Rational Design of Constrained Peptides as Protein Interface Inhibitors



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Antibody–Antigen Complex
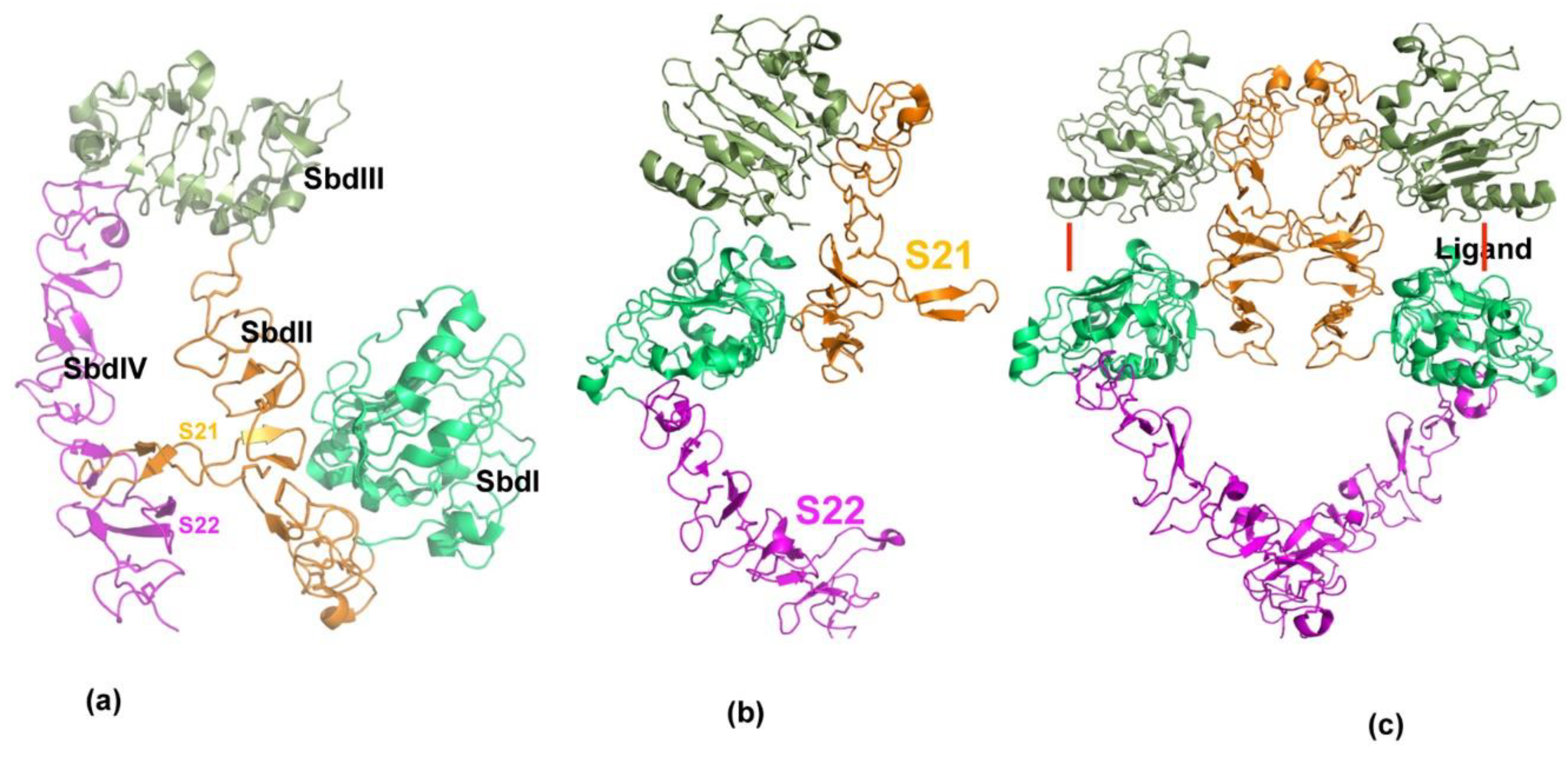
3. Structural Features of Protein–Protein Complexes
4. Receptor–Ligand Complex Peptide Inhibitor
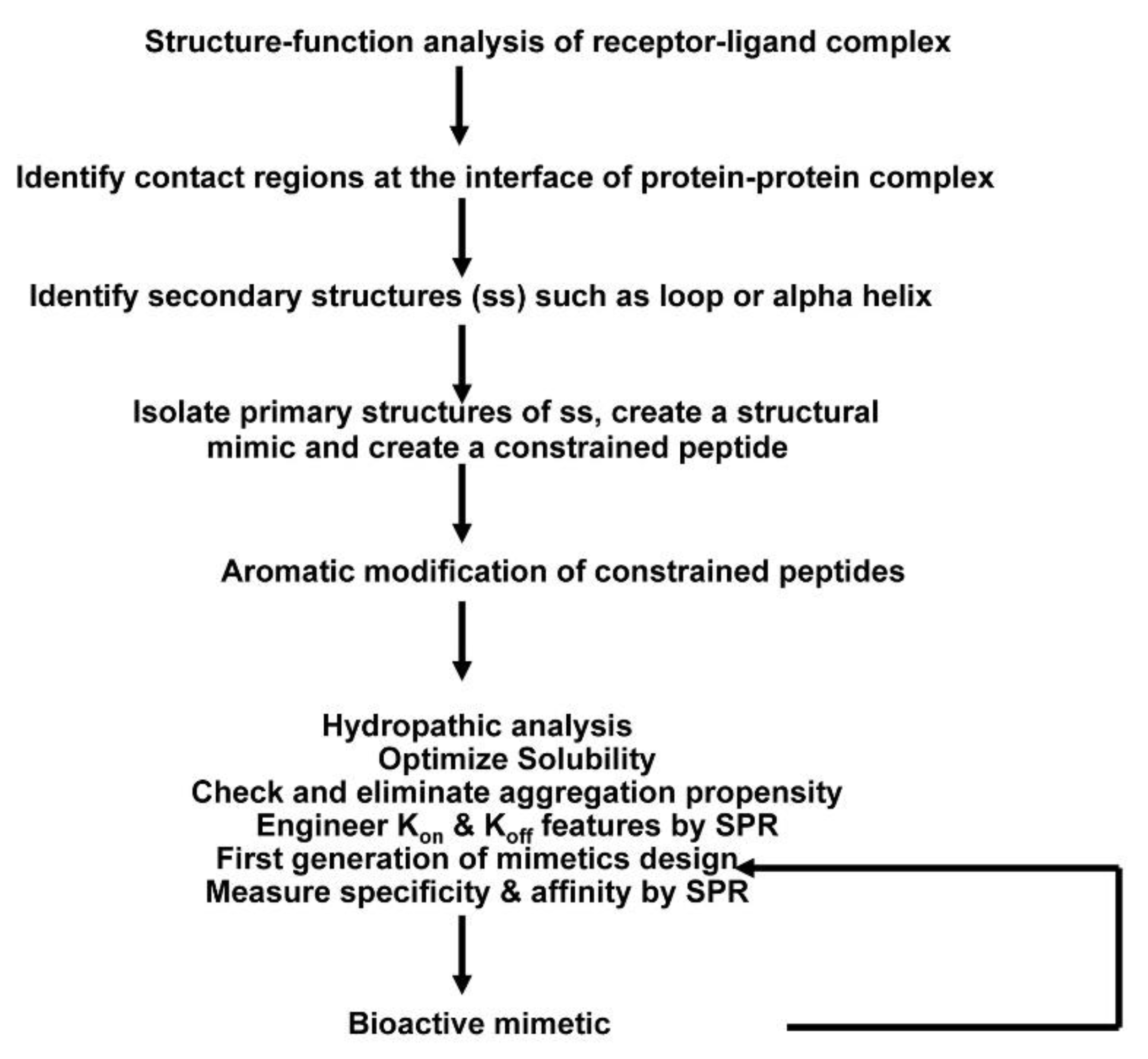
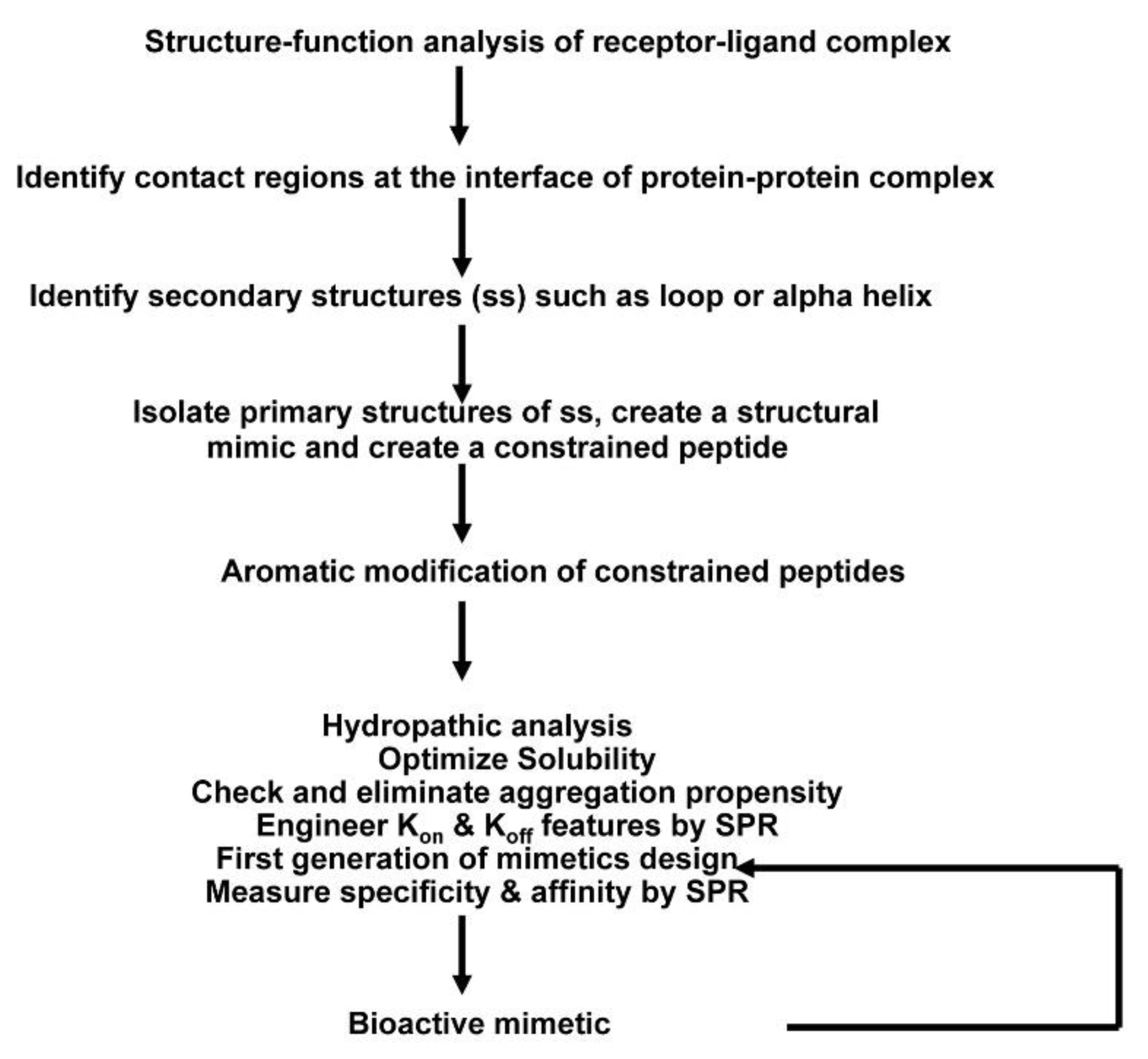
4.1. Design Concept
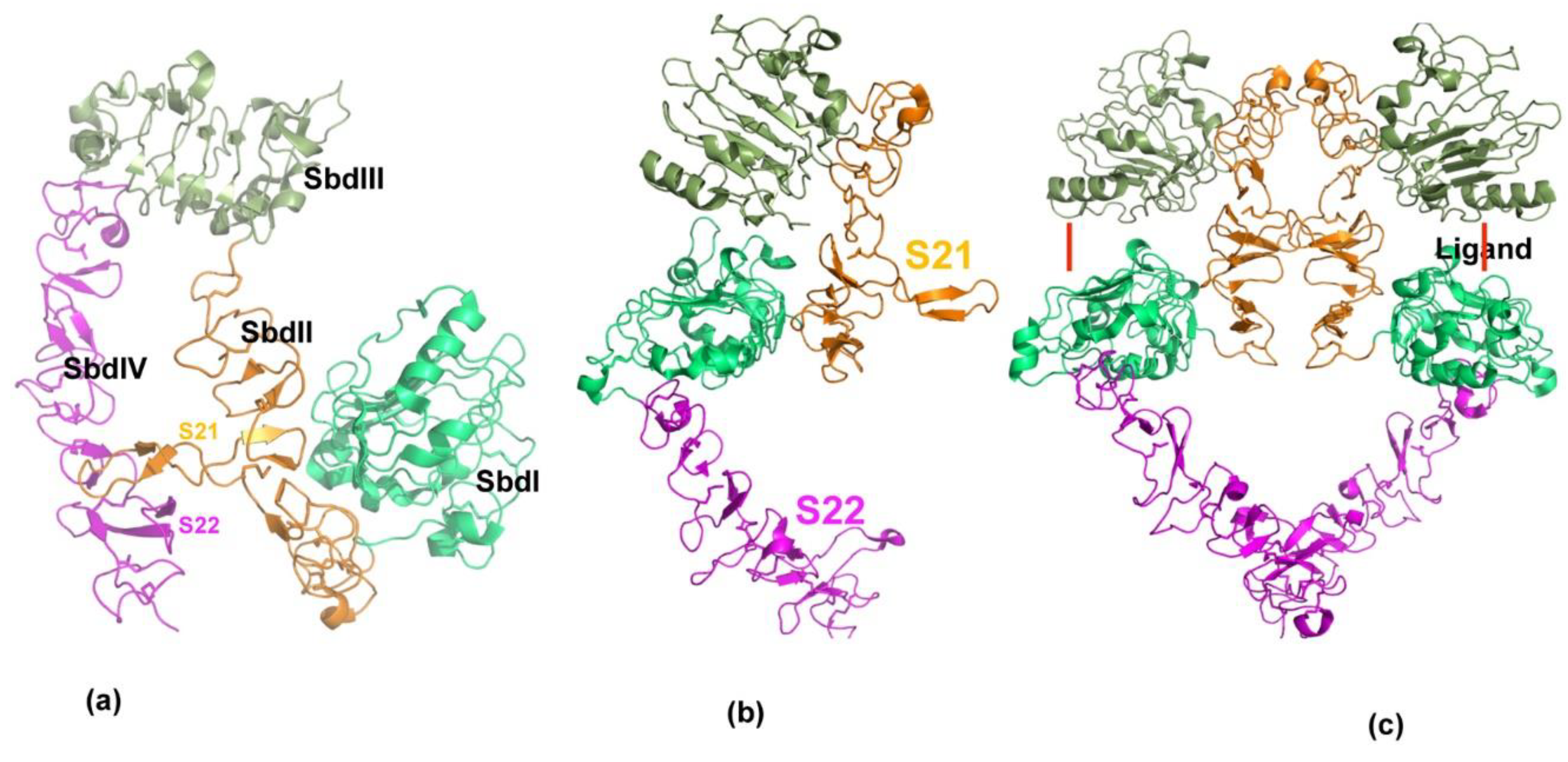
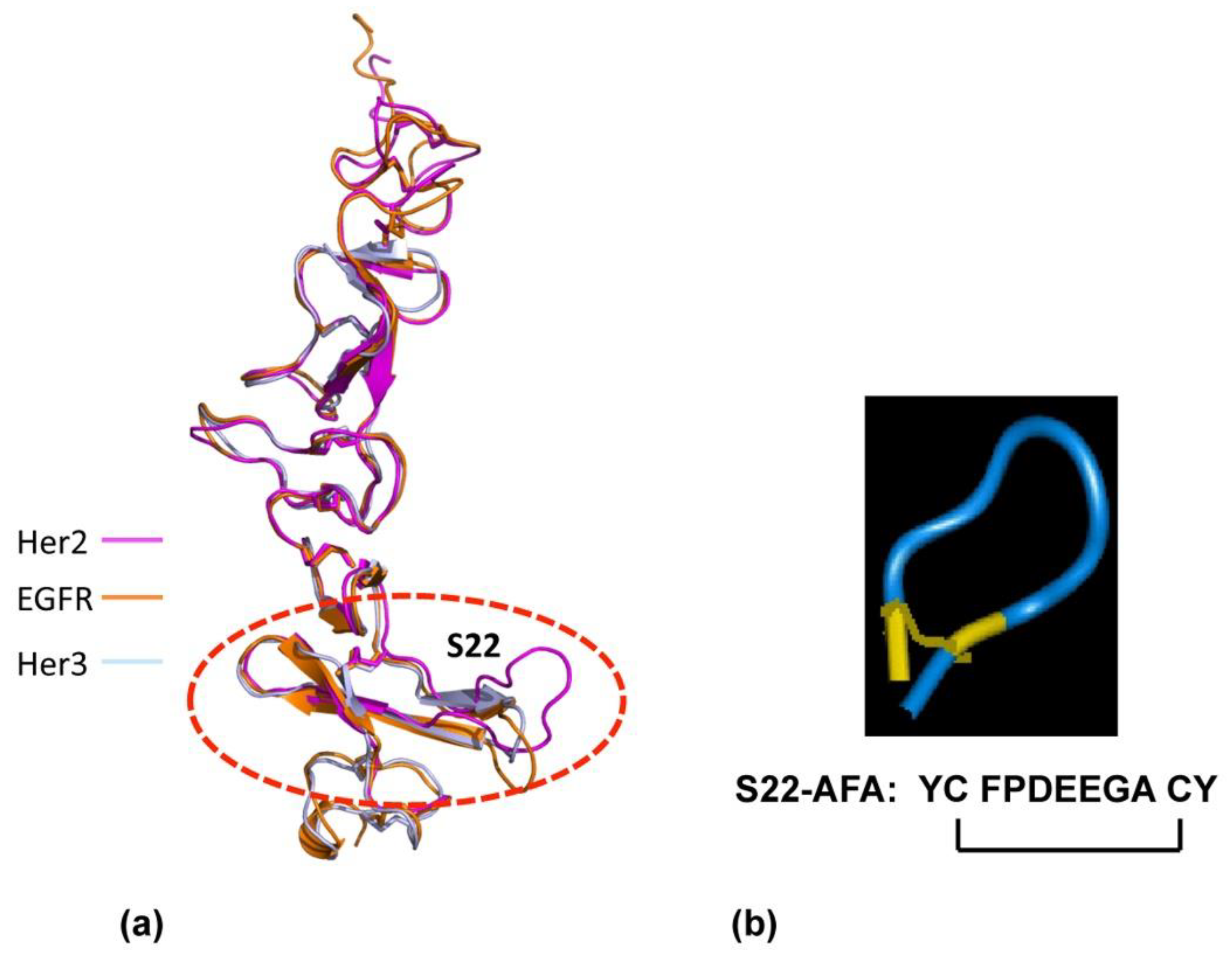
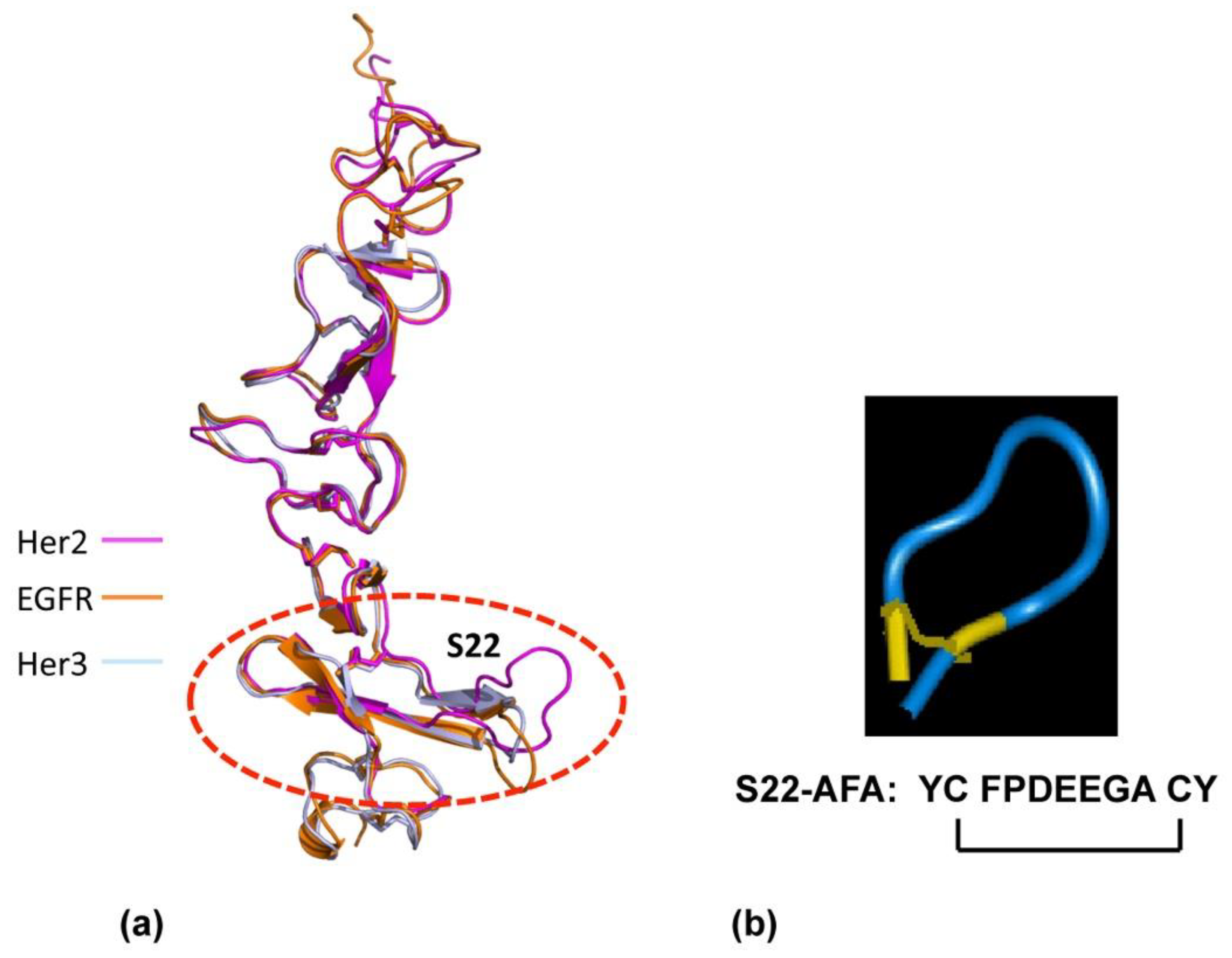
4.2. P185neu/her2 (Her2) Receptor Complex Targeted Peptidomimetics
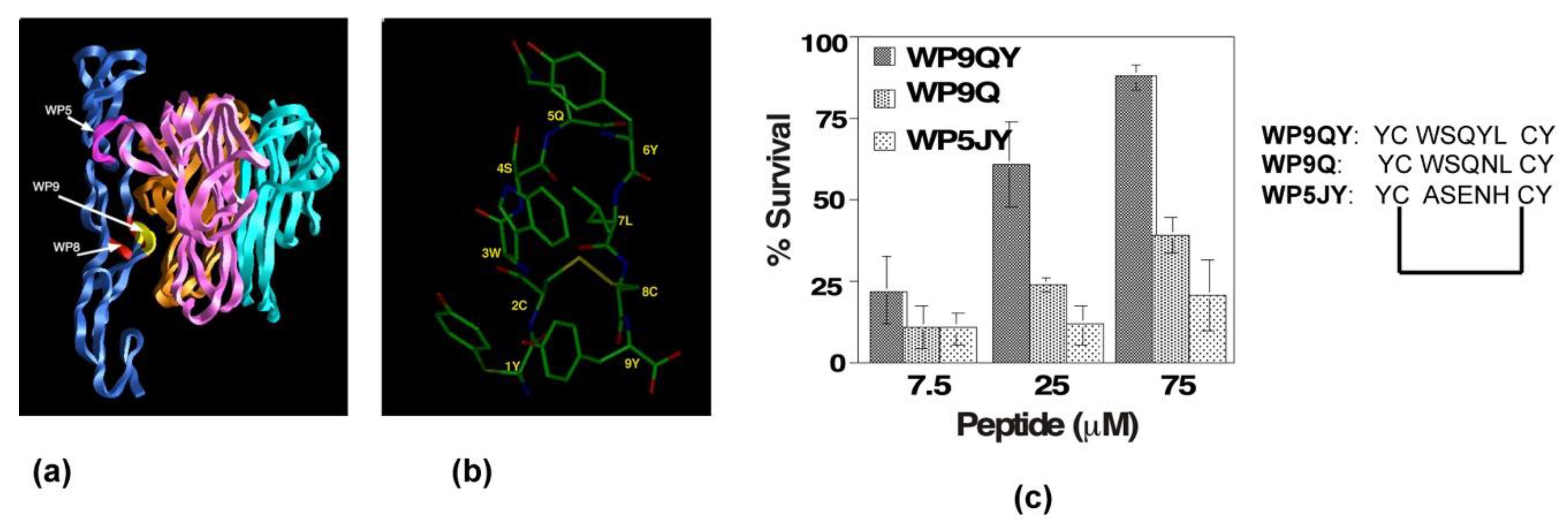
4.3. TNFR Receptor Complex Targeted of Peptidomimetics
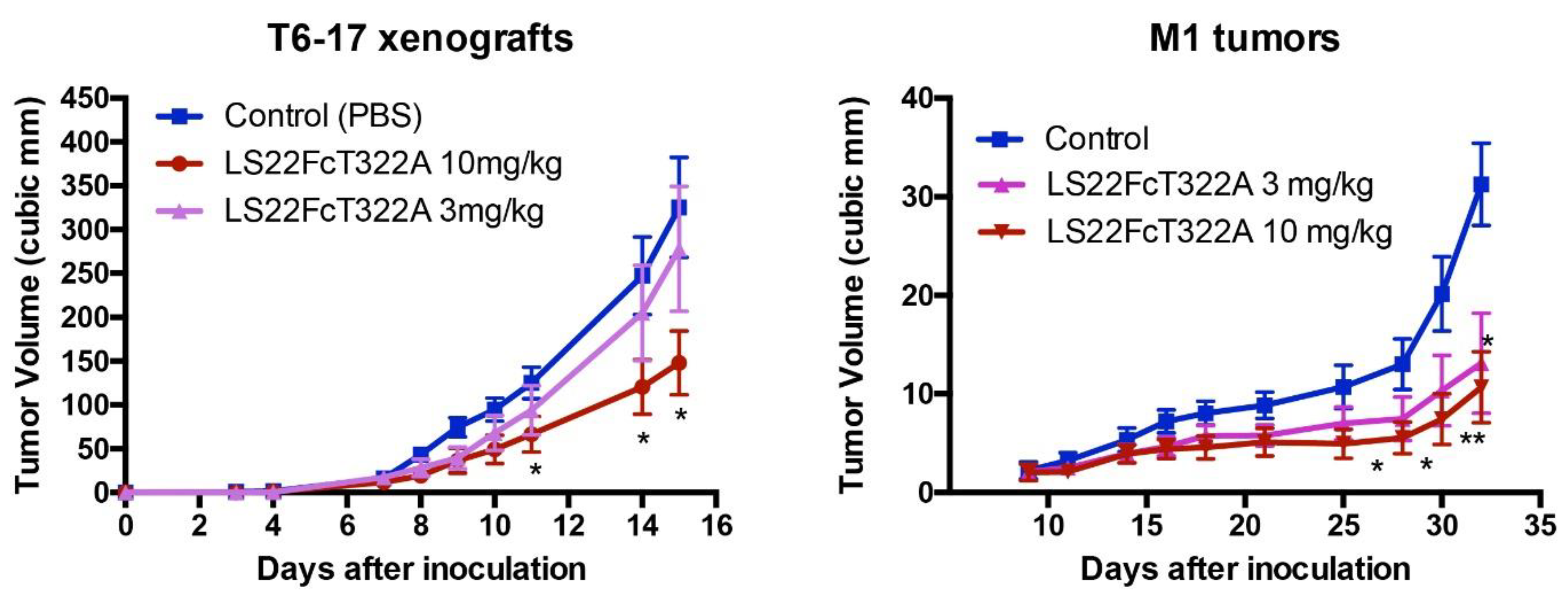
5. Applications of Constrained Peptidomimetics and Creation of Immunoadhesins
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Makley, L.N.; Gestwicki, J.E. Expanding the number of ‘druggable’ targets: Non-enzymes and protein-protein interactions. Chem. Biol. Drug Des. 2013, 81, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Findlay, G.M.; Snyder, M.W. Genomic Medicine-Progress, Pitfalls, and Promise. Cell 2019, 177, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Hennemann, H.; Wirths, S.; Carl, C. Cell-based peptide screening to access the undruggable target space. Eur. J. Med. Chem. 2015, 94, 489–496. [Google Scholar] [CrossRef]
- Neklesa, T.K.; Winkler, J.D.; Crews, C.M. Targeted protein degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Ran, X.; Gestwicki, J.E. Inhibitors of protein-protein interactions (PPIs): An analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 2018, 44, 75–86. [Google Scholar] [CrossRef]
- Ali, A.M.; Atmaj, J.; Van Oosterwijk, N.; Groves, M.R.; Dömling, A. Stapled Peptides Inhibitors: A New Window for Target Drug Discovery. Comput. Struct. Biotechnol. J. 2019, 17, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Kessler, D.; Gmachl, M.; Mantoulidis, A.; Martin, L.J.; Zoephel, A.; Mayer, M.; Gollner, A.; Covini, D.; Fischer, S.; Gerstberger, T.; et al. Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. USA 2019, 116, 15823–15829. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2020, 19, 23–38. [Google Scholar] [CrossRef]
- Cunningham, A.D.; Qvit, N.; Mochly-Rosen, D. Peptides and peptidomimetics as regulators of protein-protein interactions. Curr. Opin. Struct. Biol. 2017, 44, 59–66. [Google Scholar] [CrossRef]
- Cochran, A.G. Antagonists of protein-protein interactions. Chem. Biol. 2000, 7, R85–R94. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.A.; Engelmann, B.W.; Nash, P.D. High-throughput analysis of peptide-binding modules. Proteomics 2012, 12, 1527–1546. [Google Scholar] [CrossRef]
- Lien, S.; Lowman, H.B. Therapeutic peptides. Trends Biotechnol. 2003, 21, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef]
- Morrison, C. Constrained peptides’ time to shine? Nat. Rev. Drug Discov. 2018, 17, 531–533. [Google Scholar] [CrossRef]
- Rastogi, S.; Shukla, S.; Kalaivani, M.; Singh, G.N. Peptide-based therapeutics: Quality specifications, regulatory considerations, and prospects. Drug Discov. Today 2019, 24, 148–162. [Google Scholar] [CrossRef]
- Nagai, Y.; Lam, L.; Greene, M.I.; Zhang, H. FOXP3 and Its Cofactors as Targets of Immunotherapies. Engineering 2019, 5, 115–121. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The Single Global Macromolecular Structure Archive. Methods Mol. Biol. 2017, 1607, 627–641. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Vallat, B.; Lawson, C.L. The data universe of structural biology. IUCrJ 2020, 7, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Thornton, J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Sundberg, E.J.; Mariuzza, R.A. Molecular recognition in antibody-antigen complexes. Adv. Protein Chem. 2002, 61, 119–160. [Google Scholar] [CrossRef]
- Lafont, V.; Schaefer, M.; Stote, R.H.; Altschuh, D.; Dejaegere, A. Protein-protein recognition and interaction hot spots in an antigen-antibody complex: Free energy decomposition identifies “efficient amino acids”. Proteins 2007, 67, 418–434. [Google Scholar] [CrossRef] [PubMed]
- Strong, R.K.; Campbell, R.; Rose, D.R.; Petsko, G.A.; Sharon, J.; Margolies, M.N. Three-dimensional structure of murine anti-p-azophenylarsonate Fab 36-71. 1. X-ray crystallography, site-directed mutagenesis, and modeling of the complex with hapten. Biochemistry 1991, 30, 3739–3748. [Google Scholar] [CrossRef]
- Parkkinen, T.; Nevanen, T.K.; Koivula, A.; Rouvinen, J. Crystal structures of an enantioselective fab-fragment in free and complex forms. J. Mol. Biol. 2006, 357, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Park, B.W.; Zhang, H.T.; Wu, C.; Berezov, A.; Zhang, X.; Dua, R.; Wang, Q.; Kao, G.; O’Rourke, D.M.; Greene, M.I.; et al. Rationally designed anti-HER2/neu peptide mimetic disables P185HER2/neu tyrosine kinases in vitro and in vivo. Nat. Biotechnol. 2000, 18, 194–198. [Google Scholar] [CrossRef]
- Murali, R.; Greene, M.I. Structure based antibody-like peptidomimetics. Pharmaceuticals 2012, 5, 209–235. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Marin, A.; Thornton, J.M. Protein domain interfaces: Characterization and comparison with oligomeric protein interfaces. Protein Eng. 2000, 13, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, C.K.; Buckle, A.M.; Fersht, A.R. Structural response to mutation at a protein-protein interface. J. Mol. Biol. 1999, 286, 1487–1506. [Google Scholar] [CrossRef] [PubMed]
- Bhat, T.N.; Bentley, G.A.; Boulot, G.; Greene, M.I.; Tello, D.; Dall’Acqua, W.; Souchon, H.; Schwarz, F.P.; Mariuzza, R.A.; Poljak, R.J. Bound water molecules and conformational stabilization help mediate an antigen-antibody association. Proc. Natl. Acad. Sci. USA 1994, 91, 1089–1093. [Google Scholar] [CrossRef] [Green Version]
- Braden, B.C.; Poljak, R.J. Structural features of the reactions between antibodies and protein antigens. FASEB J. 1995, 9, 9–16. [Google Scholar] [CrossRef]
- Yan, C.; Wu, F.; Jernigan, R.L.; Dobbs, D.; Honavar, V. Characterization of protein-protein interfaces. Protein J. 2008, 27, 59–70. [Google Scholar] [CrossRef]
- Visscher, K.M.; Kastritis, P.L.; Bonvin, A.M. Non-interacting surface solvation and dynamics in protein-protein interactions. Proteins 2015, 83, 445–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieber-Emmons, T.; Murali, R.; Greene, M.I. Therapeutic peptides and peptidomimetics. Curr. Opin. Biotechnol. 1997, 8, 435–441. [Google Scholar] [CrossRef]
- Murali, R.; Greene, M.I. Structure-based design of immunologically active therapeutic peptides. Immunol. Res. 1998, 17, 163–169. [Google Scholar] [CrossRef] [PubMed]
- von Grafenstein, S.; Wallnoefer, H.G.; Kirchmair, J.; Fuchs, J.E.; Huber, R.G.; Schmidtke, M.; Sauerbrei, A.; Rollinger, J.M.; Liedl, K.R. Interface dynamics explain assembly dependency of influenza neuraminidase catalytic activity. J. Biomol. Struct. Dyn. 2015, 33, 104–120. [Google Scholar] [CrossRef] [Green Version]
- Watkins, A.M.; Wuo, M.G.; Arora, P.S. Protein-Protein Interactions Mediated by Helical Tertiary Structure Motifs. J. Am. Chem. Soc. 2015, 137, 11622–11630. [Google Scholar] [CrossRef]
- Franklin, M.C.; Carey, K.D.; Vajdos, F.F.; Leahy, D.J.; de Vos, A.M.; Sliwkowski, M.X. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 2004, 5, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Garrett, T.P.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Kofler, M.; Jorissen, R.N.; Nice, E.C.; Burgess, A.W.; et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol. Cell 2003, 11, 495–505. [Google Scholar] [CrossRef]
- Garrett, T.P.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Zhu, H.J.; Walker, F.; Frenkel, M.J.; Hoyne, P.A.; et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell 2002, 110, 763–773. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.S.; Leahy, D.J. Structure of the extracellular region of HER3 reveals an interdomain tether. Science 2002, 297, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.B.; Denney, D.W., Jr.; Leahy, D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760. [Google Scholar] [CrossRef]
- Berezov, A.; Chen, J.; Liu, Q.; Zhang, H.T.; Greene, M.I.; Murali, R. Disabling receptor ensembles with rationally designed interface peptidomimetics. J. Biol. Chem. 2002, 277, 28330–28339. [Google Scholar] [CrossRef] [Green Version]
- Naismith, J.H.; Devine, T.Q.; Kohno, T.; Sprang, S.R. Structures of the extracellular domain of the type I tumor necrosis factor receptor. Structure 1996, 4, 1251–1262. [Google Scholar] [CrossRef] [Green Version]
- Naismith, J.H.; Sprang, S.R. Modularity in the TNF-receptor family. Trends Biochem. Sci. 1998, 23, 74–79. [Google Scholar] [CrossRef]
- Baeyens, K.J.; De Bondt, H.L.; Raeymaekers, A.; Fiers, W.; De Ranter, C.J. The structure of mouse tumour-necrosis factor at 1.4 A resolution: Towards modulation of its selectivity and trimerization. Acta Cryst. D Biol. Cryst. 1999, 55, 772–778. [Google Scholar] [CrossRef] [Green Version]
- Ono, M.; Horita, S.; Sato, Y.; Nomura, Y.; Iwata, S.; Nomura, N. Structural basis for tumor necrosis factor blockade with the therapeutic antibody golimumab. Protein Sci. 2018, 27, 1038–1046. [Google Scholar] [CrossRef]
- Takasaki, W.; Kajino, Y.; Kajino, K.; Murali, R.; Greene, M.I. Structure-based design and characterization of exocyclic peptidomimetics that inhibit TNF.alpha. binding to its receptor. Nat. Biotechnol. 1997, 15, 1266–1270. [Google Scholar] [CrossRef]
- Kojima, T.; Aoki, K.; Nonaka, K.; Saito, H.; Azuma, M.; Iwai, H.; Varghese, B.J.; Yoshimasu, H.; Baron, R.; Ohya, K.; et al. Subcutaneous injections of a TNF-alpha antagonistic peptide inhibit both inflammation and bone resorption in collagen-induced murine arthritis. J. Med. Dent. Sci. 2005, 52, 91–99. [Google Scholar] [PubMed]
- Cheng, X.; Kinosaki, M.; Takami, M.; Choi, Y.; Zhang, H.; Murali, R. Disabling of receptor activator of nuclear factor-kappaB (RANK) receptor complex by novel osteoprotegerin-like peptidomimetics restores bone loss in vivo. J. Biol. Chem. 2004, 279, 8269–8277. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, A.; Cheng, X.; Kajino, K.; Berezov, A.; Murata, K.; Nakayama, T.; Yagita, H.; Murali, R.; Greene, M.I. Fas-disabling small exocyclic peptide mimetics limit apoptosis by an unexpected mechanism. Proc. Natl. Acad. Sci. USA 2004, 101, 6599–6604. [Google Scholar] [CrossRef] [Green Version]
- Masuda, K.; Richter, M.; Song, X.; Berezov, A.; Masuda, K.; Murali, R.; Greene, M.I.; Zhang, H. AHNP-streptavidin: A tetrameric bacterially produced antibody surrogate fusion protein against p185her2/neu. Oncogene 2006, 25, 7740–7746. [Google Scholar] [CrossRef] [Green Version]
- Mitran, B.; Andersson, K.G.; Lindström, E.; Garousi, J.; Rosestedt, M.; Tolmachev, V.; Ståhl, S.; Orlova, A.; Löfblom, J. Affibody-mediated imaging of EGFR expression in prostate cancer using radiocobalt-labeled DOTA-ZEGFR:2377. Oncol. Rep. 2019, 41, 534–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simeon, R.; Chen, Z. In vitro-engineered non-antibody protein therapeutics. Protein Cell 2018, 9, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanack, K.; Messerschmidt, K.; Listek, M. Antibodies and Selection of Monoclonal Antibodies. Adv. Exp. Med. Biol. 2016, 917, 11–22. [Google Scholar] [CrossRef]
- Thakur, A.; Huang, M.; Lum, L.G. Bispecific antibody based therapeutics: Strengths and challenges. Blood Rev. 2018, 32, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Fu, T.; Nagai, Y.; Lam, L.; Yee, M.; Zhu, Z.; Zhang, H. scFv-based “Grababody” as a general strategy to improve recruitment of immune effector cells to antibody-targeted tumors. Cancer Res. 2013, 73, 2619–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sazinsky, S.L.; Ott, R.G.; Silver, N.W.; Tidor, B.; Ravetch, J.V.; Wittrup, K.D. Aglycosylated immunoglobulin G1 variants productively engage activating Fc receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 20167–20172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berezov, A.; Zhang, H.T.; Greene, M.I.; Murali, R. Disabling erbB receptors with rationally designed exocyclic mimetics of antibodies: Structure-function analysis. J. Med. Chem. 2001, 44, 2565–2574. [Google Scholar] [CrossRef]
- Kato, G.; Shimizu, Y.; Arai, Y.; Suzuki, N.; Sugamori, Y.; Maeda, M.; Takahashi, M.; Tamura, Y.; Wakabayashi, N.; Murali, R.; et al. The inhibitory effects of a RANKL-binding peptide on articular and periarticular bone loss in a murine model of collagen-induced arthritis: A bone histomorphometric study. Arthritis Res. Ther. 2015, 17, 251. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Gangalum, P.R.; Galstyan, A.; Fox, I.; Patil, R.; Hubbard, P.; Murali, R.; Ljubimova, J.Y.; Holler, E. HER2-positive breast cancer targeting and treatment by a peptide-conjugated mini nanodrug. Nanomedicine 2017, 13, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque Bhuyan, M.Z.; Tamura, Y.; Sone, E.; Yoshinari, Y.; Maeda, C.; Takahashi, M.; Tabata, Y.; Murali, R.; Waki, Y.; Aoki, K. The intra-articular injection of RANKL-binding peptides inhibits cartilage degeneration in a murine model of osteoarthritis. J. Pharmacol. Sci. 2017, 134, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Idress, M.; Milne, B.F.; Thompson, G.S.; Trembleau, L.; Jaspars, M.; Houssen, W.E. Structure-Based Design, Synthesis and Bioactivity of a New Anti-TNFα Cyclopeptide. Molecules 2020, 25, 922. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murali, R.; Zhang, H.; Cai, Z.; Lam, L.; Greene, M. Rational Design of Constrained Peptides as Protein Interface Inhibitors. Antibodies 2021, 10, 32. https://doi.org/10.3390/antib10030032
Murali R, Zhang H, Cai Z, Lam L, Greene M. Rational Design of Constrained Peptides as Protein Interface Inhibitors. Antibodies. 2021; 10(3):32. https://doi.org/10.3390/antib10030032
Chicago/Turabian StyleMurali, Ramachandran, Hongtao Zhang, Zheng Cai, Lian Lam, and Mark Greene. 2021. "Rational Design of Constrained Peptides as Protein Interface Inhibitors" Antibodies 10, no. 3: 32. https://doi.org/10.3390/antib10030032
APA StyleMurali, R., Zhang, H., Cai, Z., Lam, L., & Greene, M. (2021). Rational Design of Constrained Peptides as Protein Interface Inhibitors. Antibodies, 10(3), 32. https://doi.org/10.3390/antib10030032