
A Review of Acquired Autoimmune Blistering Diseases in Inherited Epidermolysis Bullosa: Implications for the Future of Gene Therapy

 , ,
, ,
Abstract
1. Introduction
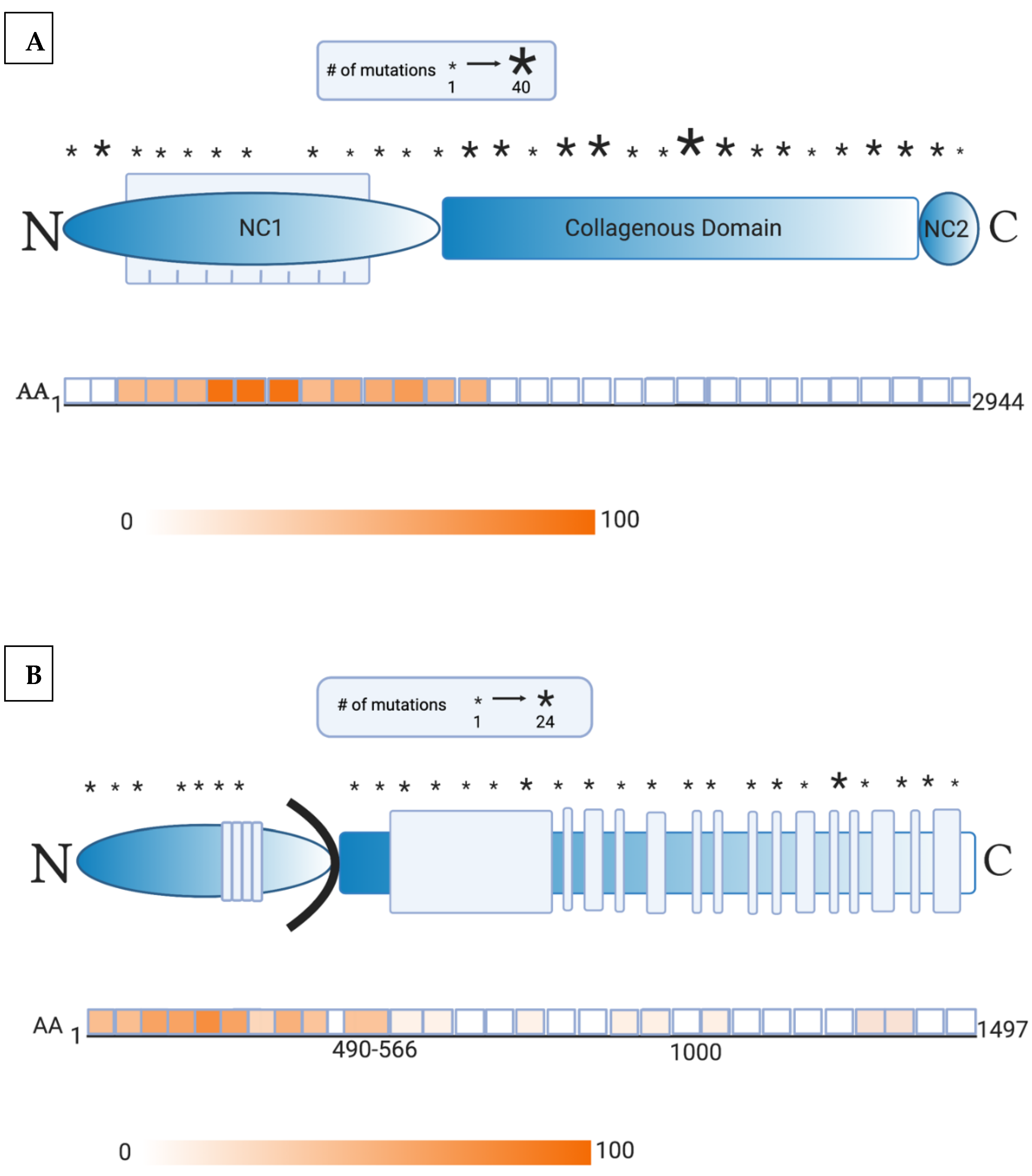
2. Presence of Anti-Skin Antibodies in EB
3. Altered ECM Properties
4. Eosinophilia and Elevated IgE
5. Pro-Inflammatory Microenvironment Due to Cytokine Dysregulation
6. Dysregulated Inflammatory Response and Blister Formation
7. Bacterial Infections
8. Implications of Autoantibodies in Gene Therapy
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patel, P.M.; Jones, V.A.; Murray, T.N.; Amber, K.T. A Review Comparing International Guidelines for the Management of Bullous Pemphigoid, Pemphigoid Gestationis, Mucous Membrane Pemphigoid, and Epidermolysis Bullosa Acquisita. Am. J. Clin. Dermatol. 2020, 21, 557–565. [Google Scholar] [CrossRef]
- Jones, V.A.; Patel, P.M.; Amber, K.T. Eosinophils in Bullous Pemphigoid. Panminerva Med. 2020. [Google Scholar] [CrossRef]
- Marinkovich, M.P.; Tang, J.Y. Gene Therapy for Epidermolysis Bullosa. J. Investig. Dermatol. 2019, 139, 1221–1226. [Google Scholar] [CrossRef] [PubMed]
- Has, C.; Nyström, A.; Saeidian, A.H.; Bruckner-Tuderman, L.; Uitto, J. Epidermolysis bullosa: Molecular pathology of connective tissue components in the cutaneous basement membrane zone. Matrix Biol. 2018, 71, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Mayr, E.; Koller, U.; Bauer, J.W. Gene Therapy for the COL7A1 Gene. In Gene Therapy; Molina, F.M., Ed.; IntechOpen: Rijeka, Croatia, 2013. [Google Scholar]
- Lapiere, J.C.; Woodley, D.T.; Parente, M.G.; Iwasaki, T.; Wynn, K.C.; Christiano, A.M.; Uitto, J. Epitope mapping of type VII collagen. Identification of discrete peptide sequences recognized by sera from patients with acquired epidermolysis bullosa. J. Clin. Investig. 1993, 92, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- Dang, N.; Murrell, D.F. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp. Dermatol. 2008, 17, 553–568. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Furukawa, F.; Imamura, S. Epitope mapping for epidermolysis bullosa acquisita autoantibody by molecularly cloned cDNA for type VII collagen. J. Investig. Dermatol. 1994, 102, 706–709. [Google Scholar] [CrossRef]
- Dresow, S.K.; Sitaru, C.; Recke, A.; Oostingh, G.J.; Zillikens, D.; Gibbs, B.F. IgE autoantibodies against the intracellular domain of BP180. Br. J. Dermatol. 2009, 160, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Di Zenzo, G.; Grosso, F.; Terracina, M.; Mariotti, F.; De Pità, O.; Owaribe, K.; Mastrogiacomo, A.; Sera, F.; Borradori, L.; Zambruno, G. Characterization of the anti-BP180 autoantibody reactivity profile and epitope mapping in bullous pemphigoid patients. J. Investig. Dermatol. 2004, 122, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.W.; Lanschuetzer, C. Type XVII collagen gene mutations in junctional epidermolysis bullosa and prospects for gene therapy. Clin. Exp. Dermatol. 2003, 28, 53–60. [Google Scholar] [CrossRef]
- Condrat, I.; He, Y.; Cosgarea, R.; Has, C. Junctional Epidermolysis Bullosa: Allelic Heterogeneity and Mutation Stratification for Precision Medicine. Front. Med. 2018, 5, 363. [Google Scholar] [CrossRef] [PubMed]
- Pasmooij, A.M.G.; Pas, H.H.; Jansen, G.H.L.; Lemmink, H.H.; Jonkman, M.F. Localized and generalized forms of blistering in junctional epidermolysis bullosa due to COL17A1 mutations in the Netherlands. Br. J. Dermatol. 2007, 156, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Gay, S.; Fine, J.D.; Storer, J.S. Autoantibodies to extracellular collagen matrix components in epidermolysis bullosa and other bullous diseases. Arch. Dermatol. Res. 1988, 280, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Guez, S.; Orenti, A.; Tadini, G.; Scuvera, G.; Corti, L.; Scala, A.; Biganzoli, E.; Berti, E.; Principi, N. Autoimmunity and Cytokine Imbalance in Inherited Epidermolysis Bullosa. Int. J. Mol. Sci. 2016, 17, 1625. [Google Scholar] [CrossRef] [PubMed]
- Tampoia, M.; Bonamonte, D.; Filoni, A.; Garofalo, L.; Morgese, M.G.; Brunetti, L.; Di Giorgio, C.; Annicchiarico, G. Prevalence of specific anti-skin autoantibodies in a cohort of patients with inherited epidermolysis bullosa. Orphanet J. Rare Dis. 2013, 8, 132. [Google Scholar] [CrossRef]
- Annicchiarico, G.; Morgese, M.G.; Esposito, S.; Lopalco, G.; Lattarulo, M.; Tampoia, M.; Bonamonte, D.; Brunetti, L.; Vitale, A.; Lapadula, G.; et al. Proinflammatory Cytokines and Antiskin Autoantibodies in Patients With Inherited Epidermolysis Bullosa. Medicine 2015, 94, e1528. [Google Scholar] [CrossRef]
- Kasperkiewicz, M.; Hirose, M.; Recke, A.; Schmidt, E.; Zillikens, D.; Ludwig, R.J. Clearance rates of circulating and tissue-bound autoantibodies to type VII collagen in experimental epidermolysis bullosa acquisita. Br. J. Dermatol. 2010, 162, 1064–1070. [Google Scholar] [CrossRef]
- Licarete, E.; Ganz, S.; Recknagel, M.J.; Di Zenzo, G.; Hashimoto, T.; Hertl, M.; Zambruno, G.; Hundorfean, G.; Mudter, J.; Neurath, M.F.; et al. Prevalence of collagen VII-specific autoantibodies in patients with autoimmune and inflammatory diseases. BMC Immunol. 2012, 13, 16. [Google Scholar] [CrossRef]
- Hayashi, R.; Natsuga, K.; Watanabe, M.; Iwata, H.; Shinkuma, S.; Ito, A.; Masui, Y.; Ito, M.; Shimomura, Y. Epidermolysis Bullosa Acquisita Develops in Dominant Dystrophic Epidermolysis Bullosa. J. Investig. Dermatol. 2016, 136, 320–323. [Google Scholar] [CrossRef][Green Version]
- Gammon, W.R.; Briggaman, R.A.; Woodley, D.T.; Heald, P.W.; Wheeler, E.C., Jr. Epidermolysis bullosa acquisita—a pemphigoid-like disease. J. Am. Acad. Dermatol. 1984, 11, 820–832. [Google Scholar] [CrossRef]
- Stevens, N.E.; Cowin, A.J.; Kopecki, Z. Skin Barrier and Autoimmunity-Mechanisms and Novel Therapeutic Approaches for Autoimmune Blistering Diseases of the Skin. Front. Immunol. 2019, 10, 1089. [Google Scholar] [CrossRef] [PubMed]
- Mihai, S.; Hirose, M.; Wang, Y.; Thurman, J.M.; Holers, V.M.; Morgan, B.P.; Kohl, J.; Zillikens, D.; Ludwig, R.J.; Nimmerjahn, F. Specific Inhibition of Complement Activation Significantly Ameliorates Autoimmune Blistering Disease in Mice. Front. Immunol. 2018, 9, 535. [Google Scholar] [CrossRef] [PubMed]
- Mihai, S.; Chiriac, M.T.; Takahashi, K.; Thurman, J.M.; Holers, V.M.; Zillikens, D.; Botto, M.; Sitaru, C. The alternative pathway of complement activation is critical for blister induction in experimental epidermolysis bullosa acquisita. J. Immunol. 2007, 178, 6514–6521. [Google Scholar] [CrossRef]
- Sitaru, C.; Kromminga, A.; Hashimoto, T.; Brocker, E.B.; Zillikens, D. Autoantibodies to type VII collagen mediate Fcgamma-dependent neutrophil activation and induce dermal-epidermal separation in cryosections of human skin. Am. J. Pathol. 2002, 161, 301–311. [Google Scholar] [CrossRef]
- Bieber, K.; Witte, M.; Sun, S.; Hundt, J.E.; Kalies, K.; Drager, S.; Kasprick, A.; Twelkmeyer, T.; Manz, R.A.; Konig, P.; et al. T cells mediate autoantibody-induced cutaneous inflammation and blistering in epidermolysis bullosa acquisita. Sci. Rep. 2016, 6, 38357. [Google Scholar] [CrossRef]
- Liu, Z.; Giudice, G.J.; Swartz, S.J.; Fairley, J.A.; Till, G.O.; Troy, J.L.; Diaz, L.A. The role of complement in experimental bullous pemphigoid. J. Clin. Investig. 1995, 95, 1539–1544. [Google Scholar] [CrossRef]
- Iwata, H.; Ujiie, H. Complement-independent blistering mechanisms in bullous pemphigoid. Exp. Dermatol. 2017, 26, 1235–1239. [Google Scholar] [CrossRef]
- Mihai, S.; Chiriac, M.T.; Herrero-González, J.E.; Goodall, M.; Jefferis, R.; Savage, C.O.S.; Zillikens, D.; Sitaru, C. IgG4 autoantibodies induce dermal-epidermal separation. J. Cell. Mol. Med. 2007, 11, 1117–1128. [Google Scholar] [CrossRef]
- Liu, Y.; Li, L.; Xia, Y. BP180 Is Critical in the Autoimmunity of Bullous Pemphigoid. Front. Immunol. 2017, 8, 1752. [Google Scholar] [CrossRef]
- Liu, Y.-D.; Wang, Y.-H.; Ye, Y.-C.; Zhao, W.-L.; Li, L. Prognostic factors for mortality in patients with bullous pemphigoid: A meta-analysis. Arch. Dermatol. Res. 2017, 309, 335–347. [Google Scholar] [CrossRef] [PubMed]
- van Beek, N.; Lüttmann, N.; Huebner, F.; Recke, A.; Karl, I.; Schulze, F.S.; Zillikens, D.; Schmidt, E. Correlation of Serum Levels of IgE Autoantibodies Against BP180 With Bullous Pemphigoid Disease Activity. JAMA Dermatol. 2017, 153, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Wieland, C.N.; Comfere, N.I.; Gibson, L.E.; Weaver, A.L.; Krause, P.K.; Murray, J.A. Anti-bullous pemphigoid 180 and 230 antibodies in a sample of unaffected subjects. Arch Dermatol. 2010, 146, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Málaga, S.; Toral, J.F.; Santos, F.; Riesgo, I.; Crespo, M. Renal amyloidosis complicating a recessive epidermolysis bullosa in childhood. Helv. Paediatr. Acta 1983, 38, 167–170. [Google Scholar] [PubMed]
- Mann, J.F.; Zeier, M.; Zilow, E.; Schärer, K.; Anton-Lamprecht, I.; Waldherr, R.; Andrassy, K.; Ritz, E. The spectrum of renal involvement in epidermolysis bullosa dystrophica hereditaria: Report of two cases. Am. J. Kidney Dis. 1988, 11, 437–441. [Google Scholar] [CrossRef]
- Chen, C.C.; Isomoto, H.; Hayashi, T. Gastrointestinal amyloidosis secondary to inherited skin disorder. Gastroenterology 2012, 142, e9–e10. [Google Scholar] [CrossRef] [PubMed]
- Annicchiarico, G.; Morgese, M.G.; Brunetti, L.; Tampoia, M.; Garofalo, L.; Aceto, G.; Fiore, T.; Mauro, S.; Minelli, M. Improvement of renal function in epidermolysis bullosa patients after gluten free diet: Two cases. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 138–141. [Google Scholar]
- Guerra, L.; Condorelli, A.G.; Fortugno, P.; Calabresi, V.; Pedicelli, C.; Di Zenzo, G.; Castiglia, D. Epidermolysis Bullosa (EB) Acquisita in an Adult Patient with Previously Unrecognized Mild Dystrophic EB and Biallelic COL7A1 Mutations. Acta Derm. Venereol. 2018, 98, 411–415. [Google Scholar] [CrossRef]
- Fania, L.; Provini, A.; Salemme, A.; Sinagra, J.L.; Guerra, L.; Mazzanti, C.; Didona, B.; Castiglia, D.; Di Zenzo, G. Development of bullous pemphigoid in junctional epidermolysis bullosa. J. Eur. Acad. Dermatol. Venereol. 2020, 34, e146–e148. [Google Scholar] [CrossRef]
- Mittapalli, V.R.; Madl, J.; Löffek, S.; Kiritsi, D.; Kern, J.S.; Römer, W.; Nyström, A.; Bruckner-Tuderman, L. Injury-Driven Stiffening of the Dermis Expedites Skin Carcinoma Progression. Cancer Res. 2016, 76, 940–951. [Google Scholar] [CrossRef]
- Kumawat, K.; Gosens, R. WNT-5A: Signaling and functions in health and disease. Cell. Mol. Life Sci. 2016, 73, 567–587. [Google Scholar] [CrossRef]
- Atanasova, V.S.; Russell, R.J.; Webster, T.G.; Cao, Q.; Agarwal, P.; Lim, Y.Z.; Krishnan, S.; Fuentes, I.; Guttmann-Gruber, C.; McGrath, J.A.; et al. Thrombospondin-1 Is a Major Activator of TGF-β Signaling in Recessive Dystrophic Epidermolysis Bullosa Fibroblasts. J. Investig. Dermatol. 2019, 139, 1497–1505.e1495. [Google Scholar] [CrossRef]
- Küttner, V.; Mack, C.; Rigbolt, K.T.G.; Kern, J.S.; Schilling, O.; Busch, H.; Bruckner-Tuderman, L.; Dengjel, J. Global remodelling of cellular microenvironment due to loss of collagen VII. Mol. Syst. Biol. 2013, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Küttner, V.; Mack, C.; Gretzmeier, C.; Bruckner-Tuderman, L.; Dengjel, J. Loss of collagen VII is associated with reduced transglutaminase 2 abundance and activity. J. Investig. Dermatol. 2014, 134, 2381–2389. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Talele, N.P.; Boo, S.; Koehler, A.; Knee-Walden, E.; Balestrini, J.L.; Speight, P.; Kapus, A.; Hinz, B. MicroRNA-21 preserves the fibrotic mechanical memory of mesenchymal stem cells. Nat. Mater. 2017, 16, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Nyström, A.; Bruckner-Tuderman, L. Injury- and inflammation-driven skin fibrosis: The paradigm of epidermolysis bullosa. Matrix Biol. 2018, 68, 547–560. [Google Scholar] [CrossRef]
- Windler, C.; Hermsdorf, U.; Brinckmann, J.; Seeger, K. A type VII collagen subdomain mutant is thermolabile and shows enhanced proteolytic degradability—Implications for the pathogenesis of recessive dystrophic epidermolysis bullosa? Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 52–59. [Google Scholar] [CrossRef]
- Ruocco, V.; Ruocco, E.; Piccolo, V.; Brunetti, G.; Guerrera, L.P.; Wolf, R. The immunocompromised district in dermatology: A unifying pathogenic view of the regional immune dysregulation. Clin. Dermatol. 2014, 32, 569–576. [Google Scholar] [CrossRef]
- Rotunda, A.M.; Bhupathy, A.R.; Dye, R.; Soriano, T.T. Pemphigus foliaceus masquerading as postoperative wound infection: Report of a case and review of the Koebner and related phenomenon following surgical procedures. Dermatol. Surg. 2005, 31, 226–231. [Google Scholar] [CrossRef]
- Sen, B.B.; Ekiz, Ö.; Rifaioglu, E.N.; Sen, T.; Atik, E.; Dogramaci, A.Ç. Localized bullous pemphigoid occurring on surgical scars. Indian J. Dermatol. Venereol. Leprol. 2013, 79, 554. [Google Scholar] [CrossRef]
- Sanchez-Palacios, C.; Chan, L.S. Development of pemphigus herpetiformis in a patient with psoriasis receiving UV-light treatment. J. Cutan. Pathol. 2004, 31, 346–349. [Google Scholar] [CrossRef]
- Baroero, L.; Coppo, P.; Bertolino, L.; Maccario, S.; Savino, F. Three case reports of post immunization and post viral Bullous Pemphigoid: Looking for the right trigger. BMC Pediatr. 2017, 17, 60. [Google Scholar] [CrossRef] [PubMed]
- Sachet, M.; Liang, Y.Y.; Oehler, R. The immune response to secondary necrotic cells. Apoptosis 2017, 22, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- Moro, F.; Fania, L.; Sinagra, J.L.M.; Salemme, A.; Zenzo, G.D. Bullous Pemphigoid: Trigger and Predisposing Factors. Biomolecules 2020, 10, 1432. [Google Scholar] [CrossRef] [PubMed]
- Kopecki, Z.; Cowin, A.J. Flightless I: An actin-remodelling protein and an important negative regulator of wound repair. Int. J. Biochem. Cell Biol. 2008, 40, 1415–1419. [Google Scholar] [CrossRef]
- Kopecki, Z.; Ruzehaji, N.; Turner, C.; Iwata, H.; Ludwig, R.J.; Zillikens, D.; Murrell, D.F.; Cowin, A.J. Topically applied flightless I neutralizing antibodies improve healing of blistered skin in a murine model of epidermolysis bullosa acquisita. J. Investig. Dermatol. 2013, 133, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Kopecki, Z.; Yang, G.N.; Arkell, R.M.; Jackson, J.E.; Melville, E.; Iwata, H.; Ludwig, R.J.; Zillikens, D.; Murrell, D.F.; Cowin, A.J. Flightless I over-expression impairs skin barrier development, function and recovery following skin blistering. J. Pathol. 2014, 232, 541–552. [Google Scholar] [CrossRef]
- Kopecki, Z.; Yang, G.; Treloar, S.; Mashtoub, S.; Howarth, G.S.; Cummins, A.G.; Cowin, A.J. Flightless I exacerbation of inflammatory responses contributes to increased colonic damage in a mouse model of dextran sulphate sodium-induced ulcerative colitis. Sci. Rep. 2019, 9, 12792. [Google Scholar] [CrossRef]
- Kopecki, Z.; Stevens, N.E.; Chong, H.T.; Yang, G.N.; Cowin, A.J. Flightless I Alters the Inflammatory Response and Autoantibody Profile in an OVA-Induced Atopic Dermatitis Skin-Like Disease. Front. Immunol. 2018, 9, 1833. [Google Scholar] [CrossRef] [PubMed]
- Saraiya, A.; Yang, C.S.; Kim, J.; Bercovitch, L.; Robinson-Bostom, L.; Telang, G. Dermal eosinophilic infiltrate in junctional epidermolysis bullosa. J. Cutan. Pathol. 2015, 42, 559–563. [Google Scholar] [CrossRef]
- Grunwald, M.H.; Amichai, B.; Avinoach, I.; Kedar, T.; Bergman, R. Dystrophic epidermolysis bullosa associated with eosinophilic infiltrate and elevated serum IgE. Pediatr. Dermatol. 1999, 16, 16–18. [Google Scholar] [CrossRef]
- Roth, R.R.; Smith, K.J.; James, W.D. Eosinophilic infiltrates in epidermolysis bullosa. Arch. Dermatol. 1990, 126, 1191–1194. [Google Scholar] [CrossRef]
- Anton-Lamprecht, I.; Schnyder, U.W. Epidermolysis bullosa herpetiformis Dowling-Meara. Report of a case and pathomorphogenesis. Dermatologica 1982, 164, 221–235. [Google Scholar] [CrossRef]
- Nomura, M.; Hamasaki, Y.-I.; Katayama, I.; Abe, K.; Niikawa, N.; Yoshiura, K.-I. Eosinophil infiltration in three patients with generalized atrophic benign epidermolysis bullosa from a Japanese family: Molecular genetic and immunohistochemical studies. J. Hum. Genet. 2005, 50, 483–489. [Google Scholar] [CrossRef]
- Drury, R.E.; Prieto, A., Jr. Epidermolysis bullosa dystrophica. report of two cases within a family group. Oral Surg. Oral Med. Oral Pathol. 1964, 18, 544–551. [Google Scholar] [CrossRef]
- Vivehanantha, S.; Carr, R.A.; McGrath, J.A.; Taibjee, S.M.; Madhogaria, S.; Ilchyshyn, A. Epidermolysis bullosa pruriginosa: A case with prominent histopathologic inflammation. JAMA Dermatol. 2013, 149, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Tasanen, K.; Tunggal, L.; Chometon, G.; Bruckner-Tuderman, L.; Aumailley, M. Keratinocytes from patients lacking collagen XVII display a migratory phenotype. Am. J. Pathol. 2004, 164, 2027–2038. [Google Scholar] [CrossRef][Green Version]
- Amber, K.T.; Valdebran, M.; Kridin, K.; Grando, S.A. The Role of Eosinophils in Bullous Pemphigoid: A Developing Model of Eosinophil Pathogenicity in Mucocutaneous Disease. Front. Med. 2018, 5, 201. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Hwang, B.-J.; Culton, D.A.; Li, N.; Burette, S.; Koller, B.H.; Messingham, K.A.; Fairley, J.A.; Lee, J.J.; Hall, R.P.; et al. Eosinophils Mediate Tissue Injury in the Autoimmune Skin Disease Bullous Pemphigoid. J. Investig. Dermatol. 2018, 138, 1032–1043. [Google Scholar] [CrossRef]
- Kasahara-Imamura, M.; Hosokawa, H.; Maekawa, N.; Horio, T. Activation of Fc epsilon RI-positive eosinophils in bullous pemphigoid. Int. J. Mol. Med. 2001, 7, 249–253. [Google Scholar]
- Romagnani, S. Immunologic influences on allergy and the TH1/TH2 balance. J. Allergy Clin. Immunol. 2004, 113, 395–400. [Google Scholar] [CrossRef]
- de Graauw, E.; Sitaru, C.; Horn, M.; Borradori, L.; Yousefi, S.; Simon, H.U.; Simon, D. Evidence for a role of eosinophils in blister formation in bullous pemphigoid. Allergy 2017, 72, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Amber, K.T.; Chernyavsky, A.; Agnoletti, A.F.; Cozzani, E.; Grando, S.A. Mechanisms of pathogenic effects of eosinophil cationic protein and eosinophil-derived neurotoxin on human keratinocytes. Exp. Dermatol. 2018, 27, 1322–1327. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Kursewicz, C.D.; Fayne, R.A.; Nanda, S.; Shah, S.M.; Nattkemper, L.; Yokozeki, H.; Yosipovitch, G. Pathophysiologic mechanisms of itch in bullous pemphigoid. J. Am. Acad. Dermatol. 2020, 83, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, B.F.; Patsinakidis, N.; Raap, U. Role of the Pruritic Cytokine IL-31 in Autoimmune Skin Diseases. Front. Immunol. 2019, 10, 1383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hwang, B.-J.; Liu, Z.; Li, N.; Lough, K.; Williams, S.E.; Chen, J.; Burette, S.W.; Diaz, L.A.; Su, M.A.; et al. BP180 dysfunction triggers spontaneous skin inflammation in mice. Proc. Natl. Acad. Sci. USA 2018, 115, 6434–6439. [Google Scholar] [CrossRef]
- Koga, H.; Teye, K.; Yamashita, K.; Ishii, N.; Tsuruta, D.; Nakama, T. Detection of anti-type VII collagen IgE antibodies in epidermolysis bullosa acquisita. Br. J. Dermatol. 2019, 180, 1107–1113. [Google Scholar] [CrossRef]
- Freire, P.C.; Muñoz, C.H.; Stingl, G. IgE autoreactivity in bullous pemphigoid: Eosinophils and mast cells as major targets of pathogenic immune reactants. Br. J. Dermatol. 2017, 177, 1644–1653. [Google Scholar] [CrossRef]
- Kawakami, Y.; Oyama, N.; Ohtsuka, M.; Nakamura, K.; Kaneko, F. Increased serum levels of interleukin-6, immunoglobulin and acute phase protein in patients with the severe clinical form of inherited epidermolysis bullosa. J. Dermatol. 2005, 32, 503–505. [Google Scholar] [CrossRef] [PubMed]
- Odorisio, T.; Di Salvio, M.; Orecchia, A.; Di Zenzo, G.; Piccinni, E.; Cianfarani, F.; Travaglione, A.; Uva, P.; Bellei, B.; Conti, A.; et al. Monozygotic twins discordant for recessive dystrophic epidermolysis bullosa phenotype highlight the role of TGF-β signalling in modifying disease severity. Hum. Mol. Genet. 2014, 23, 3907–3922. [Google Scholar] [CrossRef]
- Komiyama, Y.; Nakae, S.; Matsuki, T.; Nambu, A.; Ishigame, H.; Kakuta, S.; Sudo, K.; Iwakura, Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J. Immunol. 2006, 177, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Ishigame, H.; Kakuta, S.; Nagai, T.; Kadoki, M.; Nambu, A.; Komiyama, Y.; Fujikado, N.; Tanahashi, Y.; Akitsu, A.; Kotaki, H.; et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 2009, 30, 108–119. [Google Scholar] [CrossRef]
- Samavedam, U.K.S.R.L.; Kalies, K.; Scheller, J.; Sadeghi, H.; Gupta, Y.; Jonkman, M.F.; Schmidt, E.; Westermann, J.; Zillikens, D.; Rose-John, S.; et al. Recombinant IL-6 treatment protects mice from organ specific autoimmune disease by IL-6 classical signalling-dependent IL-1ra induction. J. Autoimmun. 2013, 40, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Kothari, P.; Pestana, R.; Mesraoua, R.; Elchaki, R.; Khan, K.M.F.; Dannenberg, A.J.; Falcone, D.J. IL-6-mediated induction of matrix metalloproteinase-9 is modulated by JAK-dependent IL-10 expression in macrophages. J. Immunol. 2014, 192, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Lettner, T.; Lang, R.; Bauer, J.W.; Wally, V. Increased levels of matrix metalloproteinase-9 and interleukin-8 in blister fluids of dystrophic and junctional epidermolysis bullosa patients. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Lettner, T.; Lang, R.; Klausegger, A.; Hainzl, S.; Bauer, J.W.; Wally, V. MMP-9 and CXCL8/IL-8 are potential therapeutic targets in epidermolysis bullosa simplex. PLoS ONE 2013, 8, e70123. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Patel, K.D. Regulation of matrix metalloproteinase-9 release from IL-8-stimulated human neutrophils. J. Leukoc. Biol. 2005, 78, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, N.; Lanza, M.; Rossiello, L.; Gombos, F.; Lanza, A. Defining the involvement of proteinases in pemphigus vulgaris: Evidence of matrix metalloproteinase-9 overexpression in experimental models of disease. J. Cell Physiol. 2007, 212, 36–41. [Google Scholar] [CrossRef]
- Petrof, G.; Abdul-Wahab, A.; Proudfoot, L.; Pramanik, R.; Mellerio, J.E.; McGrath, J.A. Serum levels of high mobility group box 1 correlate with disease severity in recessive dystrophic epidermolysis bullosa. Exp. Dermatol. 2013, 22, 433–435. [Google Scholar] [CrossRef]
- Tamai, K.; Yamazaki, T.; Chino, T.; Ishii, M.; Otsuru, S.; Kikuchi, Y.; Iinuma, S.; Saga, K.; Nimura, K.; Shimbo, T.; et al. PDGFRalpha-positive cells in bone marrow are mobilized by high mobility group box 1 (HMGB1) to regenerate injured epithelia. Proc. Natl. Acad. Sci. USA 2011, 108, 6609–6614. [Google Scholar] [CrossRef]
- Ujiie, I.; Fujita, Y.; Nakayama, C.; Matsumura, W.; Suzuki, S.; Shinkuma, S.; Nomura, T.; Abe, R.; Shimizu, H. Altered balance of epidermis-related chemokines in epidermolysis bullosa. J. Dermatol. Sci. 2017, 86, 37–45. [Google Scholar] [CrossRef]
- Burman, A.; Haworth, O.; Hardie, D.L.; Amft, E.N.; Siewert, C.; Jackson, D.G.; Salmon, M.; Buckley, C.D. A chemokine-dependent stromal induction mechanism for aberrant lymphocyte accumulation and compromised lymphatic return in rheumatoid arthritis. J. Immunol. 2005, 174, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.; Homey, B.; Vicari, A.P.; Hudak, S.; Oldham, E.; Hedrick, J.; Orozco, R.; Copeland, N.G.; Jenkins, N.A.; McEvoy, L.M.; et al. CTACK, a skin-associated chemokine that preferentially attracts skin-homing memory T cells. Proc. Natl. Acad. Sci. USA 1999, 96, 14470–14475. [Google Scholar] [CrossRef] [PubMed]
- Jaerve, A.; Schira, J.; Müller, H.W. Concise review: The potential of stromal cell-derived factor 1 and its receptors to promote stem cell functions in spinal cord repair. Stem Cells Transl. Med. 2012, 1, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Straino, S.; Di Carlo, A.; Mangoni, A.; De Mori, R.; Guerra, L.; Maurelli, R.; Panacchia, L.; Di Giacomo, F.; Palumbo, R.; Di Campli, C.; et al. High-mobility group box 1 protein in human and murine skin: Involvement in wound healing. J. Investig. Dermatol. 2008, 128, 1545–1553. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Hauschild, R.; Schwarz, J.; Moussion, C.; de Vries, I.; Legler, D.F.; Luther, S.A.; Bollenbach, T.; Sixt, M. Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science 2013, 339, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Giomi, B.; Caproni, M.; Calzolari, A.; Bianchi, B.; Fabbri, P. Th1, Th2 and Th3 cytokines in the pathogenesis of bullous pemphigoid. J. Dermatol. Sci. 2002, 30, 116–128. [Google Scholar] [CrossRef]
- Takiguchi, Y.; Itoh, N.; Suzuki, M.; Kaneko, F.; Minagawa, T. The role of gamma-interferon in blister formation of bullous pemphigoid. Nihon Hifuka Gakkai Zasshi Jpn. J. Dermatol. 1991, 101, 121–127. [Google Scholar]
- Kowalski, E.H.; Kneibner, D.; Kridin, K.; Amber, K.T. Serum and blister fluid levels of cytokines and chemokines in pemphigus and bullous pemphigoid. Autoimmun. Rev. 2019, 18, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Guez, S.; Manzoni, F.; Bosco, A.; Rigante, D. Epidermolysis bullosa and the partnership with autoimmunity: What should we assimilate? Immunol. Res. 2015, 61, 63–69. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Suda, T.; Suda, J.; Eguchi, M.; Miura, Y.; Harada, N.; Tominaga, A.; Takatsu, K. Purified interleukin 5 supports the terminal differentiation and proliferation of murine eosinophilic precursors. J. Exp. Med. 1988, 167, 43–56. [Google Scholar] [CrossRef]
- Lopez, A.F.; Sanderson, C.J.; Gamble, J.R.; Campbell, H.D.; Young, I.G.; Vadas, M.A. Recombinant human interleukin 5 is a selective activator of human eosinophil function. J. Exp. Med. 1988, 167, 219–224. [Google Scholar] [CrossRef] [PubMed]
- D’Auria, L.; Pietravalle, M.; Mastroianni, A.; Ferraro, C.; Mussi, A.; Bonifati, C.; Giacalone, B.; Ameglio, F. IL-5 levels in the serum and blister fluid of patients with bullous pemphigoid: Correlations with eosinophil cationic protein, RANTES, IgE and disease severity. Arch. Dermatol. Res. 1998, 290, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Tran, G.T.; Hodgkinson, S.J.; Carter, N.M.; Verma, N.D.; Plain, K.M.; Boyd, R.; Robinson, C.M.; Nomura, M.; Killingsworth, M.; Hall, B.M. IL-5 promotes induction of antigen-specific CD4+CD25+ T regulatory cells that suppress autoimmunity. Blood 2012, 119, 4441–4450. [Google Scholar] [CrossRef] [PubMed]
- Balighi, K.; Daneshpazhooh, M.; Azizpour, A.; Lajevardi, V.; Mohammadi, F.; Chams-Davatchi, C. Koebner phenomenon in pemphigus vulgaris patients. JAAD Case Rep. 2016, 2, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Jonkman, M.F.; de Jong, M.C.; Heeres, K.; Pas, H.H.; van der Meer, J.B.; Owaribe, K.; de Velasco, A.M.M.; Niessen, C.M.; Sonnenberg, A. 180-kD bullous pemphigoid antigen (BP180) is deficient in generalized atrophic benign epidermolysis bullosa. J. Clin. Investig. 1995, 95, 1345–1352. [Google Scholar] [CrossRef]
- McGrath, J.A.; Gatalica, B.; Christiano, A.M.; Li, K.; Owaribe, K.; McMillan, J.R.; Eady, R.A.; Uitto, J. Mutations in the 180-kD bullous pemphigoid antigen (BPAG2), a hemidesmosomal transmembrane collagen (COL17A1), in generalized atrophic benign epidermolysis bullosa. Nat. Genet. 1995, 11, 83–86. [Google Scholar] [CrossRef]
- Jones, V.A.; Patel, P.M.; Gibson, F.T.; Cordova, A.; Amber, K.T. The Role of Collagen XVII in Cancer: Squamous Cell Carcinoma and Beyond. Front. Oncol. 2020, 10, 352. [Google Scholar] [CrossRef] [PubMed]
- Van den Bergh, F.; Eliason, S.L.; Burmeister, B.T.; Giudice, G.J. Collagen XVII (BP180) modulates keratinocyte expression of the proinflammatory chemokine, IL-8. Exp. Dermatol. 2012, 21, 605–611. [Google Scholar] [CrossRef]
- Liu, Z.; Giudice, G.J.; Zhou, X.; Swartz, S.J.; Troy, J.L.; Fairley, J.A.; Till, G.O.; Diaz, L.A. A major role for neutrophils in experimental bullous pemphigoid. J. Clin. Investig. 1997, 100, 1256–1263. [Google Scholar] [CrossRef]
- Iwata, H.; Kamaguchi, M.; Ujiie, H.; Nishimura, M.; Izumi, K.; Natsuga, K.; Shinkuma, S.; Nishie, W.; Shimizu, H. Macropinocytosis of type XVII collagen induced by bullous pemphigoid IgG is regulated via protein kinase C. Lab. Investig. 2016, 96, 1301–1310. [Google Scholar] [CrossRef]
- Cusick, M.F.; Libbey, J.E.; Fujinami, R.S. Molecular mimicry as a mechanism of autoimmune disease. Clin. Rev. Allergy Immunol. 2012, 42, 102–111. [Google Scholar] [CrossRef]
- Levin, L.E.; Shayegan, L.H.; Lucky, A.W.; Hook, K.P.; Bruckner, A.L.; Feinstein, J.A.; Whittier, S.; Lauren, C.T.; Pope, E.; Lara-Corrales, I.; et al. Characterization of wound microbes in epidermolysis bullosa: Results from the epidermolysis bullosa clinical characterization and outcomes database. Pediatr. Dermatol. 2020, 38, 119–124. [Google Scholar] [CrossRef] [PubMed]
- van der Kooi-Pol, M.M.; Veenstra-Kyuchukova, Y.K.; Duipmans, J.C.; Pluister, G.N.; Schouls, L.M.; de Neeling, A.J.; Grundmann, H.; Jonkman, M.F.; van Dijl, J.M. High genetic diversity of Staphylococcus aureus strains colonizing patients with epidermolysis bullosa. Exp. Dermatol. 2012, 21, 463–466. [Google Scholar] [CrossRef]
- Föll, M.C.; Fahrner, M.; Gretzmeier, C.; Thoma, K.; Biniossek, M.L.; Kiritsi, D.; Meiss, F.; Schilling, O.; Nyström, A.; Kern, J.S. Identification of tissue damage, extracellular matrix remodeling and bacterial challenge as common mechanisms associated with high-risk cutaneous squamous cell carcinomas. Matrix Biol. 2018, 66, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Nyström, A.; Bornert, O.; Kühl, T.; Gretzmeier, C.; Thriene, K.; Dengjel, J.; Pfister-Wartha, A.; Kiritsi, D.; Bruckner-Tuderman, L. Impaired lymphoid extracellular matrix impedes antibacterial immunity in epidermolysis bullosa. Proc. Natl. Acad. Sci. USA 2018, 115, E705–E714. [Google Scholar] [CrossRef] [PubMed]
- Nyström, A.; Thriene, K.; Mittapalli, V.; Kern, J.S.; Kiritsi, D.; Dengjel, J.; Bruckner-Tuderman, L. Losartan ameliorates dystrophic epidermolysis bullosa and uncovers new disease mechanisms. EMBO Mol. Med. 2015, 7, 1211–1228. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, J.R.; Wojnarowska, F.; Kirtschig, G.; Nunn, A.J.; Bratton, D.J.; Mason, J.; Foster, K.A.; Whitham, D.; Williams, H.C.; on behalf of the BLISTER study group. A randomized controlled trial to compare the safety and effectiveness of doxycycline (200 mg daily) with oral prednisolone (0.5 mg kg−1 daily) for initial treatment of bullous pemphigoid: A protocol for the Bullous Pemphigoid Steroids and Tetracyclines (BLISTER) Trial. Br. J. Dermatol. 2015, 173, 227–234. [Google Scholar] [CrossRef]
- Bauer, J.W.; Koller, J.; Murauer, E.M.; De Rosa, L.; Enzo, E.; Carulli, S.; Bondanza, S.; Recchia, A.; Muss, W.; Diem, A.; et al. Closure of a Large Chronic Wound through Transplantation of Gene-Corrected Epidermal Stem Cells. J. Investig. Dermatol. 2017, 137, 778–781. [Google Scholar] [CrossRef]
- Hirsch, T.; Rothoeft, T.; Teig, N.; Bauer, J.W.; Pellegrini, G.; De Rosa, L.; Scaglione, D.; Reichelt, J.; Klausegger, A.; Kneisz, D.; et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature 2017, 551, 327–332. [Google Scholar] [CrossRef]
- Domloge-Hultsch, N.; Gammon, W.R.; Briggaman, R.A.; Gil, S.G.; Carter, W.G.; Yancey, K.B. Epiligrin, the major human keratinocyte integrin ligand, is a target in both an acquired autoimmune and an inherited subepidermal blistering skin disease. J. Clin. Investig. 1992, 90, 1628–1633. [Google Scholar] [CrossRef]
- Amber, K.T.; Bloom, R.; Hertl, M. A systematic review with pooled analysis of clinical presentation and immunodiagnostic testing in mucous membrane pemphigoid: Association of anti-laminin-332 IgG with oropharyngeal involvement and the usefulness of ELISA. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Christiano, A.M.; Greenspan, D.S.; Lee, S.; Uitto, J. Cloning of human type VII collagen. Complete primary sequence of the alpha 1(VII) chain and identification of intragenic polymorphisms. J. Biol. Chem. 1994, 269, 20256–20262. [Google Scholar] [CrossRef]
- Siprashvili, Z.; Nguyen, N.T.; Bezchinsky, M.Y.; Marinkovich, M.P.; Lane, A.T.; Khavari, P.A. Long-term type VII collagen restoration to human epidermolysis bullosa skin tissue. Hum. Gene Ther. 2010, 21, 1299–1310. [Google Scholar] [CrossRef]
- Conget, P.; Rodriguez, F.; Kramer, S.; Allers, C.; Simon, V.; Palisson, F.; Gonzalez, S.; Yubero, M.J. Replenishment of type VII collagen and re-epithelialization of chronically ulcerated skin after intradermal administration of allogeneic mesenchymal stromal cells in two patients with recessive dystrophic epidermolysis bullosa. Cytotherapy 2010, 12, 429–431. [Google Scholar] [CrossRef]
- Siprashvili, Z.; Nguyen, N.T.; Gorell, E.S.; Loutit, K.; Khuu, P.; Furukawa, L.K.; Lorenz, H.P.; Leung, T.H.; Keene, D.R.; Rieger, K.E.; et al. Safety and Wound Outcomes Following Genetically Corrected Autologous Epidermal Grafts in Patients With Recessive Dystrophic Epidermolysis Bullosa. JAMA 2016, 316, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Eichstadt, S.; Barriga, M.; Ponakala, A.; Teng, C.; Nguyen, N.T.; Siprashvili, Z.; Nazaroff, J.; Gorell, E.S.; Chiou, A.S.; Taylor, L.; et al. Phase 1/2a clinical trial of gene-corrected autologous cell therapy for recessive dystrophic epidermolysis bullosa. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Pendaries, V.; Gasc, G.; Titeux, M.; Leroux, C.; Vitezica, Z.G.; Mejía, J.E.; Décha, A.; Loiseau, P.; Bodemer, C.; Prost-Squarcioni, C.; et al. Immune reactivity to type VII collagen: Implications for gene therapy of recessive dystrophic epidermolysis bullosa. Gene Ther. 2010, 17, 930–937. [Google Scholar] [CrossRef]
- Woodley, D.T.; Cogan, J.; Wang, X.; Hou, Y.; Haghighian, C.; Kudo, G.; Keene, D.R.; Chen, M. De novo anti-type VII collagen antibodies in patients with recessive dystrophic epidermolysis bullosa. J. Investig. Dermatol. 2014, 134, 1138–1140. [Google Scholar] [CrossRef]
- Woodley, D.T.; Cogan, J.; Hou, Y.; Lyu, C.; Marinkovich, M.P.; Keene, D.; Chen, M. Gentamicin induces functional type VII collagen in recessive dystrophic epidermolysis bullosa patients. J. Clin. Investig. 2017, 127, 3028–3038. [Google Scholar] [CrossRef]


{kind=link}
| Antigen | EB Subtype | AIBD Subtype |
|---|---|---|
| BP230 (dystonin) | EBS | Bullous pemphigoid |
| Collagen XVII (BP180) | JEB | Bullous pemphigoid, Pemphigoid gestationis |
| Laminin 332 | JEB | Mucous membrane pemphigoid |
| α6β4 integrin | JEB | Mucous membrane pemphigoid |
| Collagen VII | DEB | Epidermolysis bullosa acquisita |
| EB Subtype | n | Autoantigen | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Collagen | FN | LAM | Dsg1 | Dsg3 | Collagen XVII/ BP180 | BP230 | |||||||||
| I | II | III | IV | V | VI | VII | |||||||||
| [14] | EBA | 2 | 0.0% | 0.0% | 100.0% | 0.0% | 100.0% | 0.0% | 0.0% | 50.0% | |||||
| EBS | 20 | 0.0% | 0.0% | 85.0% | 60.0% | 85.0% | 0.0% | 0.0% | 40.0% | ||||||
| JEB | 4 | 0.0% | 0.0% | 100.0% | 50.0% | 100.0% | 25.0% | 0.0% | 0.0% | ||||||
| DEB | 6 | 0.0% | 16.7% | 83.3% | 33.3% | 100.0% | 16.7% | 16.7% | 66.7% | ||||||
| Total | 32 | 0.0% | 3.1% | 87.5% | 50.0% | 90.6% | 6.3% | 3.1% | 40.6% | ||||||
| [15] | RDEB | 19 | 4.96 U/mL | 5.62 U/mL | 6.14 U/mL | 14.2 U/mL | 12.7 U/mL | ||||||||
| Other EB | 23 | 1.08 U/mL | 2.67 U/mL | 2.8 U/mL | 5.7 U/mL | 3.7 U/mL | |||||||||
| Healthy Controls | 38 | 0.26 U/mL | 2.12 U/mL | 1.58 U/mL | 1.82 U/mL | 1.68 U/mL | |||||||||
| [16] | RDEB | 17 | 88% | combined percentage of 88% | |||||||||||
| EBS | 10 | 10% | combined percentage of 50% | ||||||||||||
| Year | Author | EB Type | AIBD Type | Workup |
|---|---|---|---|---|
| 2016 | Hayashi | DDEB | EBA | DIF: Linear deposits of IgG and C3 at the DEJ IIF: Linear deposition of IgG at the dermal side of the DEJ Immunoblot analysis: Reactive to collagen type VII and its NC1 domain. Non-reactive to laminin 322 Mutations: c.7868G > A in the COL7A1 gene |
| 2018 | Guerra | RDEB | EBA | DIF: Linear deposition of IgG with a u-serrated pattern along the cutaneous BMZ IIF: IgG binding to the dermal side of the salt-split skin ELISA: Positive for anti-collagen type VII, anti-BP180, and anti-BP230 Immunoblot Analysis: Reactive to laminin 332 Mutations: c.410G > A and c.3674C > T in the COL7A1 gene |
| 2019 | Fania | JEB | BP | DIF: Linear IgG and C3 deposits in an n-serrated pattern at the DEJ IIF: Epidermal staining of the salt-split skin ELISA: Positive for anti-BP180. Negative for anti-BP230 Immunoblot analysis: Reactive to BP180 and its LAD-1 domain. Not reactive to laminin 332 Mutations: c.1132 + 5G > A in the LAMB3 gene |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, P.M.; Jones, V.A.; Behnam, C.T.; Di Zenzo, G.; Amber, K.T. A Review of Acquired Autoimmune Blistering Diseases in Inherited Epidermolysis Bullosa: Implications for the Future of Gene Therapy. Antibodies 2021, 10, 19. https://doi.org/10.3390/antib10020019
Patel PM, Jones VA, Behnam CT, Di Zenzo G, Amber KT. A Review of Acquired Autoimmune Blistering Diseases in Inherited Epidermolysis Bullosa: Implications for the Future of Gene Therapy. Antibodies. 2021; 10(2):19. https://doi.org/10.3390/antib10020019
Chicago/Turabian StylePatel, Payal M., Virginia A. Jones, Christy T. Behnam, Giovanni Di Zenzo, and Kyle T. Amber. 2021. "A Review of Acquired Autoimmune Blistering Diseases in Inherited Epidermolysis Bullosa: Implications for the Future of Gene Therapy" Antibodies 10, no. 2: 19. https://doi.org/10.3390/antib10020019
APA StylePatel, P. M., Jones, V. A., Behnam, C. T., Di Zenzo, G., & Amber, K. T. (2021). A Review of Acquired Autoimmune Blistering Diseases in Inherited Epidermolysis Bullosa: Implications for the Future of Gene Therapy. Antibodies, 10(2), 19. https://doi.org/10.3390/antib10020019