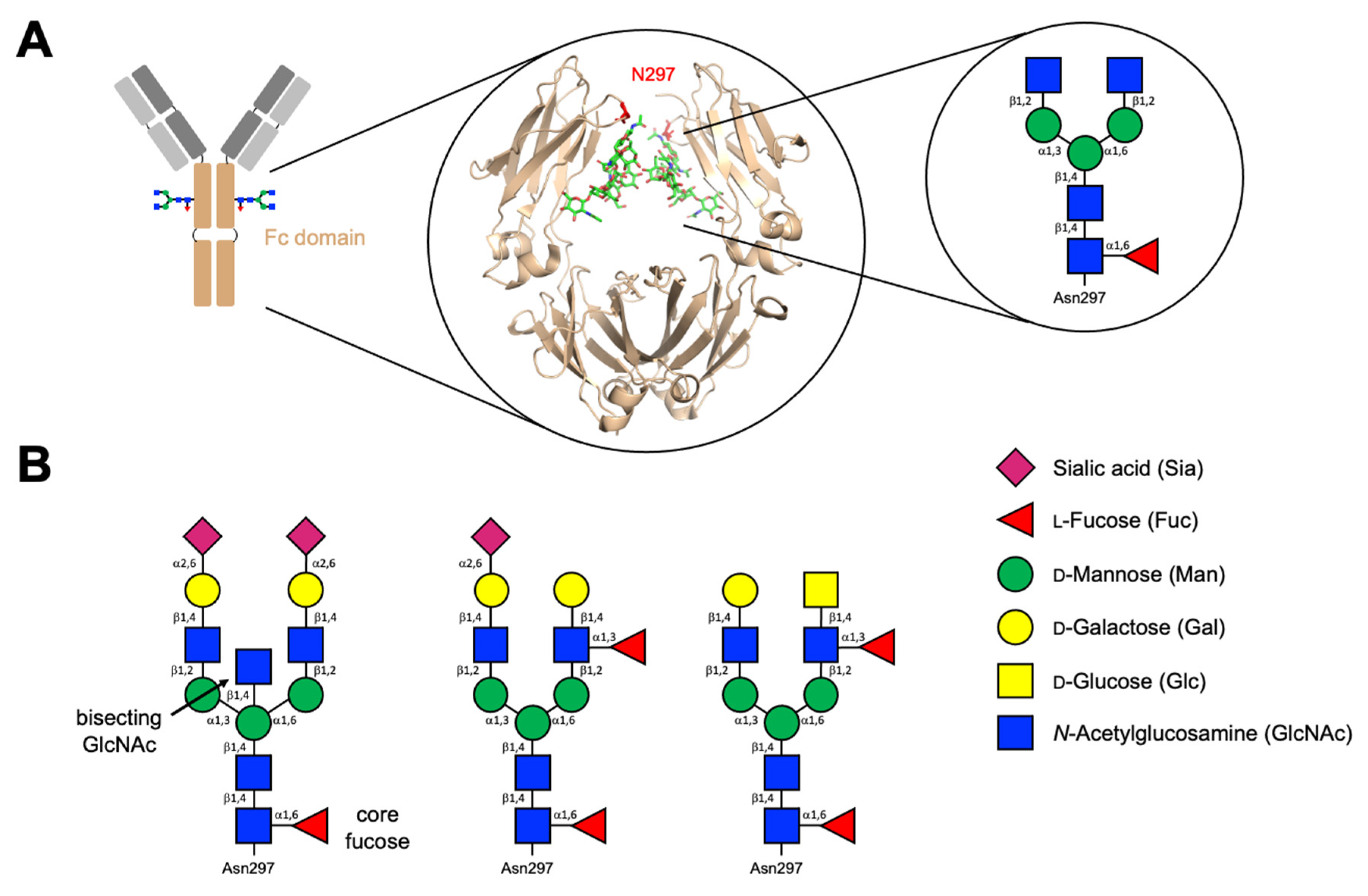
2.2. Immunoglobulins and Their Glycans
2.3. mAb Glycoengineering
3. Small Molecule Inhibitors for the Glycoengineering of Monoclonal Antibodies
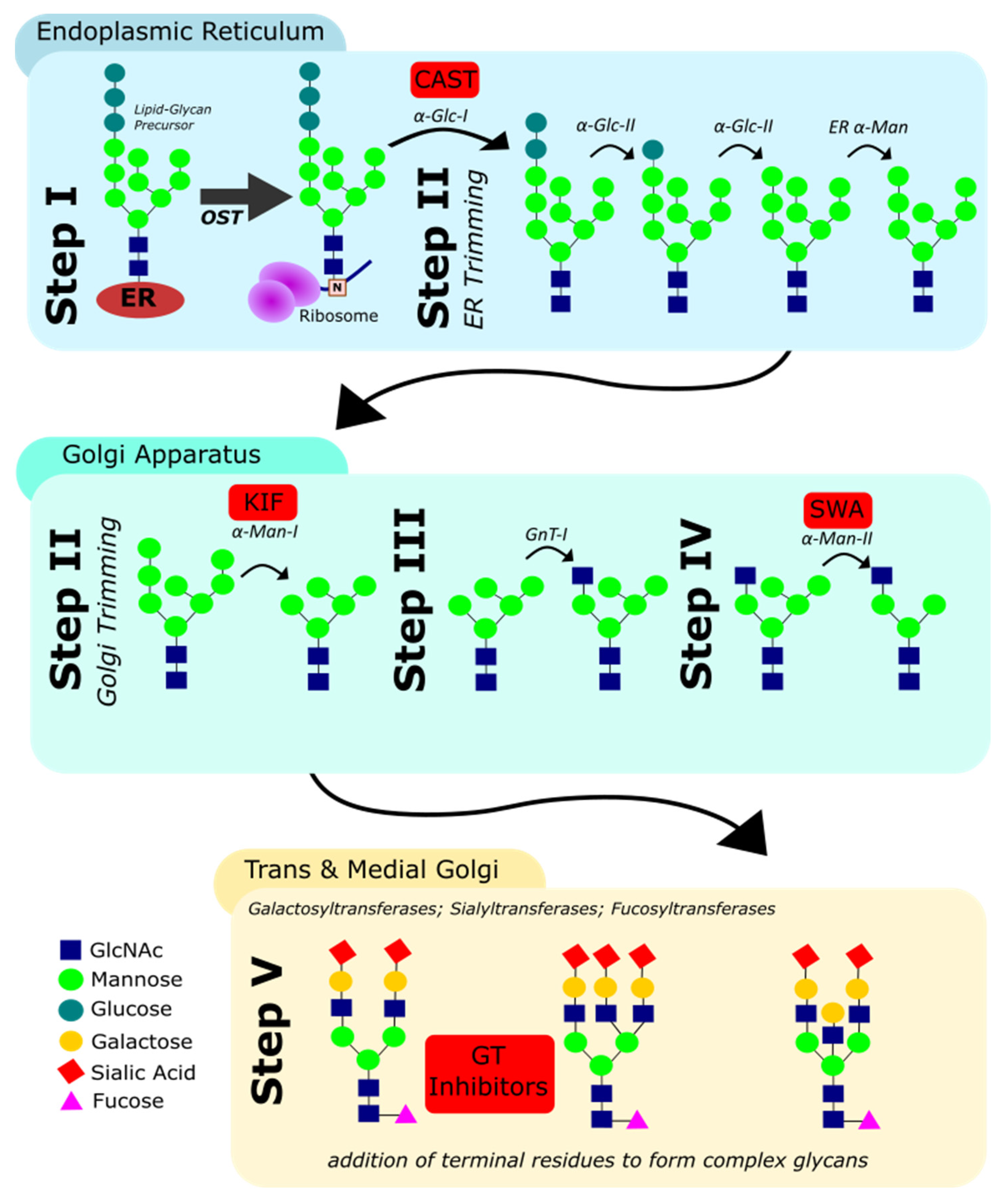
3.1. N-Glycan Biosynthesis in Eukaryotes
- (i)
- The en bloc transfer of an oligosaccharide to the asparagine residue in N-X-ST sequons of the nascent polypeptide by the oligosaccharyl transferase (OST);
- (ii)
- The trimming of terminal d-glucose (Glc) and d-mannose (Man) residues by glucosidases and mannosidases in the ER and cis-Golgi;
- (iii)
- The addition of a N-acetyl d-glucosamine (GlcNAc) residue onto the intermediate Man5GlcNAc2 structure;
- (iv)
- The further removal of Man residues by mannosidases in the medial-Golgi; and finally;
- (v)
- The elaboration of the resulting hexasaccharide by addition of GlcNAc, d-galactose (Gal), L-fucose (Fuc) and sialic acid (Sia) residues. These final steps, including the addition of terminal residues, are catalysed by different glycosyltransferases in the medial- and trans-Golgi.
3.2. mAb Expression Systems
3.3. Small Molecule Inhibitors: Advantages and Challenges
- -
- High potency against its molecular target;
- -
- Target specificity (or known off-target profile);
- -
- Good cellular uptake and activity in cell culture;
- -
- Chemical and enzymatic stability in the culture medium;
- -
- No cell toxicity;
- -
- No detrimental effect on antibody yield.
- -
- While many inhibitors of carbohydrate-active enzymes such as glycosidases and glycosyltransferases have been reported, the number of inhibitors with suitable properties for applications in cell culture is still limited.
3.4. Inhibitors of Carbohydrate-Active Enzymes for mAb Glycoengineering
3.4.1. Glycosidase Inhibitors
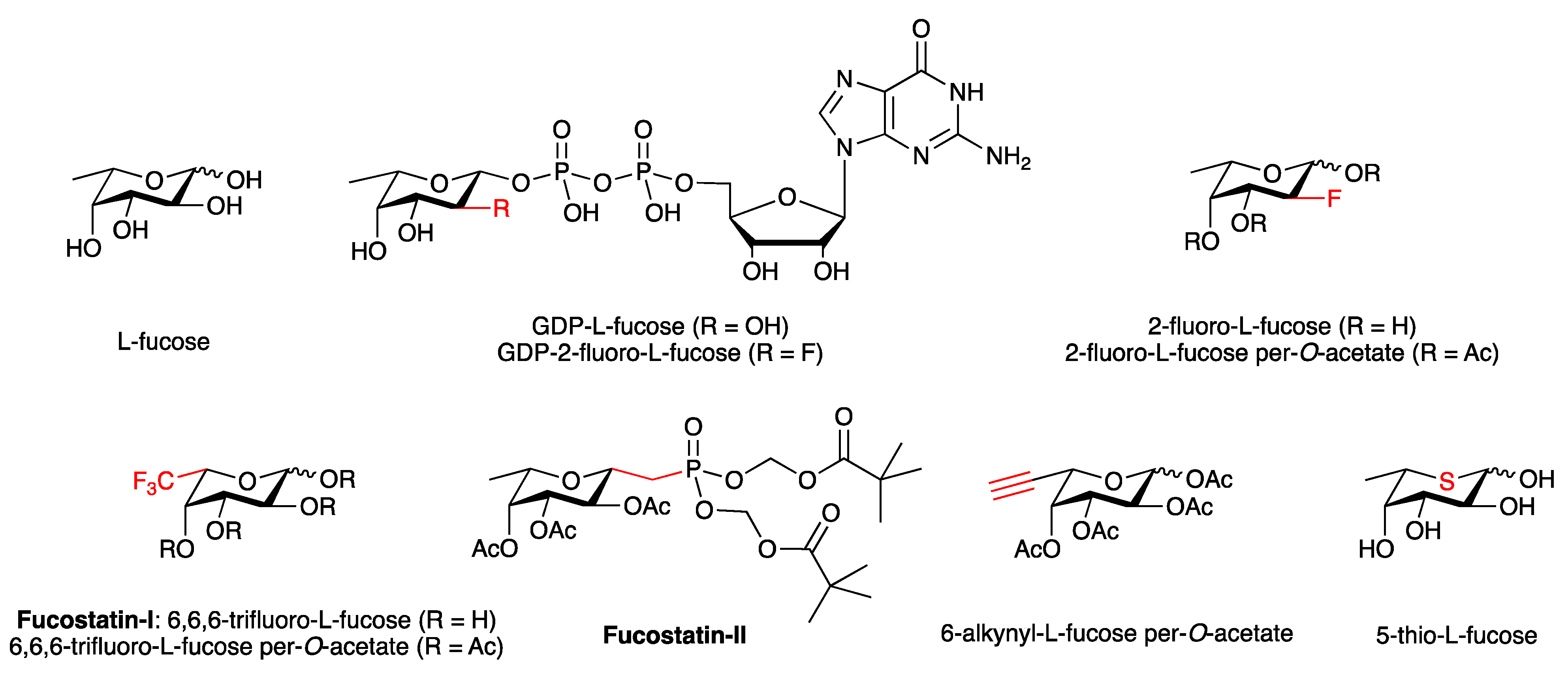
3.4.2. Fucosylation Inhibitors
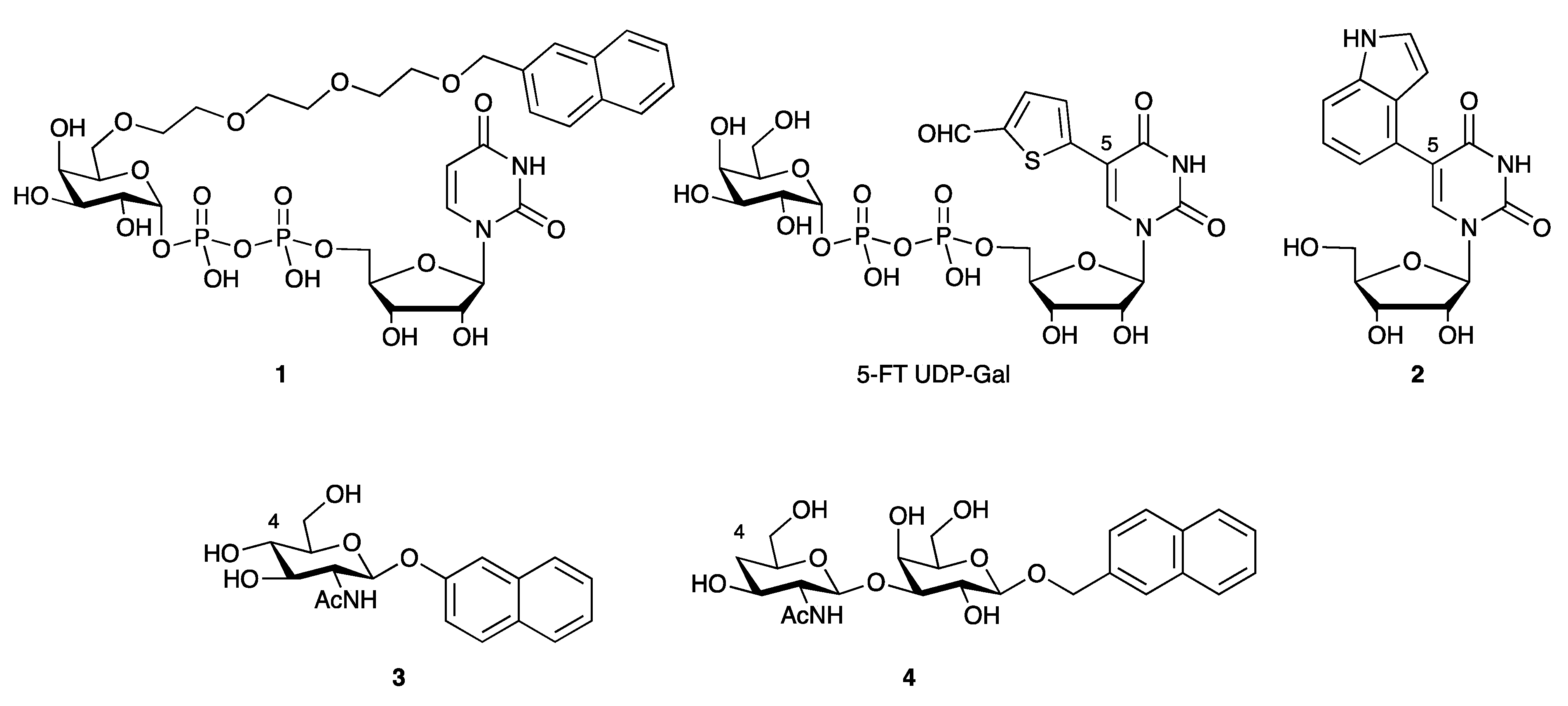
3.4.3. Galactosyltransferase Inhibitors
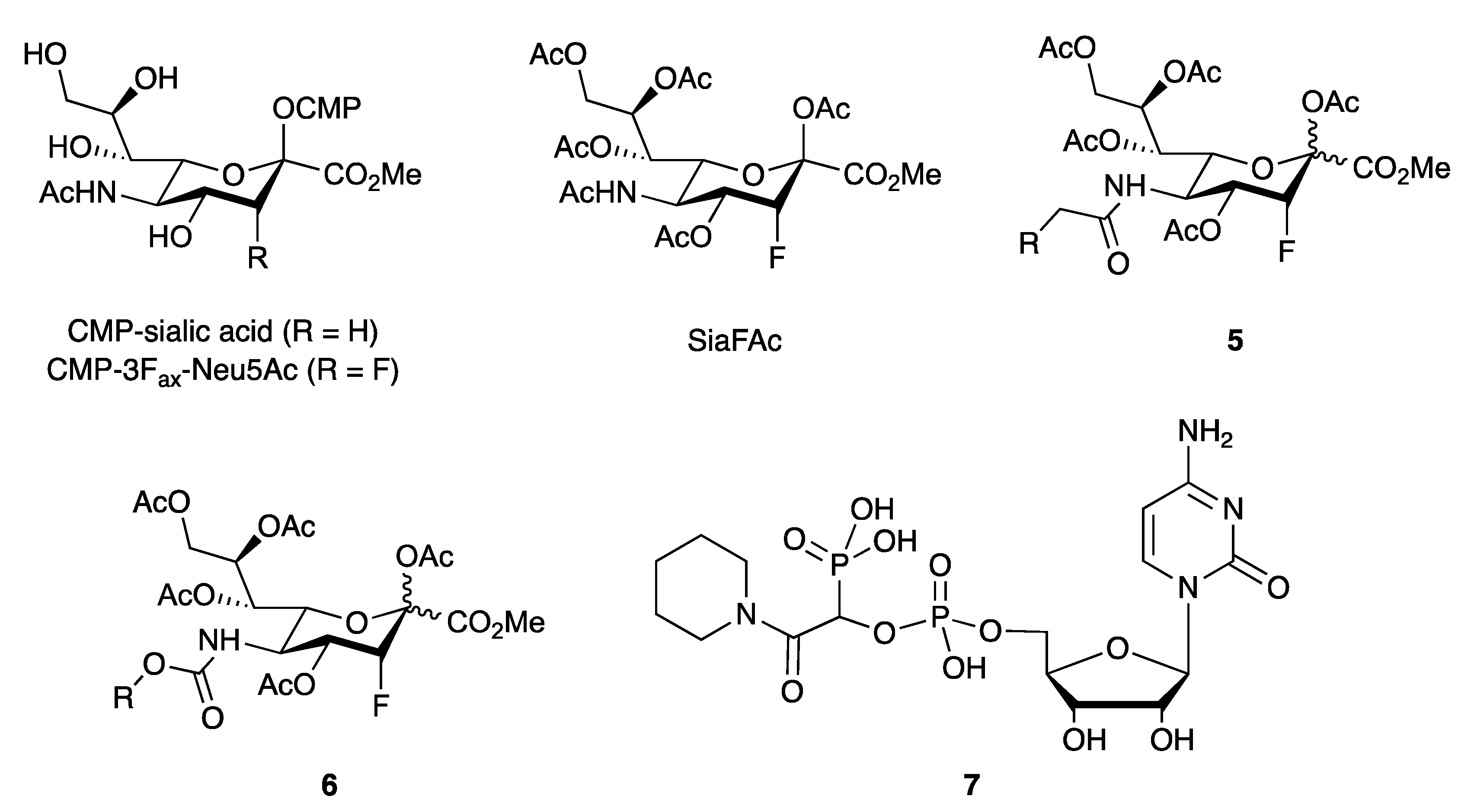
3.4.4. Sialyltransferase Inhibitors
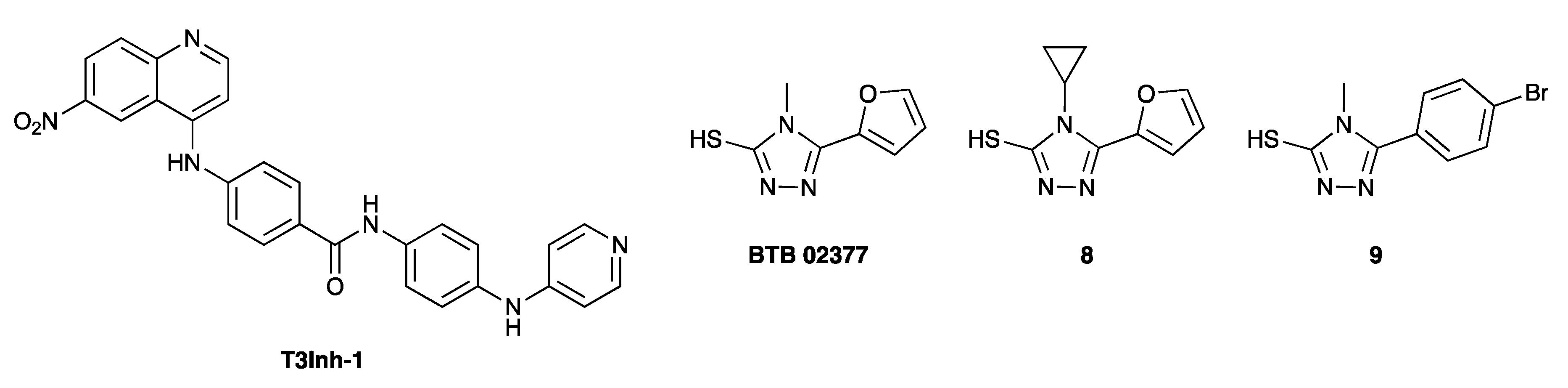
3.4.5. Non-Substrate-like Inhibitor Chemotypes
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Levin, M.; Silberstein, S.D.; Gilbert, R.; Lucas, S.; Munsie, L.; Garrelts, A.; Kennedy, K.; Everman, N.; Pearlman, E. Basic Considerations for the Use of Monoclonal Antibodies in Migraine. Headache 2018, 58, 1689–1696. [Google Scholar] [CrossRef]
- Schmidt, A.F.; Pearce, L.S.; Wilkins, J.T.; Overington, J.P.; Hingorani, A.D.; Casas, J.P. PCSK9 monoclonal antibodies for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2017, 4, Cd011748. [Google Scholar] [CrossRef]
- Edris, A.; De Feyter, S.; Maes, T.; Joos, G.; Lahousse, L. Monoclonal antibodies in type 2 asthma: A systematic review and network meta-analysis. Respir. Res. 2019, 20, 179. [Google Scholar] [CrossRef] [Green Version]
- Cruz, E.; Kayser, V. Monoclonal antibody therapy of solid tumors: Clinical limitations and novel strategies to enhance treatment efficacy. Biologics 2019, 13, 33–51. [Google Scholar] [CrossRef] [Green Version]
- Grilo, A.L.; Mantalaris, A. The Increasingly Human and Profitable Monoclonal Antibody Market. Trends Biotechnol. 2019, 37, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, H.W., Jr.; Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010, 125 (Suppl. S2), S41–S52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Sousa-Pereira, P.; Woof, J.M. IgA: Structure, Function, and Developability. Antibodies 2019, 8, 57. [Google Scholar] [CrossRef] [Green Version]
- Josephs, D.H.; Spicer, J.F.; Karagiannis, P.; Gould, H.J.; Karagiannis, S.N. IgE immunotherapy: A novel concept with promise for the treatment of cancer. MAbs 2014, 6, 54–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.X.; Tong, X.; Li, C.; Giddens, J.P.; Li, T. Glycoengineering of Antibodies for Modulating Functions. Annu. Rev. Biochem. 2019, 88, 433–459. [Google Scholar] [CrossRef]
- Jensen-Jarolim, E.; Turner, M.C.; Karagiannis, S.N. AllergoOncology: IgE- and IgG(4)-mediated immune mechanisms linking allergy with cancer and their translational implications. J. Allergy Clin. Immunol. 2017, 140, 982–984. [Google Scholar] [CrossRef] [Green Version]
- Maverakis, E.; Kim, K.; Shimoda, M.; Gershwin, M.E.; Patel, F.; Wilken, R.; Raychaudhuri, S.; Ruhaak, L.R.; Lebrilla, C.B. Glycans in the immune system and The Altered Glycan Theory of Autoimmunity: A critical review. J. Autoimmun. 2015, 57, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damelang, T.; Rogerson, S.J.; Kent, S.J.; Chung, A.W. Role of IgG3 in Infectious Diseases. Trends Immunol. 2019, 40, 197–211. [Google Scholar] [CrossRef]
- Davies, A.M.; Sutton, B.J. Human IgG4: A structural perspective. Immunol. Rev. 2015, 268, 139–159. [Google Scholar] [CrossRef] [Green Version]
- Steffen, U.; Koeleman, C.A.; Sokolova, M.V.; Bang, H.; Kleyer, A.; Rech, J.; Unterweger, H.; Schicht, M.; Garreis, F.; Hahn, J.; et al. IgA subclasses have different effector functions associated with distinct glycosylation profiles. Nat. Commun. 2020, 11, 120. [Google Scholar] [CrossRef] [Green Version]
- Gould, H.J.; Sutton, B.J.; Beavil, A.J.; Beavil, R.L.; McCloskey, N.; Coker, H.A.; Fear, D.; Smurthwaite, L. The biology of IGE and the basis of allergic disease. Annu. Rev. Immunol. 2003, 21, 579–628. [Google Scholar] [CrossRef] [PubMed]
- Blandino, R.; Baumgarth, N. Secreted IgM: New tricks for an old molecule. J. Leukoc. Biol. 2019, 106, 1021–1034. [Google Scholar] [CrossRef]
- Sathe, A.; Cusick, J.K. Biochemistry, Immunoglobulin M (IgM). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Chen, K.; Xu, W.; Wilson, M.; He, B.; Miller, N.W.; Bengtén, E.; Edholm, E.S.; Santini, P.A.; Rath, P.; Chiu, A.; et al. Immunoglobulin D enhances immune surveillance by activating antimicrobial, proinflammatory and B cell-stimulating programs in basophils. Nat. Immunol. 2009, 10, 889–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutzeit, C.; Chen, K.; Cerutti, A. The enigmatic function of IgD: Some answers at last. Eur. J. Immunol. 2018, 48, 1101–1113. [Google Scholar] [CrossRef]
- Griggs, J.; Zinkewich-Peotti, K. The state of the art: Immune-mediated mechanisms of monoclonal antibodies in cancer therapy. Br. J. Cancer 2009, 101, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.L.; Suscovich, T.J.; Fortune, S.M.; Alter, G. Beyond binding: Antibody effector functions in infectious diseases. Nat. Rev. Immunol. 2018, 18, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.M.; Wormald, M.R.; Rudd, P.M.; Davey, G.P. Fc gamma receptors: Glycobiology and therapeutic prospects. J. Inflamm. Res. 2016, 9, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Bruhns, P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012, 119, 5640–5649. [Google Scholar] [CrossRef] [PubMed]
- Swisher, J.F.; Feldman, G.M. The many faces of FcγRI: Implications for therapeutic antibody function. Immunol. Rev. 2015, 268, 160–174. [Google Scholar] [CrossRef]
- Qiao, J.; Al-Tamimi, M.; Baker, R.I.; Andrews, R.K.; Gardiner, E.E. The platelet Fc receptor, FcγRIIa. Immunol. Rev. 2015, 268, 241–252. [Google Scholar] [CrossRef]
- Bournazos, S.; Ravetch, J.V. Fcγ Receptor Function and the Design of Vaccination Strategies. Immunity 2017, 47, 224–233. [Google Scholar] [CrossRef]
- De Haan, N.; Falck, D.; Wuhrer, M. Monitoring of immunoglobulin N- and O-glycosylation in health and disease. Glycobiology 2020, 30, 226–240. [Google Scholar] [CrossRef]
- Plomp, R.; Bondt, A.; de Haan, N.; Rombouts, Y.; Wuhrer, M. Recent Advances in Clinical Glycoproteomics of Immunoglobulins (Igs). Mol. Cell. Proteom. 2016, 15, 2217–2228. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.M.; Cosgrave, E.F.L.; Struwe, W.B.; Wormald, M.; Davey, G.P.; Jefferis, R.; Rudd, P.M. Glycosylation and Fc receptors. Curr. Top. Microbiol. Immunol. 2014, 382, 165–199. [Google Scholar] [PubMed]
- Jennewein, M.F.; Alter, G. The Immunoregulatory Roles of Antibody Glycosylation. Trends Immunol. 2017, 38, 358–372. [Google Scholar] [CrossRef]
- Van de Bovenkamp, F.S.; Hafkenscheid, L.; Rispens, T.; Rombouts, Y. The Emerging Importance of IgG Fab Glycosylation in Immunity. J. Immunol. 2016, 196, 1435–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Giddens, J.; Fan, S.Q.; Toonstra, C.; Wang, L.X. Chemoenzymatic glycoengineering of intact IgG antibodies for gain of functions. J. Am. Chem. Soc. 2012, 134, 12308–12318. [Google Scholar] [CrossRef] [Green Version]
- Raymond, C.; Robotham, A.; Spearman, M.; Butler, M.; Kelly, J.; Durocher, Y. Production of α2,6-sialylated IgG1 in CHO cells. MAbs 2015, 7, 571–583. [Google Scholar] [CrossRef] [Green Version]
- Peschke, B.; Keller, C.W.; Weber, P.; Quast, I.; Lünemann, J.D. Fc-Galactosylation of Human Immunoglobulin Gamma Isotypes Improves C1q Binding and Enhances Complement-Dependent Cytotoxicity. Front. Immunol. 2017, 8, 646. [Google Scholar] [CrossRef]
- Quast, I.; Keller, C.W.; Maurer, M.A.; Giddens, J.P.; Tackenberg, B.; Wang, L.X.; Münz, C.; Nimmerjahn, F.; Dalakas, M.C.; Lünemann, J.D. Sialylation of IgG Fc domain impairs complement-dependent cytotoxicity. J. Clin. Investig. 2015, 125, 4160–4170. [Google Scholar] [CrossRef] [Green Version]
- Sondermann, P.; Pincetic, A.; Maamary, J.; Lammens, K.; Ravetch, J.V. General mechanism for modulating immunoglobulin effector function. Proc. Natl. Acad. Sci. USA 2013, 110, 9868–9872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temming, A.R.; Dekkers, G.; van de Bovenkamp, F.S.; Plomp, H.R.; Bentlage, A.E.H.; Szittner, Z.; Derksen, N.I.L.; Wuhrer, M.; Rispens, T.; Vidarsson, G. Human DC-SIGN and CD23 do not interact with human IgG. Sci. Rep. 2019, 9, 9995. [Google Scholar] [CrossRef]
- Wang, T.T. IgG Fc Glycosylation in Human Immunity. Curr. Top. Microbiol. Immunol. 2019, 423, 63–75. [Google Scholar] [PubMed]
- Buettner, M.J.; Shah, S.R.; Saeui, C.T.; Ariss, R.; Yarema, K.J. Improving Immunotherapy Through Glycodesign. Front. Immunol. 2018, 9, 2485. [Google Scholar] [CrossRef]
- Hiatt, A.; Bohorova, N.; Bohorov, O.; Goodman, C.; Kim, D.; Pauly, M.H.; Valesco, J.; Whaley, K.J.; Piedra, P.A.; Gilbert, B.E.; et al. Glycan variants of a respiratory syncytial virus antibody with enhanced effector function and in vivo efficacy. Proc. Natl. Acad. Sci. USA 2014, 111, 5992–5997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Brown, D.; Reed, C.; Chung, S.; Lutman, J.; Stefanich, E.; Wong, A.; Stephan, J.P.; Bayer, R. Production, characterization, and pharmacokinetic properties of antibodies with N-linked mannose-5 glycans. MAbs 2012, 4, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; Gonzalez, J.; Edwards, K.; Mallajosyula, V.; Buzzanco, A.S.; Sherwood, R.; Buffone, C.; Kathale, N.; Providenza, S.; Xie, M.M.; et al. Proinflammatory IgG Fc structures in patients with severe COVID-19. Nat. Immunol. 2021, 22, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.D.; de Graaf, E.L.; Sonneveld, M.E.; Plomp, H.R.; Nouta, J.; Hoepel, W.; Chen, H.J.; Linty, F.; Visser, R.; Brinkhaus, M.; et al. Afucosylated IgG characterizes enveloped viral responses and correlates with COVID-19 severity. Science 2021, 371. [Google Scholar] [CrossRef]
- Petrović, T.; Alves, I.; Bugada, D.; Pascual, J.; Vučković, F.; Skelin, A.; Gaifem, J.; Villar-Garcia, J.; Vicente, M.M.; Fernandes, Â.; et al. Composition of the immunoglobulin G glycome associates with the severity of COVID-19. Glycobiology 2021, 31, 372–377. [Google Scholar]
- Hou, H.; Yang, H.; Liu, P.; Huang, C.; Wang, M.; Li, Y.; Zhu, M.; Wang, J.; Xu, Y.; Wang, Y.; et al. Profile of Immunoglobulin G N-Glycome in COVID-19 Patients: A Case-Control Study. Front. Immunol. 2021, 12, 748566. [Google Scholar] [CrossRef]
- Chandler, K.B.; Mehta, N.; Leon, D.R.; Suscovich, T.J.; Alter, G.; Costello, C.E. Multi-isotype Glycoproteomic Characterization of Serum Antibody Heavy Chains Reveals Isotype- and Subclass-Specific N-Glycosylation Profiles. Mol. Cell. Proteom. 2019, 18, 686–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nettleton, M.Y.; Kochan, J.P. Role of glycosylation sites in the IgE Fc molecule. Int. Arch. Allergy Immunol. 1995, 107, 328–329. [Google Scholar] [CrossRef] [Green Version]
- Plomp, R.; Hensbergen, P.J.; Rombouts, Y.; Zauner, G.; Dragan, I.; Koeleman, C.A.; Deelder, A.M.; Wuhrer, M. Site-specific N-glycosylation analysis of human immunoglobulin e. J. Proteome Res. 2014, 13, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Shade, K.T.; Platzer, B.; Washburn, N.; Mani, V.; Bartsch, Y.C.; Conroy, M.; Pagan, J.D.; Bosques, C.; Mempel, T.R.; Fiebiger, E.; et al. A single glycan on IgE is indispensable for initiation of anaphylaxis. J. Exp. Med. 2015, 212, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Shade, K.-T.C.; Conroy, M.E.; Washburn, N.; Kitaoka, M.; Huynh, D.J.; Laprise, E.; Patil, S.U.; Shreffler, W.G.; Anthony, R.M. Sialylation of immunoglobulin E is a determinant of allergic pathogenicity. Nature 2020, 582, 265–270. [Google Scholar] [CrossRef]
- Pereira, N.A.; Chan, K.F.; Lin, P.C.; Song, Z. The “less-is-more” in therapeutic antibodies: Afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. MAbs 2018, 10, 693–711. [Google Scholar] [CrossRef]
- Goede, V.; Fischer, K.; Engelke, A.; Schlag, R.; Lepretre, S.; Montero, L.F.; Montillo, M.; Fegan, C.; Asikanius, E.; Humphrey, K.; et al. Obinutuzumab as frontline treatment of chronic lymphocytic leukemia: Updated results of the CLL11 study. Leukemia 2015, 29, 1602–1604. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.; Davies, A.; Ando, K.; Klapper, W.; Opat, S.; Owen, C.; Phillips, E.; Sangha, R.; Schlag, R.; Seymour, J.F.; et al. Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N. Engl. J. Med. 2017, 377, 1331–1344. [Google Scholar] [CrossRef] [PubMed]
- Kasamon, Y.L.; Chen, H.; de Claro, R.A.; Nie, L.; Ye, J.; Blumenthal, G.M.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Mogamulizumab-kpkc for Mycosis Fungoides and Sézary Syndrome. Clin. Cancer Res. 2019, 25, 7275–7280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strasser, R.; Stadlmann, J.; Schähs, M.; Stiegler, G.; Quendler, H.; Mach, L.; Glössl, J.; Weterings, K.; Pabst, M.; Steinkellner, H. Generation of glyco-engineered Nicotiana benthamiana for the production of monoclonal antibodies with a homogeneous human-like N-glycan structure. Plant Biotechnol. J. 2008, 6, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Louie, S.; Haley, B.; Marshall, B.; Heidersbach, A.; Yim, M.; Brozynski, M.; Tang, D.; Lam, C.; Petryniak, B.; Shaw, D.; et al. FX knockout CHO hosts can express desired ratios of fucosylated or afucosylated antibodies with high titers and comparable product quality. Biotechnol. Bioeng. 2017, 114, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Sjögren, J.; Lood, R.; Nägeli, A. On enzymatic remodeling of IgG glycosylation; unique tools with broad applications. Glycobiology 2020, 30, 254–267. [Google Scholar] [CrossRef]
- Sha, S.; Agarabi, C.; Brorson, K.; Lee, D.Y.; Yoon, S. N-Glycosylation Design and Control of Therapeutic Monoclonal Antibodies. Trends Biotechnol. 2016, 34, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Blundell, P.A.; Lu, D.; Dell, A.; Haslam, S.; Pleass, R.J. Choice of Host Cell Line Is Essential for the Functional Glycosylation of the Fc Region of Human IgG1 Inhibitors of Influenza B Viruses. J. Immunol. 2020, 204, 1022–1034. [Google Scholar] [CrossRef]
- Chiang, A.W.; Li, S.; Spahn, P.N.; Richelle, A.; Kuo, C.C.; Samoudi, M.; Lewis, N.E. Modulating carbohydrate-protein interactions through glycoengineering of monoclonal antibodies to impact cancer physiology. Curr. Opin. Struct. Biol. 2016, 40, 104–111. [Google Scholar] [CrossRef] [Green Version]
- Steinke, J.W.; Platts-Mills, T.A.; Commins, S.P. The alpha-gal story: Lessons learned from connecting the dots. J. Allergy Clin. Immunol. 2015, 135, 589–596. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.H.; Mirakhur, B.; Chan, E.; Le, Q.T.; Berlin, J.; Morse, M.; Murphy, B.A.; Satinover, S.M.; Hosen, J.; Mauro, D.; et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N. Engl. J. Med. 2008, 358, 1109–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaaltiel, Y.; Tekoah, Y. Plant specific N-glycans do not have proven adverse effects in humans. Nat. Biotechnol. 2016, 34, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Dumont, J.; Euwart, D.; Mei, B.; Estes, S.; Kshirsagar, R. Human cell lines for biopharmaceutical manufacturing: History, status, and future perspectives. Crit. Rev. Biotechnol. 2016, 36, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Clausen, H.; Wandall, H.H.; Steentoft, C.; Stanley, P.; Schnaar, R.L. Glycosylation Engineering. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 713–728. [Google Scholar]
- Ilieva, K.M.; Fazekas-Singer, J.; Bax, H.J.; Crescioli, S.; Montero-Morales, L.; Mele, S.; Sow, H.S.; Stavraka, C.; Josephs, D.H.; Spicer, J.F.; et al. AllergoOncology: Expression platform development and functional profiling of an anti-HER2 IgE antibody. Allergy 2019, 74, 1985–1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero-Morales, L.; Maresch, D.; Crescioli, S.; Castilho, A.; Ilieva, K.M.; Mele, S.; Karagiannis, S.N.; Altmann, F.; Steinkellner, H. In Planta Glycan Engineering and Functional Activities of IgE Antibodies. Front. Bioeng. Biotechnol. 2019, 7, 242. [Google Scholar] [CrossRef] [PubMed]
- Shade, K.T.; Conroy, M.E.; Anthony, R.M. IgE Glycosylation in Health and Disease. Curr. Top. Microbiol. Immunol. 2019, 423, 77–93. [Google Scholar] [PubMed]
- Goetze, A.M.; Liu, Y.D.; Zhang, Z.; Shah, B.; Lee, E.; Bondarenko, P.V.; Flynn, G.C. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology 2011, 21, 949–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gloster, T.M.; Davies, G.J. Glycosidase inhibition: Assessing mimicry of the transition state. Org. Biomol. Chem. 2010, 8, 305–320. [Google Scholar] [CrossRef] [Green Version]
- Ezekowitz, R.A.; Stahl, P.D. The structure and function of vertebrate mannose lectin-like proteins. J. Cell Sci. Suppl. 1988, 121–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kommineni, V.; Markert, M.; Ren, Z.; Palle, S.; Carrillo, B.; Deng, J.; Tejeda, A.; Nandi, S.; McDonald, K.A.; Marcel, S.; et al. In Vivo Glycan Engineering via the Mannosidase I Inhibitor (Kifunensine) Improves Efficacy of Rituximab Manufactured in Nicotiana benthamiana Plants. Int. J. Mol. Sci. 2019, 20, 194. [Google Scholar] [CrossRef] [Green Version]
- Krahn, N.; Spearman, M.; Meier, M.; Dorion-Thibaudeau, J.; McDougall, M.; Patel, T.R.; De Crescenzo, G.; Durocher, Y.; Stetefeld, J.; Butler, M. Inhibition of glycosylation on a camelid antibody uniquely affects its FcγRI binding activity. Eur. J. Pharm. Sci. 2017, 96, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Hughes, T.A.; Kelkar, A.; Yu, X.; Cheng, K.; Park, S.; Huang, W.C.; Lovell, J.F.; Neelamegham, S. Inhibition of SARS-CoV-2 viral entry upon blocking N- and O-glycan elaboration. Elife 2020, 9, e61552. [Google Scholar] [CrossRef] [PubMed]
- Rajasekharan, S.; Milan Bonotto, R.; Nascimento Alves, L.; Kazungu, Y.; Poggianella, M.; Martinez-Orellana, P.; Skoko, N.; Polez, S.; Marcello, A. Inhibitors of Protein Glycosylation Are Active against the Coronavirus Severe Acute Respiratory Syndrome Coronavirus SARS-CoV-2. Viruses 2021, 13, 808. [Google Scholar] [CrossRef]
- Burkart, M.D.; Vincent, S.P.; Düffels, A.; Murray, B.W.; Ley, S.V.; Wong, C.H. Chemo-enzymatic synthesis of fluorinated sugar nucleotide: Useful mechanistic probes for glycosyltransferases. Bioorg. Med. Chem. 2000, 8, 1937–1946. [Google Scholar] [CrossRef]
- Rillahan, C.D.; Antonopoulos, A.; Lefort, C.T.; Sonon, R.; Azadi, P.; Ley, K.; Dell, A.; Haslam, S.M.; Paulson, J.C. Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nat. Chem. Biol. 2012, 8, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Okeley, N.M.; Alley, S.C.; Anderson, M.E.; Boursalian, T.E.; Burke, P.J.; Emmerton, K.M.; Jeffrey, S.C.; Klussman, K.; Law, C.L.; Sussman, D.; et al. Development of orally active inhibitors of protein and cellular fucosylation. Proc. Natl. Acad. Sci. USA 2013, 110, 5404–5409. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.G.; Mujacic, M.; Frohn, M.J.; Pickrell, A.J.; Kodama, P.; Bagal, D.; San Miguel, T.; Sickmier, E.A.; Osgood, S.; Swietlow, A.; et al. Facile Modulation of Antibody Fucosylation with Small Molecule Fucostatin Inhibitors and Cocrystal Structure with GDP-Mannose 4,6-Dehydratase. ACS Chem. Biol. 2016, 11, 2734–2743. [Google Scholar] [CrossRef]
- McKenzie, N.C.; Scott, N.E.; John, A.; White, J.M.; Goddard-Borger, E.D. Synthesis and use of 6,6,6-trifluoro-L-fucose to block core-fucosylation in hybridoma cell lines. Carbohydr. Res. 2018, 465, 4–9. [Google Scholar] [CrossRef]
- Zandberg, W.F.; Kumarasamy, J.; Pinto, B.M.; Vocadlo, D.J. Metabolic inhibition of sialyl-Lewis X biosynthesis by 5-thiofucose remodels the cell surface and impairs selectin-mediated cell adhesion. J. Biol. Chem. 2012, 287, 40021–40030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kizuka, Y.; Nakano, M.; Yamaguchi, Y.; Nakajima, K.; Oka, R.; Sato, K.; Ren, C.T.; Hsu, T.L.; Wong, C.H.; Taniguchi, N. An Alkynyl-Fucose Halts Hepatoma Cell Migration and Invasion by Inhibiting GDP-Fucose-Synthesizing Enzyme FX, TSTA3. Cell Chem. Biol. 2017, 24, 1467–1478.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashwell, G.; Harford, J. Carbohydrate-specific receptors of the liver. Annu. Rev. Biochem. 1982, 51, 531–554. [Google Scholar] [CrossRef] [PubMed]
- Hennet, T. The galactosyltransferase family. Cell. Mol. Life Sci. 2002, 59, 1081–1095. [Google Scholar] [CrossRef]
- McDonald, A.G.; Hayes, J.M.; Bezak, T.; Głuchowska, S.A.; Cosgrave, E.F.; Struwe, W.B.; Stroop, C.J.; Kok, H.; van de Laar, T.; Rudd, P.M.; et al. Galactosyltransferase 4 is a major control point for glycan branching in N-linked glycosylation. J. Cell Sci. 2014, 127 Pt 23, 5014–5026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajimoto, T.; Node, M. Synthesis of Glycosyltransferase Inhibitors. Synthesis 2009, 2009, 3179–3210. [Google Scholar] [CrossRef]
- Takaya, K.; Nagahori, N.; Kurogochi, M.; Furuike, T.; Miura, N.; Monde, K.; Lee, Y.C.; Nishimura, S. Rational design, synthesis, and characterization of novel inhibitors for human beta1,4-galactosyltransferase. J. Med. Chem. 2005, 48, 6054–6065. [Google Scholar] [CrossRef]
- Pesnot, T.; Jørgensen, R.; Palcic, M.M.; Wagner, G.K. Structural and mechanistic basis for a new mode of glycosyltransferase inhibition. Nat. Chem. Biol. 2010, 6, 321–323. [Google Scholar] [CrossRef] [Green Version]
- Descroix, K.; Pesnot, T.; Yoshimura, Y.; Gehrke, S.S.; Wakarchuk, W.; Palcic, M.M.; Wagner, G.K. Inhibition of Galactosyltransferases by a Novel Class of Donor Analogues. J. Med. Chem. 2012, 55, 2015–2024. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.K.; Pesnot, T.; Palcic, M.M.; Jørgensen, R. Novel UDP-GalNAc Derivative Structures Provide Insight into the Donor Specificity of Human Blood Group Glycosyltransferase. J. Biol. Chem. 2015, 290, 31162–31172. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Kanabar, V.; Padilla, B.; Man, F.; Pitchford, S.C.; Page, C.P.; Wagner, G.K. Uncharged nucleoside inhibitors of β-1,4-galactosyltransferase with activity in cells. Chem. Commun. 2016, 52, 3955–3958. [Google Scholar] [CrossRef] [Green Version]
- Kanabar, V.; Tedaldi, L.; Jiang, J.; Nie, X.; Panina, I.; Descroix, K.; Man, F.; Pitchford, S.C.; Page, C.P.; Wagner, G.K. Base-modified UDP-sugars reduce cell surface levels of P-selectin glycoprotein 1 (PSGL-1) on IL-1β-stimulated human monocytes. Glycobiology 2016, 26, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wagner, G.K. An acceptor analogue of β-1,4-galactosyltransferase: Substrate, inhibitor, or both? Carbohydr. Res. 2017, 450, 54–59. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.R.; Yang, F.; Sinha, A.; Ramakrishnan, B.; Tor, Y.; Qasba, P.K.; Esko, J.D. Deoxygenated disaccharide analogs as specific inhibitors of beta1-4-galactosyltransferase 1 and selectin-mediated tumor metastasis. J. Biol. Chem. 2009, 284, 4952–4959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audry, M.; Jeanneau, C.; Imberty, A.; Harduin- Lepers, A.; Delannoy, P.; Breton, C. Current trends in the structure-activity relationships of sialyltransferases. Glycobiology 2011, 21, 716–726. [Google Scholar] [CrossRef] [Green Version]
- Christie, D.R.; Shaikh, F.M.; Lucas, J.A., IV; Lucas, J.A., III; Bellis, S.L. ST6Gal-I expression in ovarian cancer cells promotes an invasive phenotype by altering integrin glycosylation and function. J. Ovarian Res. 2008, 1, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkart, M.D.; Vincent, S.P.; Wong, C.-H. An efficient synthesis of CMP-3-fluoroneuraminic acid. Chem. Commun. 1999, 16, 1525–1526. [Google Scholar] [CrossRef]
- Heise, T.; Pijnenborg, J.F.A.; Büll, C.; van Hilten, N.; Kers-Rebel, E.D.; Balneger, N.; Elferink, H.; Adema, G.J.; Boltje, T.J. Potent Metabolic Sialylation Inhibitors Based on C-5-Modified Fluorinated Sialic Acids. J. Med. Chem. 2019, 62, 1014–1021. [Google Scholar] [CrossRef]
- Montgomery, A.; Szabo, R.; Skropeta, D.; Yu, H. Computational characterisation of the interactions between human ST6Gal I and transition-state analogue inhibitors: Insights for inhibitor design. J. Mol. Recognit. 2016, 29, 210–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedaldi, L.; Wagner, G.K. Beyond substrate analogues: New inhibitor chemotypes for glycosyltransferases. MedChemComm 2014, 5, 1106–1125. [Google Scholar] [CrossRef]
- Song, L.; Linstedt, A.D. Inhibitor of ppGalNAc-T3-mediated O-glycosylation blocks cancer cell invasiveness and lowers FGF23 levels. Elife 2017, 6, e24051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Chen, F.; Petrella, A.; Chacón-Huete, F.; Covone, J.; Tsai, T.W.; Yu, C.C.; Forgione, P.; Kwan, D.H. A High-Throughput Glycosyltransferase Inhibition Assay for Identifying Molecules Targeting Fucosylation in Cancer Cell-Surface Modification. ACS Chem. Biol. 2019, 14, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Rillahan, C.D.; Brown, S.J.; Register, A.C.; Rosen, H.; Paulson, J.C. High-throughput screening for inhibitors of sialyl- and fucosyltransferases. Angew. Chem. Int. Ed. Engl. 2011, 50, 12534–12537. [Google Scholar] [CrossRef]
- Chao, L.; Jongkees, S. High-Throughput Approaches in Carbohydrate-Active Enzymology: Glycosidase and Glycosyl Transferase Inhibitors, Evolution, and Discovery. Angew. Chem. Int. Ed. Engl. 2019, 58, 12750–12760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, A.P.; Xiao, K.; Wang, X.; Skropeta, D.; Yu, H. Computational Glycobiology: Mechanistic Studies of Carbohydrate-Active Enzymes and Implication for Inhibitor Design. Adv. Protein Chem. Struct. Biol. 2017, 109, 25–76. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ig Isotype | MW (kDa) | Biological Roles | N-Glycosylation Sites |
|---|---|---|---|
| IgG1 | 146 | Most abundant IgG subclass forming the primary antibody response. Large role in response against viral infections; able to effectively drive complement-dependent cytotoxicity (CDC) [7] | N180 [13] |
| IgG2 | 146 | Predominantly responds to glycans such as bacterial capsule polysaccharides. Roles in the bacterial immune response. Poor at driving CDC and antibody-dependent cell-mediated cytotoxicity (ADCC) [7] | N176 [13] |
| IgG3 | 170 | Pro-inflammatory and highly potent mediator of effector functions such as CDC and ADCC. Large roles in the viral response. [7,14] | N227; N322 [13] |
| IgG4 | 146 | Protective roles in allergy. Does not drive ADCC or CDC [7,15] | N177 [13] |
| IgA1 | 160 (serum) 385 (secretory) | Predominant serum IgA class. Mucosal defence. Less pro-inflammatory compared to IgA2 [16] | N144; N352 [13] |
| IgA2 | Mucosal defence; cytokine production and NET formation via macrophages and neutrophils. Pro-inflammatory [16] | N47; N92; N131; N205; N327 [13] | |
| IgE | 196 | Allergy and hypersensitivity; immune response against parasitic worms [8,17] | N21; N49; N99; N146; N252; N264; N275 [13] |
| IgM | 190 | Early immune response; B cell receptor [18,19] | N46; N209; N272; N279; N439 [13] |
| IgD | 184 | Involvement in activating B cells to produce antibodies; antimicrobial response [20,21] | N225; N316; N367 [13] |
| IgG Receptor | Specific Isotypes Engaged | Cell Expression | Immune Functions |
|---|---|---|---|
| FcγRI | IgG1; IgG3; IgG4 | Monocytes/macrophages; Dendritic Cells (DCs); inducible expression on neutrophils and mast cells | Effector cell activation; phagocytosis [24] |
| FcγRIIa | N/A | Monocytes/macrophages Neutrophils; DCs; basophils; mast cells; eosinophils; platelets | Platelet activation and aggregation [27]; effector cell activation; phagocytosis; degranulation; ADCC [24]; antigen processing and presentation on DCs [28] |
| FcγRIIb | N/A | B cells; DCs; basophils; subsets of monocytes/ macrophages; subsets of neutrophils | Inhibition of effector activity [24]; limits DC maturation; opposes BCR signalling, and induces apoptosis to eliminate low affinity BCR B cells [28] |
| FcγRIIc | N/A | NK cells, monocytes/macrophages; neutrophils | Activating variant expressed in ~11% of individuals |
| FcγRIIIa | N/A | NK cells, monocytes/macrophages | Effector cell activation; ADCC; phagocytosis [24] |
| FcγRIIIb | IgG1; IgG3 | Neutrophils; subsets of basophils | Unclear [24] |
| FcRN | IgG1 | Endothelial and epithelial cells; monocytes/macrophages; neutrophils; DCs | Recycling of IgG in serum and protection from degradation; responsible for long serum half lives, transport of IgG across mucosal surfaces and placenta during pregnancy [7] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; McCraw, A.J.; Gardner, R.A.; Spencer, D.I.R.; Karagiannis, S.N.; Wagner, G.K. Glycoengineering of Therapeutic Antibodies with Small Molecule Inhibitors. Antibodies 2021, 10, 44. https://doi.org/10.3390/antib10040044
Li S, McCraw AJ, Gardner RA, Spencer DIR, Karagiannis SN, Wagner GK. Glycoengineering of Therapeutic Antibodies with Small Molecule Inhibitors. Antibodies. 2021; 10(4):44. https://doi.org/10.3390/antib10040044
Chicago/Turabian StyleLi, Shasha, Alex J. McCraw, Richard A. Gardner, Daniel I.R. Spencer, Sophia N. Karagiannis, and Gerd K. Wagner. 2021. "Glycoengineering of Therapeutic Antibodies with Small Molecule Inhibitors" Antibodies 10, no. 4: 44. https://doi.org/10.3390/antib10040044
APA StyleLi, S., McCraw, A. J., Gardner, R. A., Spencer, D. I. R., Karagiannis, S. N., & Wagner, G. K. (2021). Glycoengineering of Therapeutic Antibodies with Small Molecule Inhibitors. Antibodies, 10(4), 44. https://doi.org/10.3390/antib10040044