
Dynamics of Bacterial Community Abundance and Structure in Horizontal Subsurface Flow Wetland Mesocosms Treating Municipal Wastewater

 ,
, 


Abstract
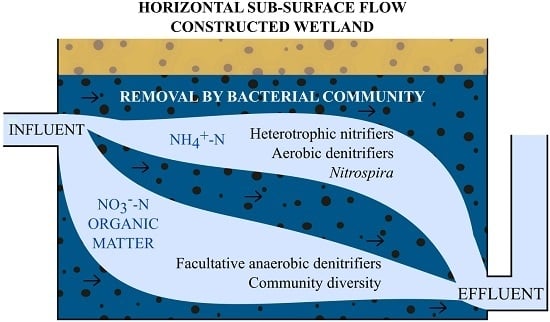
:
1. Introduction
2. Materials and Methods
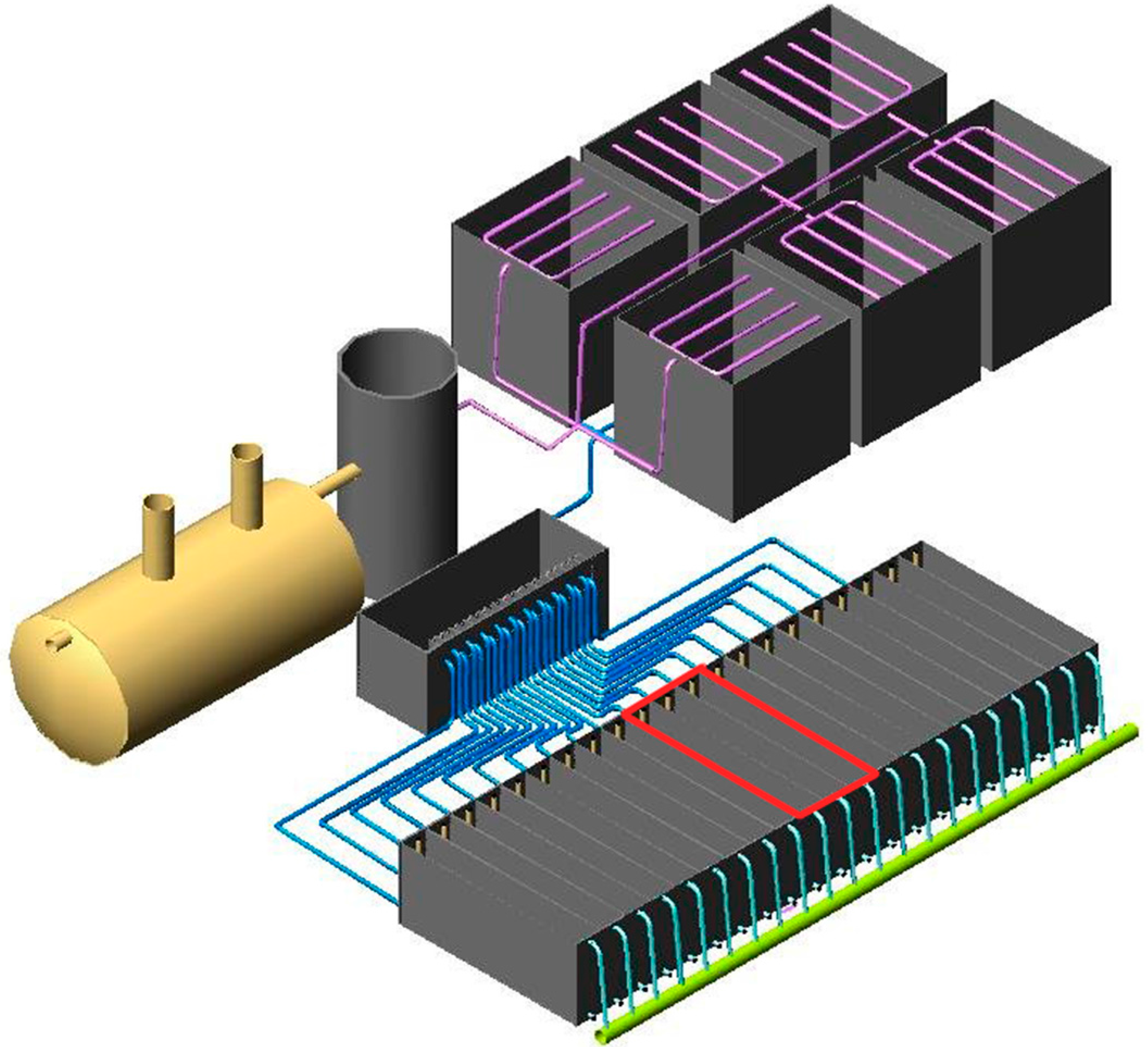
2.1. Study System Description
2.2. Sampling
2.3. DNA Extraction, PCR and Sequencing
2.4. Sequence Data Preparation and Taxonomic Assessment
2.5. Quantitative PCR
2.6. Statistical Analyses
3. Results
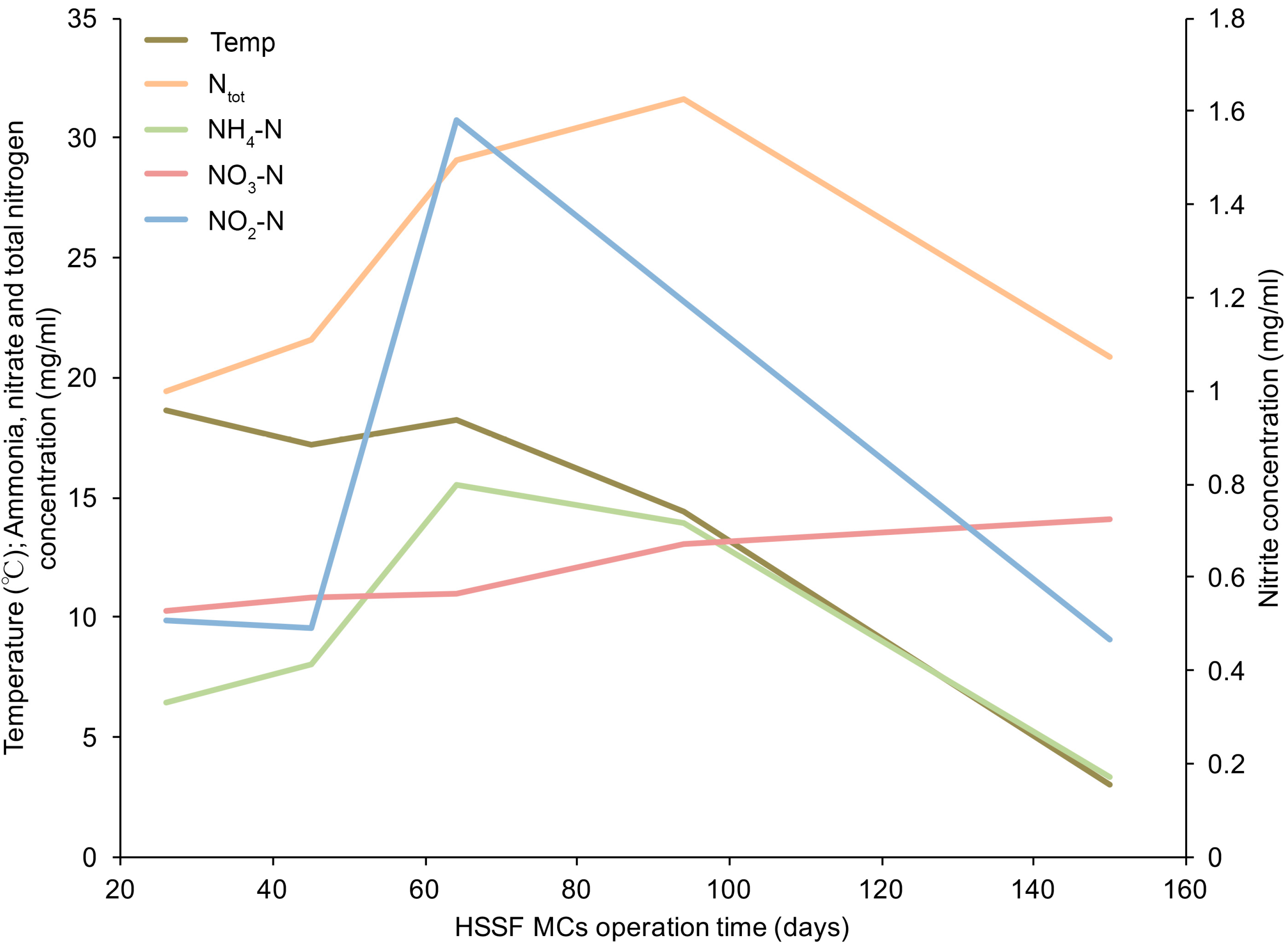
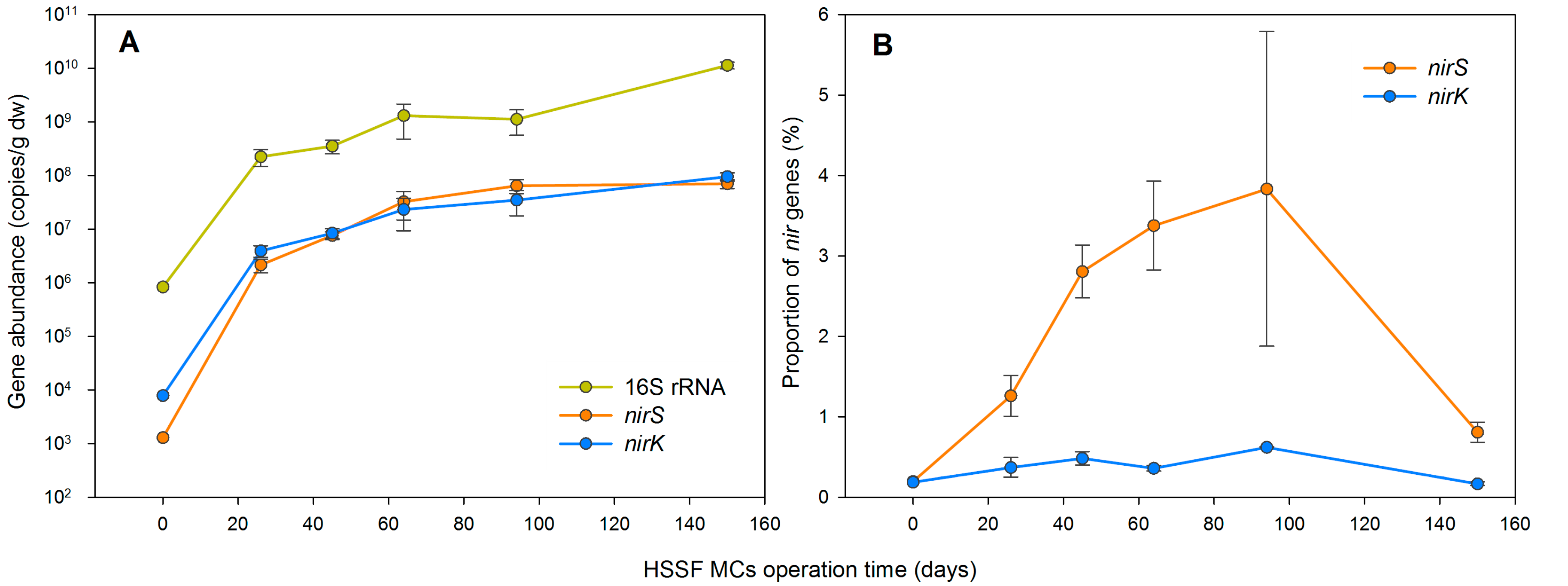
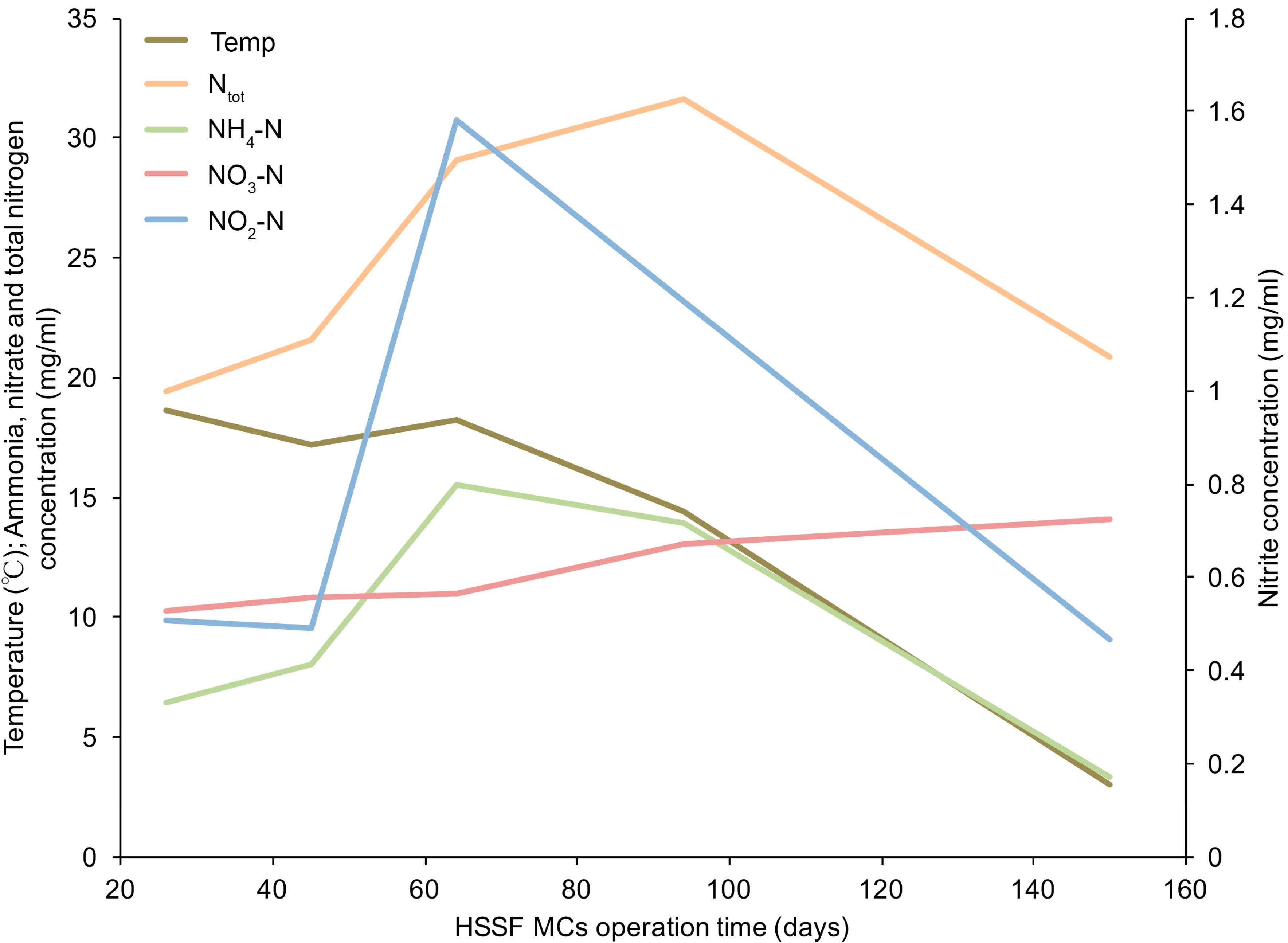
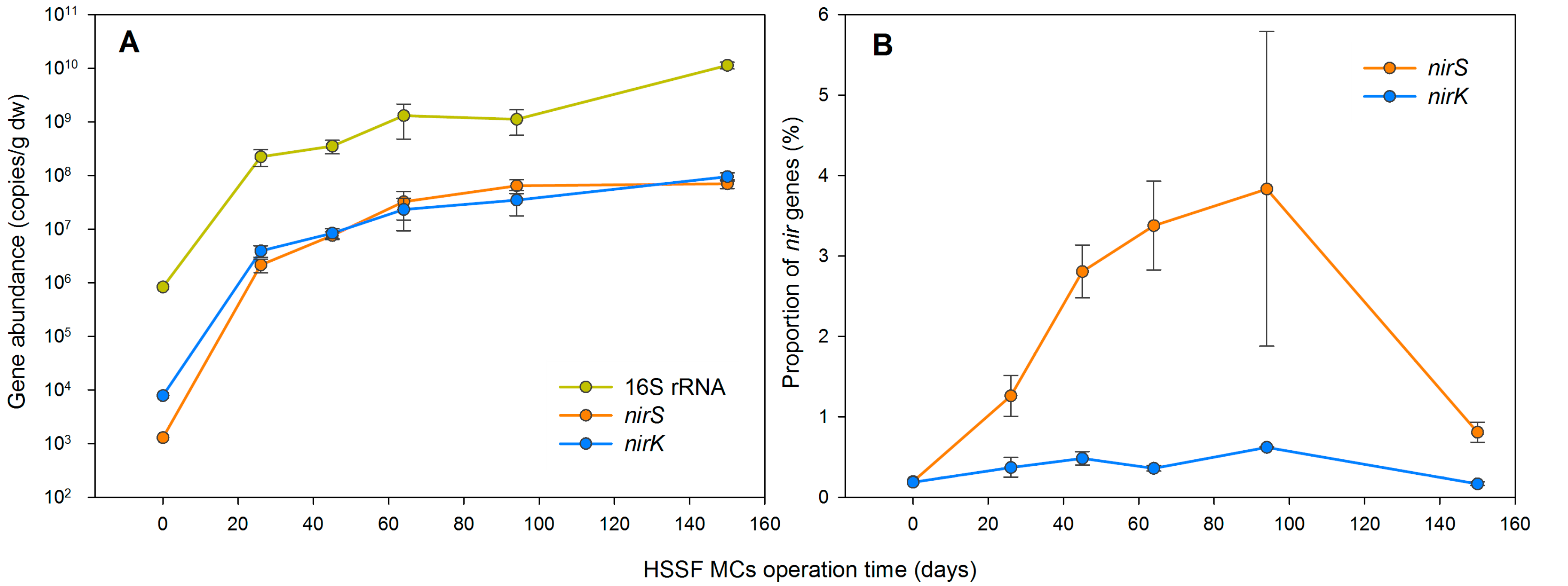
3.1. The Dynamics of Bacterial 16S rRNA Gene Abundance in HSSF MCs during the Study Period
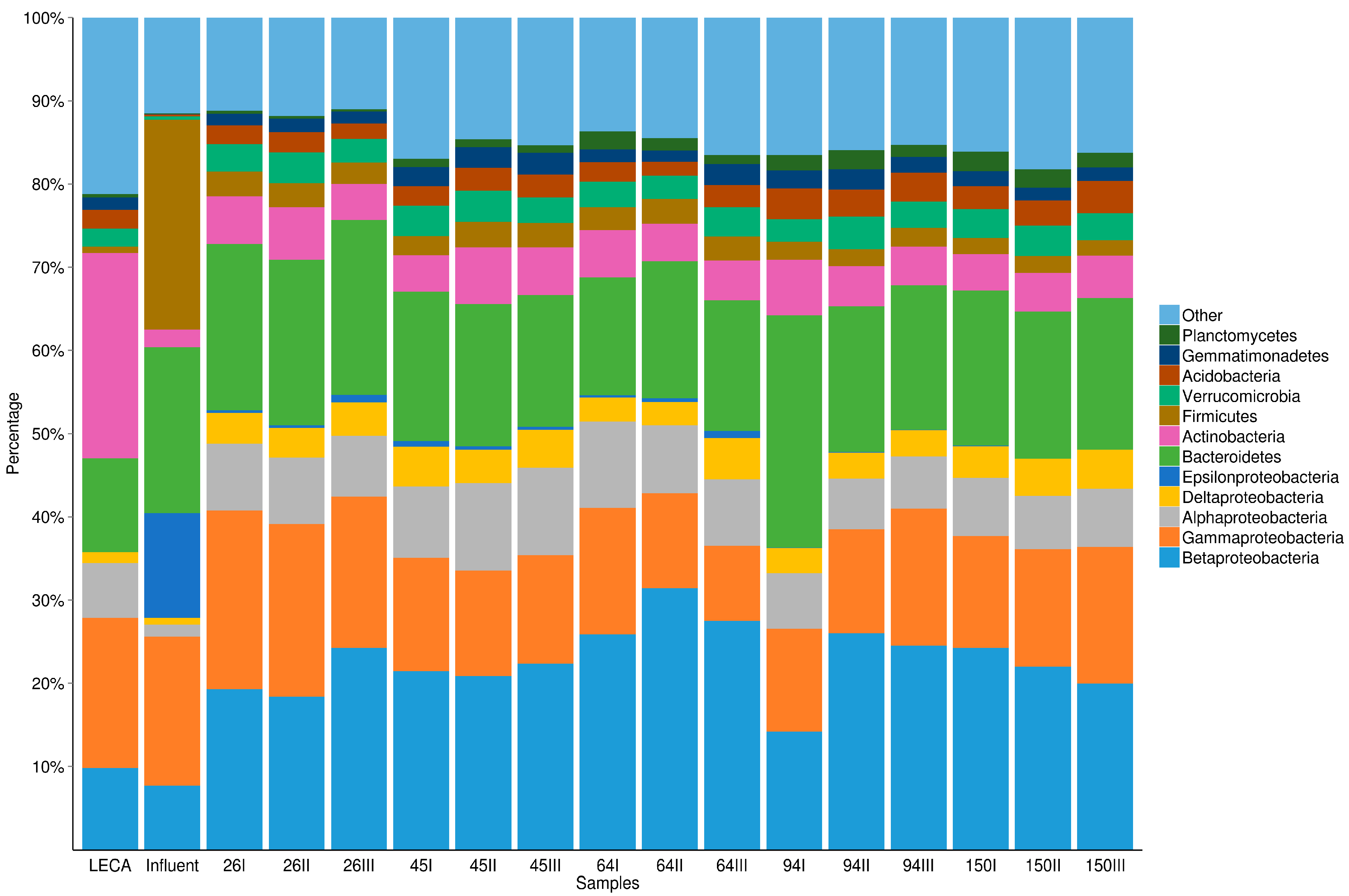
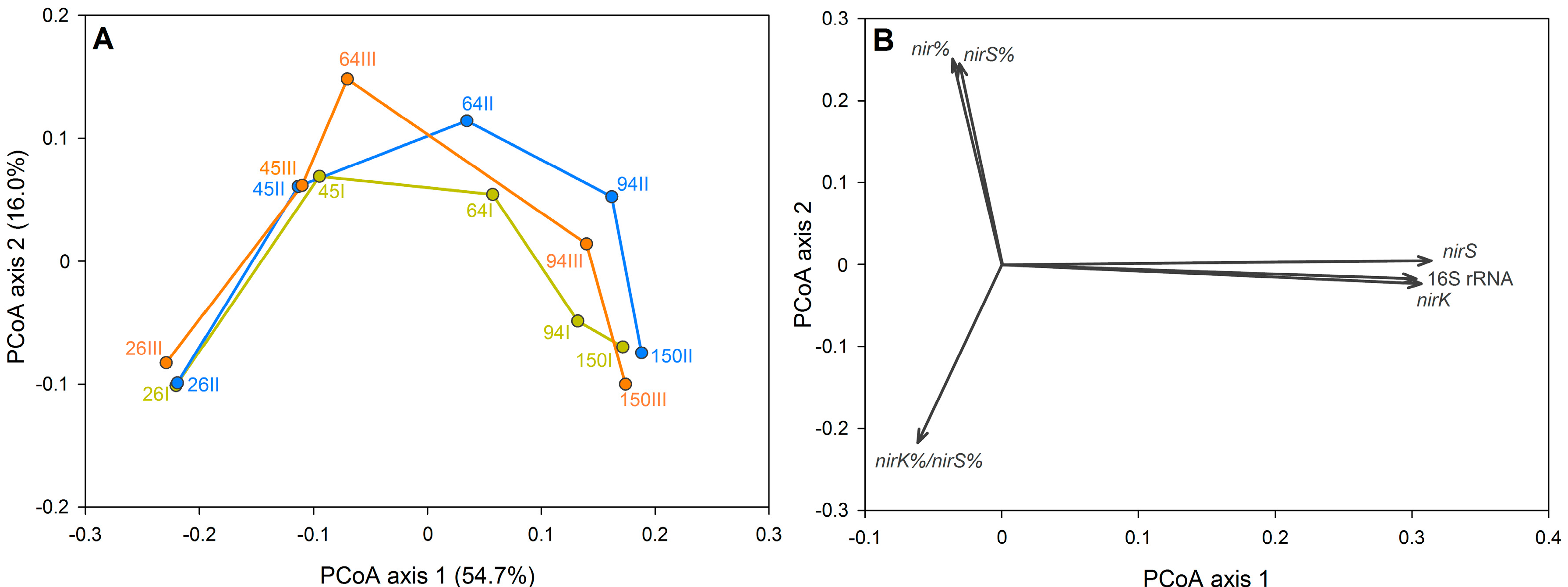
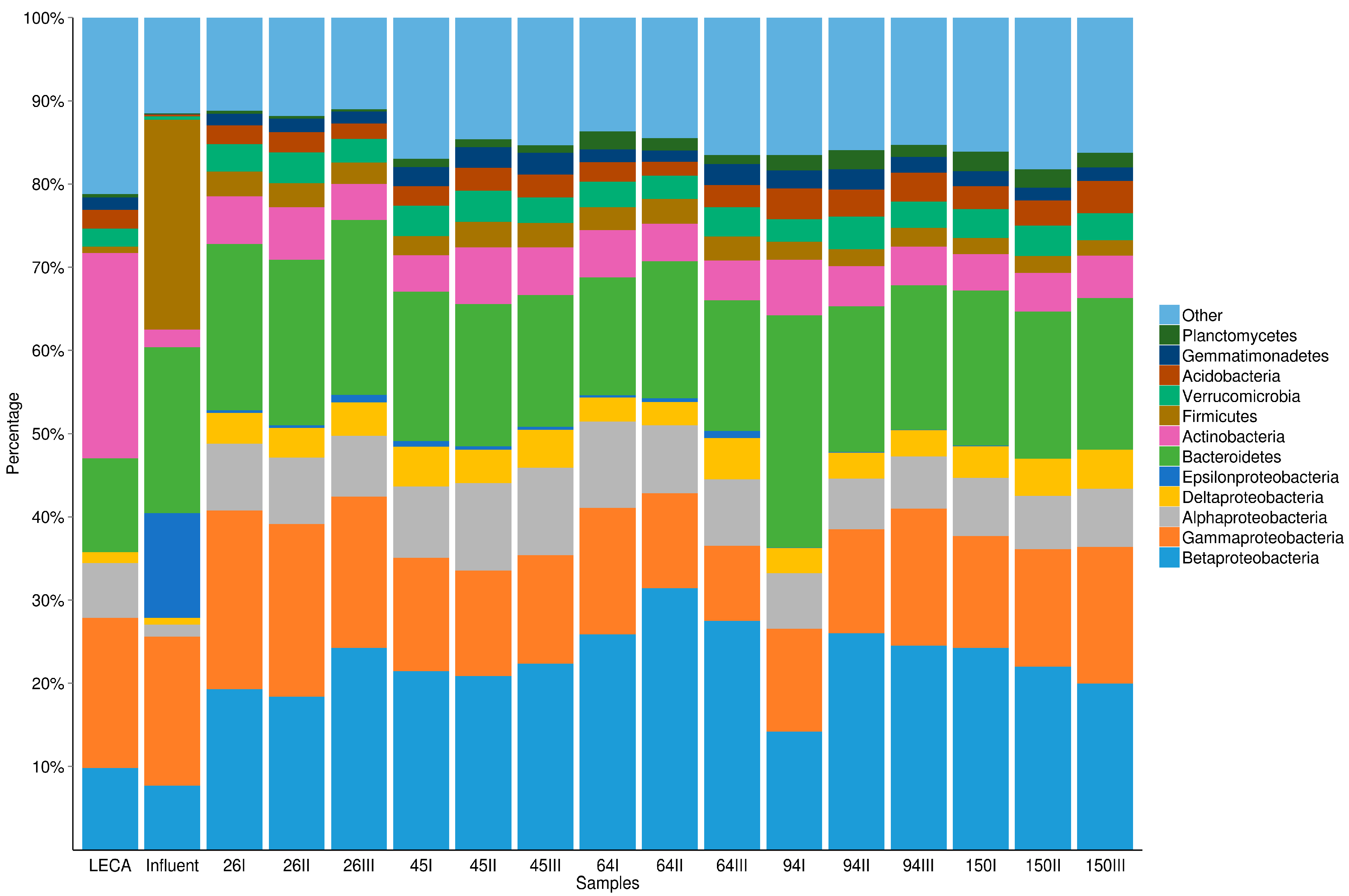
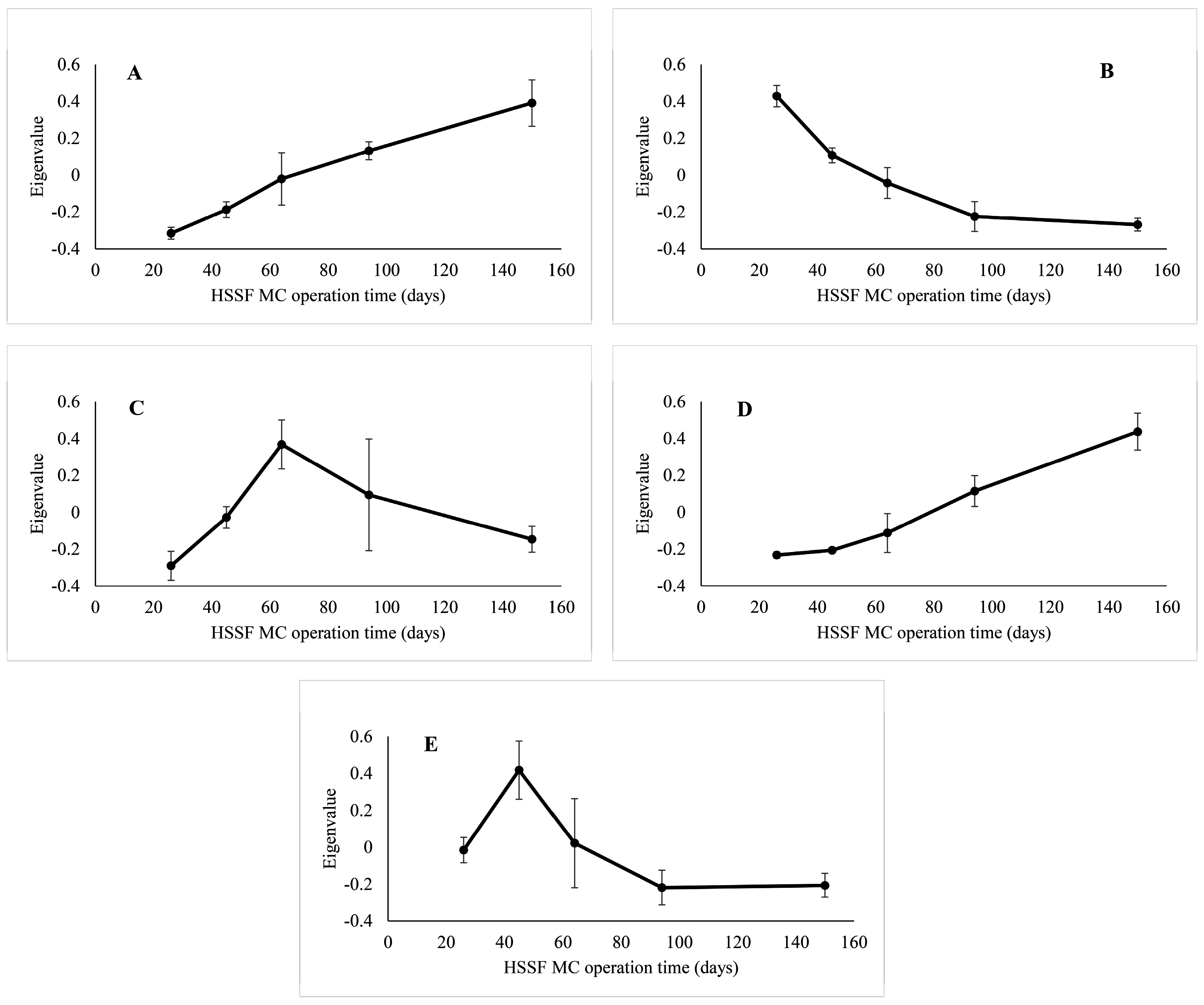
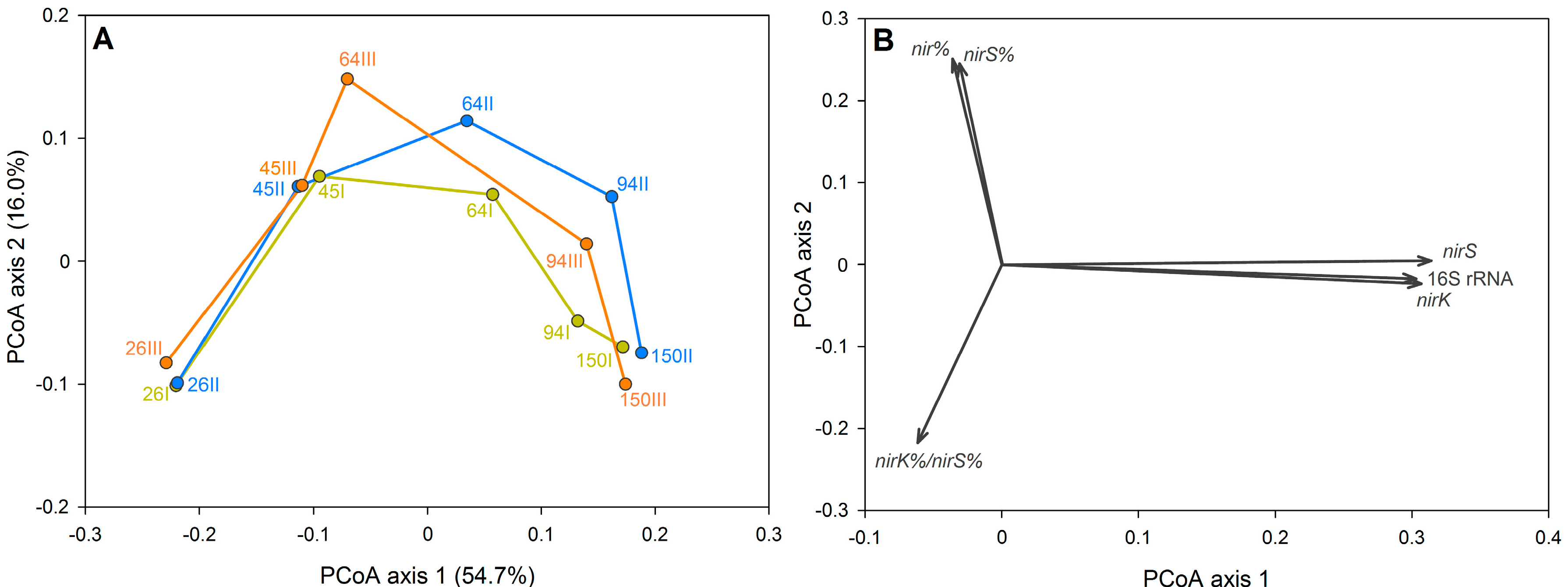
3.2. The Dynamics of Bacterial Community Structure in WMB during the Study Period
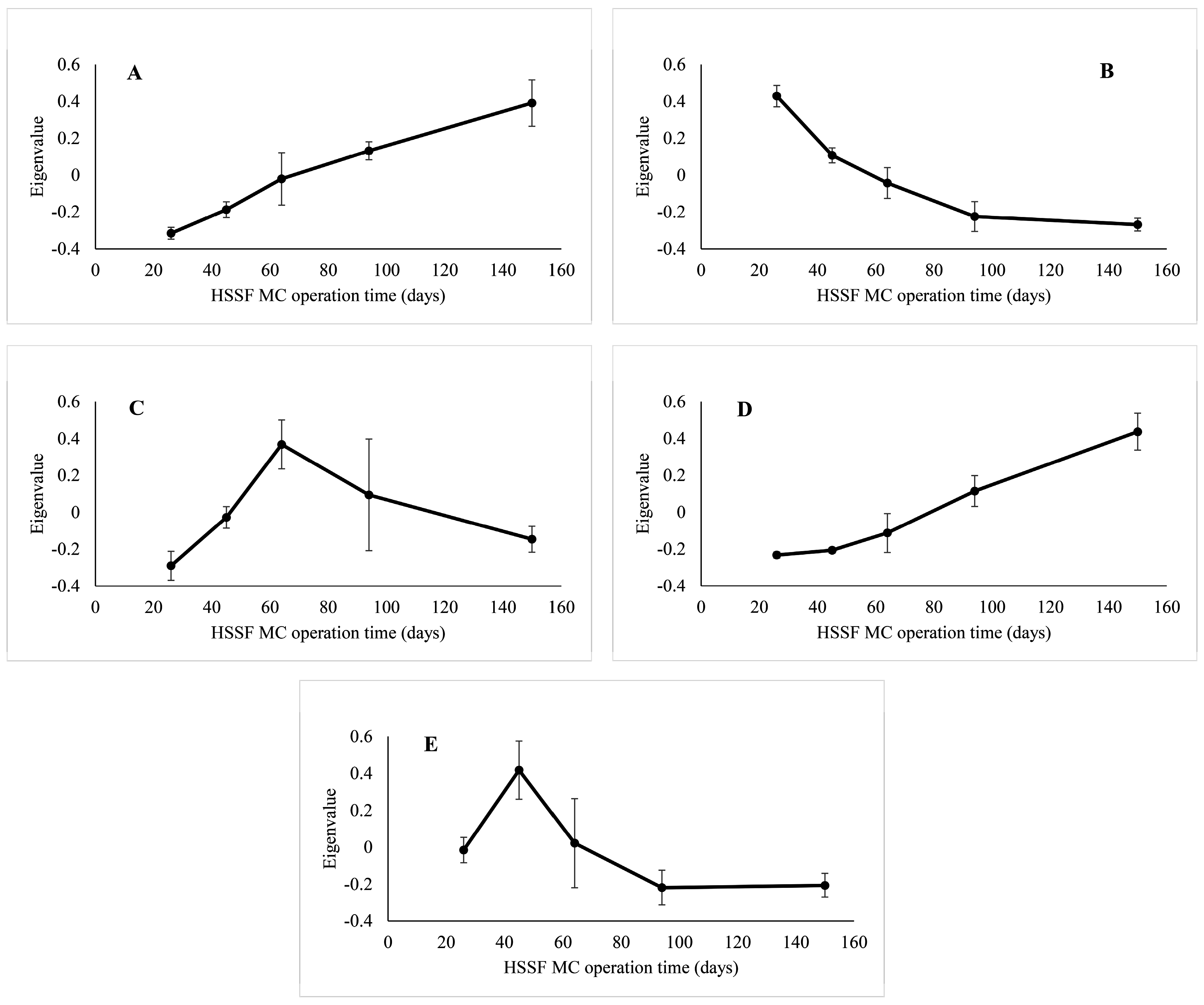
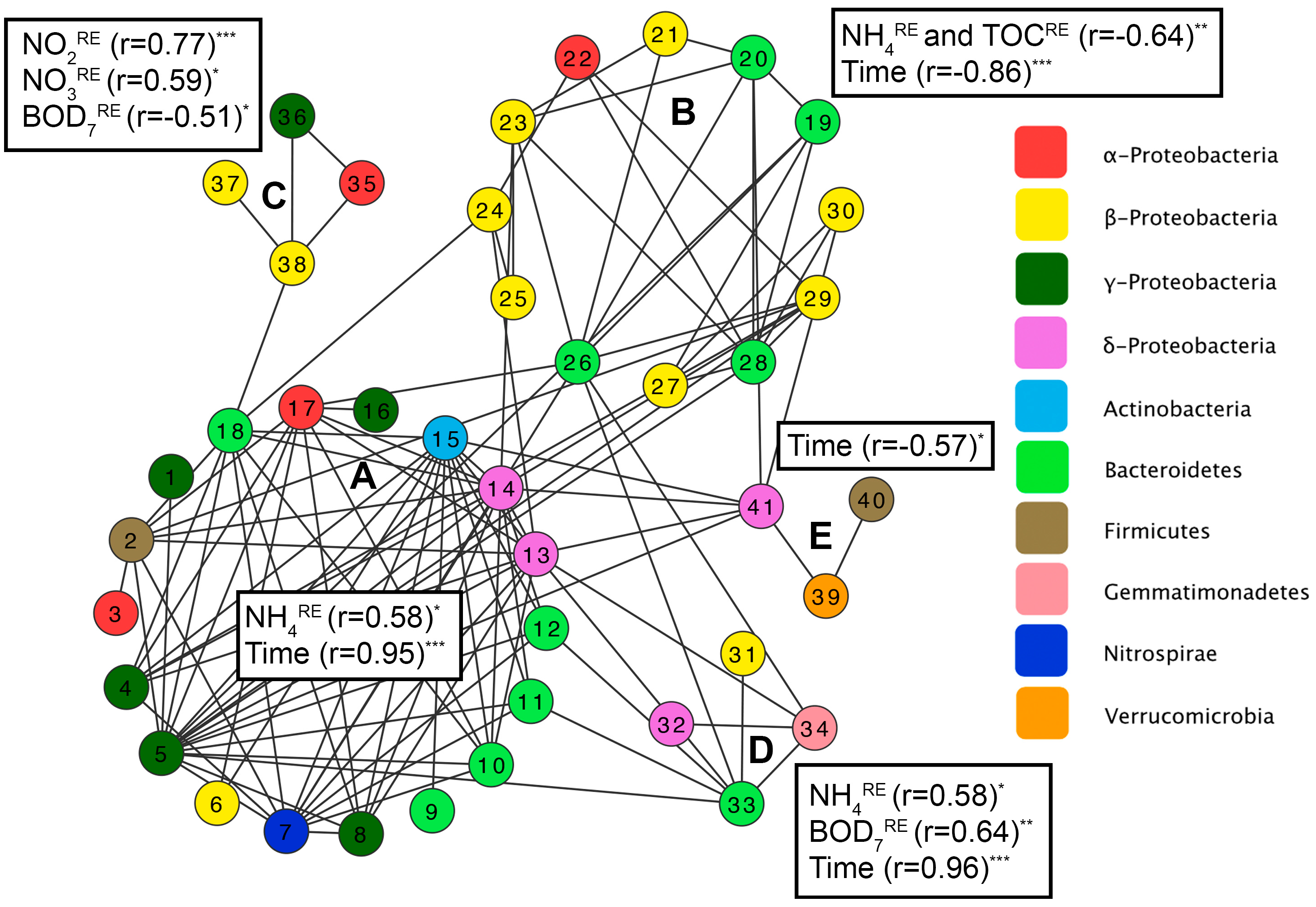
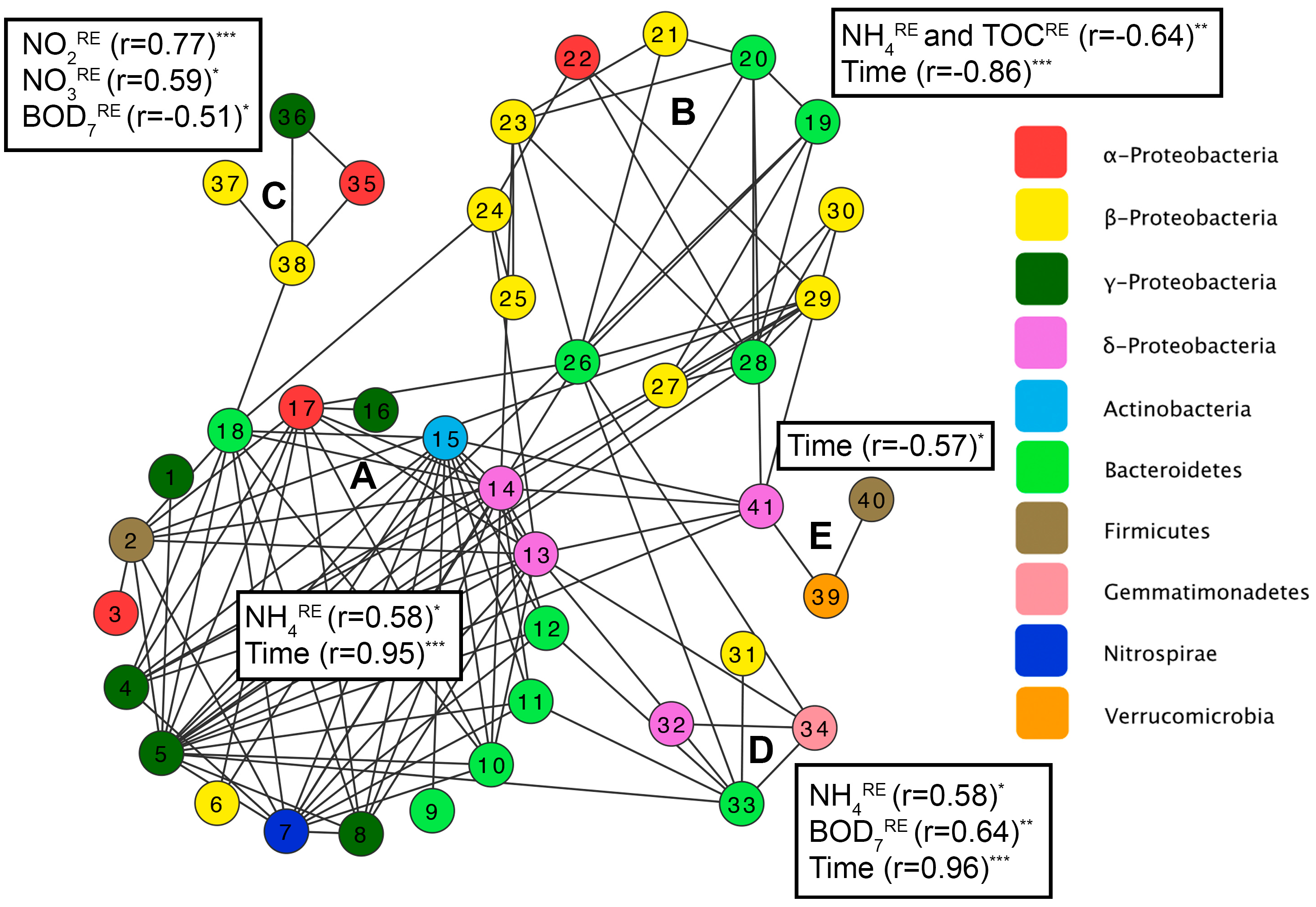
3.3. Factors Affecting Bacterial Community Composition in HSSF MCs
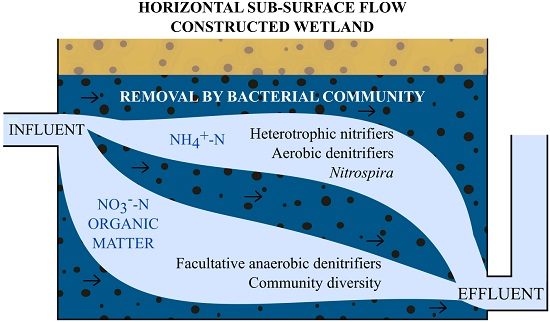
3.4. Denitrification Potential of the HSSF MCs WMB Bacterial Community
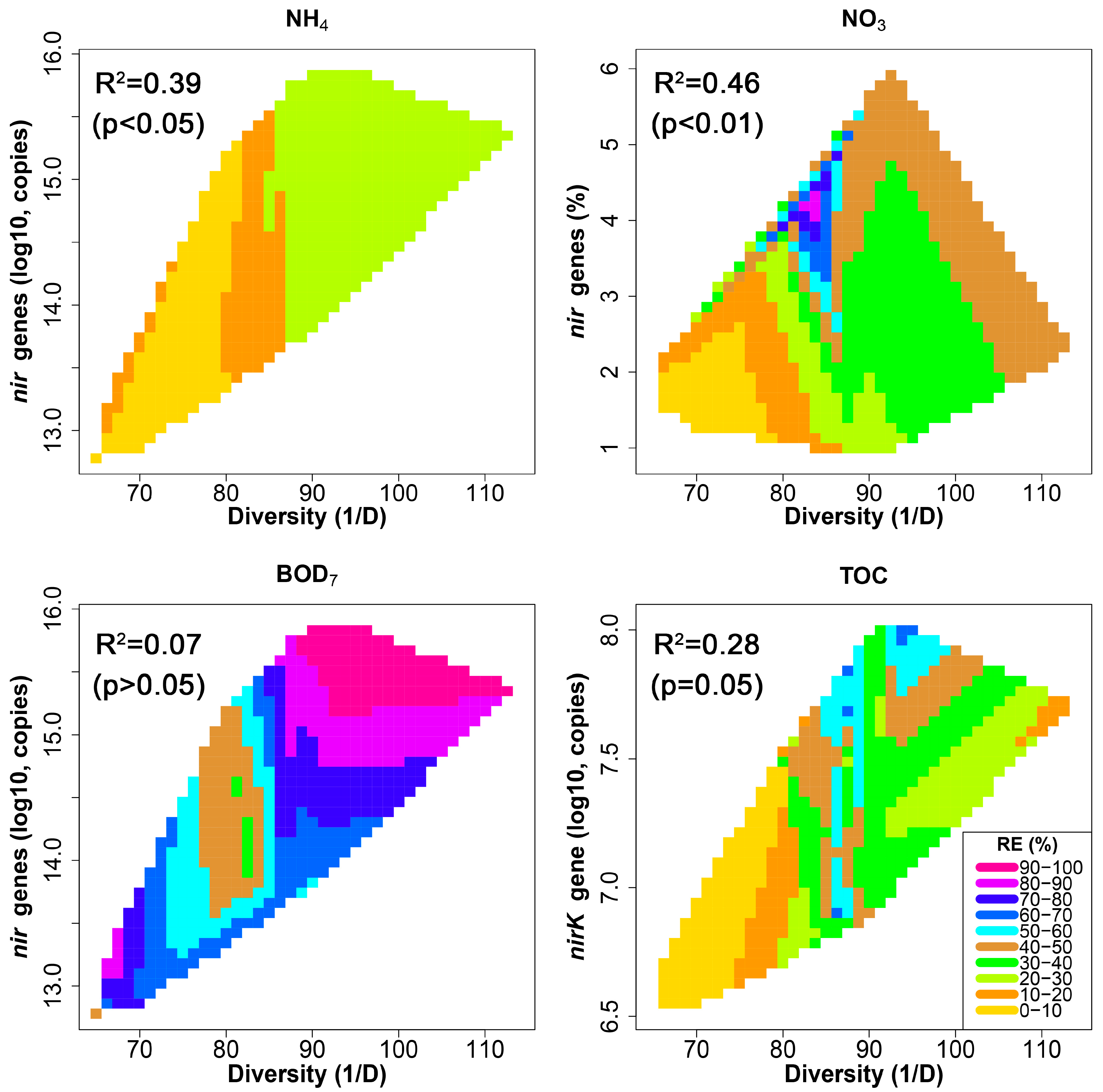
3.5. Relationship between HSSF MC Treatment Efficiency and Bacterial Community Structure
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A. Bacterial Community Structure and Dynamics in Horizontal Subsurface Flow Constructed Wetland Units Treating Municipal Wastewater
Appendix A.1. DNA Extraction, PCR and Sequencing
Appendix A.2. Quantitative PCR


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Target Gene | Amplicon Size (bp) | Primer Reference | PCR Programme for Standards | qPCR Programme | Standard Curve |
|---|---|---|---|---|---|---|
| L-V6 | 16S rRNA | 111 | [33] | 1 cycle: 95 °C 5 min; 30 cycles: 94 °C 30 s, 54 °C 30 s, 72 °C 45 s | 1 cycle: 95 °C 10 min; 40 cycles: 95 °C 15 s; 54 °C 30 s; 72 °C 30 s Melting curve: 65–90 °C | 108–102, 50, 25 |
| R-V6 | ||||||
| FlaCu | nirK | 473 | [81] | 1 cycle: 95 °C 5 min; 10 cycles: 95 °C 30 s, 60 °C 1 min a, 72 °C 1 min; 25 cycles: 95 °C 30 s, 58 °C 1 min, 72 °C 1 min; 1 cycle: 72 °C 5 min | 1 cycle: 95 °C 10 min; 6 cycles: 95 °C 15 s, 63 °C 30 s b, 72 °C 30 s, 80 °C 30 sc; 40 cycles: 95 °C 15 s, 61 °C 30 s, 72 °C 30 s, 80 °C 30 s c; 1 cycle: 95 °C 15 s, 60 °C 30 s, 72 °C 10 min Melting curve: 60–95 °C | 108–103 |
| R3Cu | ||||||
| nirSCd3aF | nirS | 425 | [82] | 1 cycle: 94 °C 2 min; 7 cycles: 94 °C 30 s, 63 °C 1 min a, 72 °C 1 min; 28 cycles: 94 °C 30 s, 57 °C 1 min, 72 °C 1 min; 1 cycle: 72 °C 10 min | 1 cycle: 95 °C 10 min; 6 cycles: 95 °C 15 s, 63 °C 30 s b, 72 °C 30 s, 80 °C 30 s c; 40 cycles: 95 °C 15 s, 58 °C 30 s, 72 °C 30 s, 80 °C 30 s c; 1 cycle: 95 °C 15 s, 60 °C 30 s, 72 °C 10 min Melting curve: 60–95 °C | 108–103 |
| nirSR3cd |
| Parameter | 16S rRNA | nirK | nirS | nir | nirK (%) | nirS (%) | nir (%) | nirK/nirS | 1/D | Richness | Evenness | NRI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Time | 0.91 *** | 0.93 *** | 0.93 *** | 0.96 *** | - | - | - | - | 0.77 *** | - | 0.70 ** | - |
| Temp | −0.76 *** | −0.87 *** | −0.82 *** | −0.87 *** | - | - | - | - | −0.89 *** | - | −0.84 *** | - |
| pH in | - | - | −0.55 * | - | - | - | - | - | - | - | - | - |
| pH eff | −0.77 *** | −0.73 ** | −0.85 *** | −0.79 *** | - | - | - | - | −0.61 * | - | −0.59 * | - |
| ΔpH | −0.68 ** | −0.67 ** | −0.63 * | −0.68 ** | - | - | - | - | −0.70 ** | - | −0.66 ** | - |
| N in | - | - | - | - | 0.61 * | 0.77 *** | 0.76 *** | −0.56 * | - | 0.61 * | - | - |
| N RE | - | - | - | - | - | - | - | - | - | 0.86 *** | 0.54 * | - |
| NO3 in | 0.91 *** | 0.93 *** | 0.93 *** | 0.96 *** | - | - | - | - | 0.77 *** | - | 0.70 ** | - |
| NO3 eff | - | - | - | - | −0.62 * | −0.72 ** | −0.74 ** | - | - | −0.81 *** | - | - |
| NO3 RE | - | - | - | - | - | 0.55 * | 0.57 * | - | 0.52 * | 0.83 *** | 0.58 * | - |
| NO2 in | - | - | - | - | - | 0.72 ** | 0.69 ** | - | - | - | - | - |
| NO2 eff | - | - | - | - | - | - | - | - | - | - | -- | - |
| NO2 RE | - | - | - | - | - | 0.71 ** | 0.69 ** | −0.62 * | 0.61 * | - | - | |
| NH4 in | - | - | - | - | 0.53 * | 0.86 *** | 0.84 *** | −0.65 ** | - | - | - | |
| NH4 eff | - | - | - | - | - | 0.78 *** | 0.75 ** | −0.57 * | - | - | - | |
| NH4 RE | 0.63 * | 0.70 ** | 0.64 * | 0.72 ** | - | - | - | - | 0.82 *** | - | 0.73 ** | −0.55 * |
| BOD7 in | - | - | - | - | 0.74 ** | - | - | - | - | - | - | |
| BOD7 eff | −0.59 * | −0.65 ** | −0.59 * | −0.67 ** | - | 0.51 * | - | - | −0.67 ** | - | −0.60 * | 0.63 * |
| BOD7 RE | - | 0.61 * | 0.53 * | 0.61 * | - | −0.53 * | −0.52 * | - | 0.65 ** | - | 0.58 * | −0.63 * |
| TOC in | - | - | - | - | 0.77 *** | 0.87 *** | 0.86 *** | - | - | - | - | |
| TOC RE | - | 0.57 * | - | 0.53 * | - | - | - | - | 0.58 * | - | 0.54 * | - |
| No. of Sequences | Phylum | Family |
|---|---|---|
| 16,642 | Bacteroidetes | Chitinophagaceae |
| 14,569 | Proteobacteria | Comamonadaceae |
| 8619 | Proteobacteria | Pseudomonadaceae |
| 7851 | Proteobacteria | Rhodocyclaceae |
| 7800 | Proteobacteria | Enterobacteriaceae |
| 7763 | Proteobacteria | Unclassified |
| 7000 | Bacteroidetes | Chitinophagaceae |
| 6751 | Proteobacteria | Burkholderiaceae |
| 6240 | Proteobacteria | Unclassified |
| 5846 | Proteobacteria | Xanthomonadaceae |
| 5836 | Proteobacteria | Rhodocyclaceae |
| 5399 | Proteobacteria | Burkholderiaceae |
| 5373 | Proteobacteria | Comamonadaceae |
| 5321 | Bacteroidetes | Flavobacteriaceae |
| 4911 | Proteobacteria | Pseudomonadaceae |
| 4731 | Proteobacteria | Comamonadaceae |
| 4663 | Bacteroidetes | Flavobacteriaceae |
| 4371 | Gemmatimonadetes | Gemmatimonadaceae |
| 4365 | Proteobacteria | Comamonadaceae |
| 3703 | Bacteroidetes | Chitinophagaceae |
| Taxon | LECA | Influent | WMB (n = 15) |
|---|---|---|---|
| Proteobacteria: | |||
| Alphaproteobacteria | 18% | 4% | 15% (±3%) |
| Betaproteobacteria | 27% | 19% | 44% (±7%) |
| Gammaproteobacteria | 49% | 43% | 29% (±6%) |
| Deltaproteobacteria | 4% | 2% | 8% (±2%) |
| Epsilonproteobacteria | <1% | 30% | 1% (±1%) |
| unclassified | 2% | 3% | 3% (±1%) |
| Bacteroidetes: | |||
| Sphingobacteria | 66% | 5% | 48% (±4%) |
| Flavobacteria | 23% | 20% | 31% (±4%) |
| Bacteroidia | 4% | 74% | 10% (±2%) |
| unclassified | 8% | 1% | 11% (±4%) |
| M | 16S rRNA | nirK | nirS | nirS (%) | nir (%) | nirK/nirS | NO2 | NO3 | Ntot | NH4 | TOC | pH |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 0.93 *** | 0.94 *** | 0.90 *** | - | - | - | - | 0.92 *** | - | - | - | - |
| B | −0.79 *** | −0.89 *** | −0.98 *** | - | - | - | - | −0.87 *** | −0.54 * | - | - | - |
| C | - | - | - | 0.82 *** | 0.79 *** | −0.8 *** | 0.77 *** | 0.73 *** | 0.68 *** | 0.64 ** | −0.55 * | |
| D | 0.93 *** | 0.91 *** | 0.79 *** | - | - | - | - | 0.95 *** | - | - | - | |
| E | −0.59 * | −0.63 ** | −0.55 * | - | - | - | - | −0.61 * | - | - | 0.63 ** |
References
- Vymazal, J. Horizontal sub-surface flow and hybrid constructed wetlands systems for wastewater treatment. Ecol. Eng. 2005, 25, 478–490. [Google Scholar] [CrossRef]
- Vymazal, J. Constructed Wetlands for Wastewater Treatment. Water 2010, 2, 530–549. [Google Scholar] [CrossRef]
- Mantovi, P.; Marmiroli, M.; Maestri, E.; Tagliavini, S.; Piccinini, S.; Marmiroli, N. Application of a horizontal subsurface flow constructed wetland on treatment of dairy parlor wastewater. Bioresour. Technol. 2003, 88, 85–94. [Google Scholar] [CrossRef]
- Matamoros, V.; Puigagut, J.; García, J.; Bayona, J.M. Behavior of selected priority organic pollutants in horizontal subsurface flow constructed wetlands: A preliminary screening. Chemosphere 2007, 69, 1374–1380. [Google Scholar] [CrossRef] [PubMed]
- Vymazal, J. Long-term performance of constructed wetlands with horizontal sub-surface flow: Ten case studies from the Czech Republic. Ecol. Eng. 2011, 37, 54–63. [Google Scholar] [CrossRef]
- Calheiros, C.S.C.; Rangel, A.O.S.S.; Castro, P.M.L. Treatment of industrial wastewater with two-stage constructed wetlands planted with Typha latifolia and Phragmites australis. Bioresour. Technol. 2009, 100, 3205–3213. [Google Scholar] [CrossRef] [PubMed]
- Ibekwe, A.M.; Grieve, C.M.; Lyon, S.R. Characterization of microbial communities and composition in constructed dairy wetland wastewater effluent. Appl. Environ. Microbiol. 2003, 69, 5060–5069. [Google Scholar] [CrossRef] [PubMed]
- Vymazal, J.; Kröpfelová, L. Removal of nitrogen in constructed wetlands with horizontal sub-surface flow: A review. Wetlands 2009, 29, 1114–1124. [Google Scholar] [CrossRef]
- Yalcuk, A.; Ugurlu, A. Comparison of horizontal and vertical constructed wetland systems for landfill leachate treatment. Bioresour. Technol. 2009, 100, 2521–2526. [Google Scholar] [CrossRef] [PubMed]
- Kadlec, R.H.; Wallace, S. Treatment Wetlands, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Truu, M.; Juhanson, J.; Truu, J. Microbial biomass, activity and community composition in constructed wetlands. Sci. Total Environ. 2009, 407, 3958–3971. [Google Scholar] [CrossRef] [PubMed]
- Faulwetter, J.L.; Gagnon, V.; Sundberg, C.; Chazarenc, F.; Burr, M.D.; Brisson, J.; Camper, A.K.; Stein, O.R. Microbial processes influencing performance of treatment wetlands: A review. Ecol. Eng. 2009, 35, 987–1004. [Google Scholar] [CrossRef]
- Van Elsas, J.D.; Boersma, F.G.H. A review of molecular methods to study the microbiota of soil and the mycosphere. Eur. J. Soil Biol. 2011, 47, 77–87. [Google Scholar] [CrossRef]
- Rastogi, G.; Sani, R.K. Molecular techniques to assess microbial community structure, function, and dynamics in the environment. In Microbes and Microbial Technology; Ahmad, I., Ahmad, F., Pichtel, J., Eds.; Springer: New York, NY, USA, 2011; pp. 29–57. [Google Scholar]
- Mackay, I.M. Real-time PCR in the microbiology laboratory. Clin. Microbiol. Infect. 2004, 10, 190–212. [Google Scholar] [CrossRef] [PubMed]
- Peralta, R.M.; Ahn, C.; Gillevet, P.M. Characterization of soil bacterial community structure and physicochemical properties in created and natural wetlands. Sci. Total Environ. 2013, 443, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Ansola, G.; Arroyo, P.; Sáenz de Miera, L.E. Characterisation of the soil bacterial community structure and composition of natural and constructed wetlands. Sci. Total Environ. 2014, 473–474, 63–71. [Google Scholar] [CrossRef] [PubMed]
- DeJournett, T.D.; Arnold, W.A.; LaPara, T.M. The characterization and quantification of methanotrophic bacterial populations in constructed wetland sediments using PCR targeting 16S rRNA gene fragments. Appl. Soil Ecol. 2007, 35, 648–659. [Google Scholar] [CrossRef]
- Song, M.; Shin, S.G.; Hwang, S. Methanogenic population dynamics assessed by real-time quantitative PCR in sludge granule in upflow anaerobic sludge blanket treating swine wastewater. Bioresour. Technol. 2010, 101, S23–S28. [Google Scholar] [CrossRef] [PubMed]
- Chon, K.; Chang, J.-S.; Lee, E.; Lee, J.; Ryu, J.; Cho, J. Abundance of denitrifying genes coding for nitrate (narG), nitrite (nirS), and nitrous oxide (nosZ) reductases in estuarine versus wastewater effluent-fed constructed wetlands. Ecol. Eng. 2011, 37, 64–69. [Google Scholar] [CrossRef]
- Wang, Q.; Xie, H.; Ngo, H.H.; Guo, W.; Zhang, J.; Liu, C.; Liang, S.; Hu, Z.; Yang, Z.; Zhao, C. Microbial abundance and community in subsurface flow constructed wetland microcosms: Role of plant presence. Environ. Sci. Pollut. Res. 2016, 23, 4036–4045. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.M.; Albertsen, M.; Vollertsen, J.; Nielsen, P.H. The activated sludge ecosystem contains a core community of abundant organisms. ISME J. 2016, 10, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Wang, X.; Wen, X.; Xia, Y. Microbial community structures in different wastewater treatment plants as revealed by 454-pyrosequencing analysis. Bioresour. Technol. 2012, 117, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, M.; Xia, Y.; Wen, X.; Ding, K. Pyrosequencing analysis of bacterial diversity in 14 wastewater treatment systems in china. Appl. Environ. Microbiol. 2012, 78, 7042–7047. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Shao, M.-F.; Ye, L. 454 Pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J. 2012, 6, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Vanier, S.; Dahab, M. Start-up performance of a subsurface-flow constructed wetland for domestic wastewater treatment. Environ. Technol. 2001, 22, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Lee, S.-H.; Kang, H. Denitrification rates and community structure of denitrifying bacteria in newly constructed wetland. Eur. J. Soil Biol. 2011, 47, 24–29. [Google Scholar] [CrossRef]
- Ramond, J.-B.; Welz, P.J.; Cowan, D.A.; Burton, S.G. Microbial community structure stability, a key parameter in monitoring the development of constructed wetland mesocosms during start-up. Res. Microbiol. 2012, 163, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.P.; Legge, R.L. Dynamics in the bacterial community-level physiological profiles and hydrological characteristics of constructed wetland mesocosms during start-up. Ecol. Eng. 2011, 37, 666–677. [Google Scholar] [CrossRef]
- Nurk, K.; Zaytsev, I.; Talpsep, I.; Truu, J.; Mander, Ü. Bioaugmentation in a newly established LECA-based horizontal flow soil filter reduces the adaptation period and enhances denitrification. Bioresour. Technol. 2009, 100, 6284–6289. [Google Scholar] [CrossRef] [PubMed]
- Zaytsev, I.; Mander, Ü.; Lõhmus, K.; Nurk, K. Enhanced denitrification in a bioaugmented horizontal subsurface flow filter. Ecol. Eng. 2011, 37, 1050–1057. [Google Scholar] [CrossRef]
- American Public Health Organisation (APHA). Standard Methods for the Examination of Water and Wastewater, 17th ed.; American Public Health Organisation: Washington, DC, USA, 1998. [Google Scholar]
- Hummelen, R.; Fernandes, A.D.; Macklaim, J.M.; Dickson, R.J.; Changalucha, J.; Gloor, G.B.; Reid, G. Deep sequencing of the vaginal microbiota of women with HIV. PLoS ONE 2010, 5, e12078. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, S.; Materna, A.C.; Timberlake, S.C.; Blackburn, M.C.; Malmstrom, R.R.; Alm, E.J.; Chisholm, S.W. Unlocking short read sequencing for metagenomics. PLoS ONE 2010, 5, e11840. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- Werner, J.J.; Koren, O.; Hugenholtz, P.; DeSantis, T.Z.; Walters, W.A.; Caporaso, J.G.; Angenent, L.T.; Knight, R.; Ley, R.E. Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J. 2012, 6, 94–103. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Howes, C.G.; Abarenkov, K.; Mohn, W.W.; Nilsson, R.H. V-Xtractor: An open-source, high-throughput software tool to identify and extract hypervariable regions of small subunit (16S/18S) ribosomal RNA gene sequences. J. Microbiol. Methods 2010, 83, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Jiang, R.; Chen, T. Clustering 16S rRNA for OTU prediction: A method of unsupervised Bayesian clustering. Bioinformatics 2011, 27, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Gobet, A.; Quince, C.; Ramette, A. Multivariate Cutoff Level Analysis (MultiCoLA) of large community data sets. Nucleic Acids Res. 2010, 38, e155. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Nõlvak, H.; Truu, M.; Truu, J. Evaluation of quantitative real-time PCR workflow modifications on 16S rRNA and tetA gene quantification in environmental samples. Sci. Total Environ. 2012, 426, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Ruijter, J.M.; Ramakers, C.; Hoogaars, W.M.H.; Karlen, Y.; Bakker, O.; van den Hoff, M.J.B.; Moorman, A.F.M. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009, 37, e45. [Google Scholar] [CrossRef] [PubMed]
- McArdle, B.H.; Anderson, M.J. Fitting multivariate models to community data: A comment on distance-based redundancy analysis. Ecology 2001, 82, 290–297. [Google Scholar] [CrossRef]
- Deng, Y.; Jiang, Y.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Kembel, S.W.; Cowan, P.D.; Helmus, M.R.; Cornwell, W.K.; Morlon, H.; Ackerly, D.D.; Blomberg, S.P.; Webb, C.O. Picante: R tools for integrating phylogenies and ecology. Bioinformatics 2010, 26, 1463–1464. [Google Scholar] [CrossRef] [PubMed]
- Gloor, G.B.; Hummelen, R.; Macklaim, J.M.; Dickson, R.J.; Fernandes, A.D.; MacPhee, R.; Reid, G. Microbiome profiling by Illumina sequencing of combinatorial sequence-tagged PCR products. PLoS ONE 2010, 5, e15406. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Huse, S.M.; Dethlefsen, L.; Huber, J.A.; Mark Welch, D.; Welch, D.M.; Relman, D.A.; Sogin, M.L. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 2008, 4, e1000255. [Google Scholar] [CrossRef]
- Liu, Z.; Lozupone, C.; Hamady, M.; Bushman, F.D.; Knight, R. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res. 2007, 35, e120. [Google Scholar] [CrossRef] [PubMed]
- McLellan, S.L.; Huse, S.M.; Mueller-Spitz, S.R.; Andreishcheva, E.N.; Sogin, M.L. Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent. Environ. Microbiol. 2010, 12, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, P.; Ansola, G.; Blanco, I.; Molleda, P.; de Luis Calabuig, E.; Sáenz de Miera, L.E. Comparative analysis of the composition of bacterial communities from two constructed wetlands for municipal and swine wastewater treatment. J. Water Health 2010, 8, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Iasur-Kruh, L.; Hadar, Y.; Milstein, D.; Gasith, A.; Minz, D. Microbial population and activity in wetland microcosms constructed for improving treated municipal wastewater. Microb. Ecol. 2010, 59, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Duan, L.; Song, Y.; Li, J.; Piceno, Y.M.; Andersen, G.L.; Alvarez-Cohen, L.; Moreno-Andrade, I.; Huang, C.-L.; Hermanowicz, S.W. Bacterial community structure in geographically distributed biological wastewater treatment reactors. Environ. Sci. Technol. 2010, 44, 7391–7396. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sheng, H.-F.; He, Y.; Wu, J.-Y.; Jiang, Y.-X.; Tam, N.F.-Y.; Zhou, H.-W. Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of Illumina tags. Appl. Environ. Microbiol. 2012, 78, 8264–8271. [Google Scholar] [CrossRef] [PubMed]
- Adrados, B.; Sánchez, O.; Arias, C.A.; Becares, E.; Garrido, L.; Mas, J.; Brix, H.; Morató, J. Microbial communities from different types of natural wastewater treatment systems: Vertical and horizontal flow constructed wetlands and biofilters. Water Res. 2014, 55, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Calheiros, C.S.C.; Duque, A.F.; Moura, A.; Henriques, I.S.; Correia, A.; Rangel, A.O.S.S.; Castro, P.M.L. Substrate effect on bacterial communities from constructed wetlands planted with Typha latifolia treating industrial wastewater. Ecol. Eng. 2009, 35, 744–753. [Google Scholar] [CrossRef]
- Koch, H.; Lücker, S.; Albertsen, M.; Kitzinger, K.; Herbold, C.; Spieck, E.; Nielsen, P.H.; Wagner, M.; Daims, H. Expanded metabolic versatility of ubiquitous nitrite-oxidizing bacteria from the genus Nitrospira. Proc. Natl. Acad. Sci. USA 2015, 112, 11371–11376. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, C.M.; Camejo, P.; Oshlag, J.Z.; Noguera, D.R. Ammonia-oxidizing microbial communities in reactors with efficient nitrification at low-dissolved oxygen. Water Res. 2015, 70, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Li, W. Study on characteristics in the removal process of ammonia nitrogen and nitrate nitrogen by an isolated heterotrophic nitrification-aerobic denitrification strain Rhodococcus sp. J. Environ. Prot. 2013, 4, 74–79. [Google Scholar] [CrossRef]
- Sun, Y.; Li, A.; Zhang, X.; Ma, F. Regulation of dissolved oxygen from accumulated nitrite during the heterotrophic nitrification and aerobic denitrification of Pseudomonas stutzeri T13. Appl. Microbiol. Biotechnol. 2015, 99, 3243–3248. [Google Scholar] [CrossRef] [PubMed]
- Ibekwe, A.M.; Ma, J.; Murinda, S.; Reddy, G.B. Bacterial community dynamics in surface flow constructed wetlands for the treatment of swine waste. Sci. Total Environ. 2016, 544, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Despland, L.M.; Clark, M.W.; Vancov, T.; Aragno, M. Nutrient removal and microbial communities’ development in a young unplanted constructed wetland using BauxsolTM pellets to treat wastewater. Sci. Total Environ. 2014, 484, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Bouali, M.; Zrafi, I.; Bakhrouf, A.; Chaussonnerie, S.; Sghir, A. Bacterial structure and spatiotemporal distribution in a horizontal subsurface flow constructed wetland. Appl. Microbiol. Biotechnol. 2014, 98, 3191–3203. [Google Scholar] [CrossRef] [PubMed]
- Zumft, W.G. Cell biology and molecular basis of denitrification. Microbiol. Mol. Biol. Rev. 1997, 61, 533–616. [Google Scholar] [PubMed]
- Jones, C.M.; Stres, B.; Rosenquist, M.; Hallin, S. Phylogenetic analysis of nitrite, nitric oxide, and nitrous oxide respiratory enzymes reveal a complex evolutionary history for denitrification. Mol. Biol. Evol. 2008, 25, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Correa-Galeote, D.; Marco, D.E.; Tortosa, G.; Bru, D.; Philippot, L.; Bedmar, E.J. Spatial distribution of N-cycling microbial communities showed complex patterns in constructed wetland sediments. FEMS Microbiol. Ecol. 2013, 83, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.M.; Hallin, S. Ecological and evolutionary factors underlying global and local assembly of denitrifier communities. ISME J. 2010, 4, 633–641. [Google Scholar] [CrossRef] [PubMed]
- García-Lledó, A.; Vilar-Sanz, A.; Trias, R.; Hallin, S.; Bañeras, L. Genetic potential for N2O emissions from the sediment of a free water surface constructed wetland. Water Res. 2011, 45, 5621–5632. [Google Scholar] [CrossRef] [PubMed]
- Coban, O.; Kuschk, P.; Kappelmeyer, U.; Spott, O.; Martienssen, M.; Jetten, M.S.M.; Knoeller, K. Nitrogen transforming community in a horizontal subsurface-flow constructed wetland. Water Res. 2015, 74, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Seshan, H.; Goyal, M.K.; Falk, M.W.; Wuertz, S. Support vector regression model of wastewater bioreactor performance using microbial community diversity indices: Effect of stress and bioaugmentation. Water Res. 2014, 53, 282–296. [Google Scholar] [PubMed]
- Paranychianakis, N.V.; Tsiknia, M.; Kalogerakis, N. Pathways regulating the removal of nitrogen in planted and unplanted subsurface flow constructed wetlands. Water Res. 2016, 102, 321–329. [Google Scholar] [PubMed]
- Chen, J.; Strous, M. Denitrification and aerobic respiration, hybrid electron transport chains and co-evolution. Biochim. Biophys. Acta 2013, 1827, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.; Yu, T.; Ning, K.; Guo, Z.; Wong, M.H. Effects of nitrogen removal microbes and partial nitrification-denitrification in the integrated vertical-flow constructed wetland. Ecol. Eng. 2016, 95, 83–89. [Google Scholar] [CrossRef]
- Shade, A. Diversity is the question, not the answer. ISME J. 2016, 4, e2287v1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Carvalho, P.N.; Lv, T.; Arias, C.; Brix, H.; Chen, Z. Microbial density and diversity in constructed wetland systems and the relation to pollutant removal efficiency. Water Sci. Technol. 2016, 73, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Truu, J.; Truu, M.; Espenberg, M.; Nõlvak, H.; Juhanson, J. Phytoremediation and plant-assisted bioremediation in soil and treatment wetlands: A review. Open Biotechnol. J. 2015, 9, 85–92. [Google Scholar] [CrossRef]
- Parameswaran, P.; Jalili, R.; Tao, L.; Shokralla, S.; Gharizadeh, B.; Ronaghi, M.; Fire, A.Z. A pyrosequencing-tailored nucleotide barcode design unveils opportunities for large-scale sample multiplexing. Nucleic Acids Res. 2007, 35, e130. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N. BARCRAWL and BARTAB: Software tools for the design and implementation of barcoded primers for highly multiplexed DNA sequencing. BMC Bioinform. 2009, 10, 362. [Google Scholar] [CrossRef] [PubMed]
- Hallin, S.; Lindgren, P.-E. PCR detection of genes encoding nitrite reductase in denitrifying bacteria. Appl. Environ. Microbiol. 1999, 65, 1652–1657. [Google Scholar] [PubMed]
- Kandeler, E.; Deiglmayr, K.; Tscherko, D.; Bru, D.; Philippot, L. Abundance of narG, nirS, nirK, and nosZ genes of denitrifying bacteria during primary successions of a glacier foreland. Appl. Environ. Microbiol. 2006, 72, 5957–5962. [Google Scholar] [CrossRef] [PubMed]
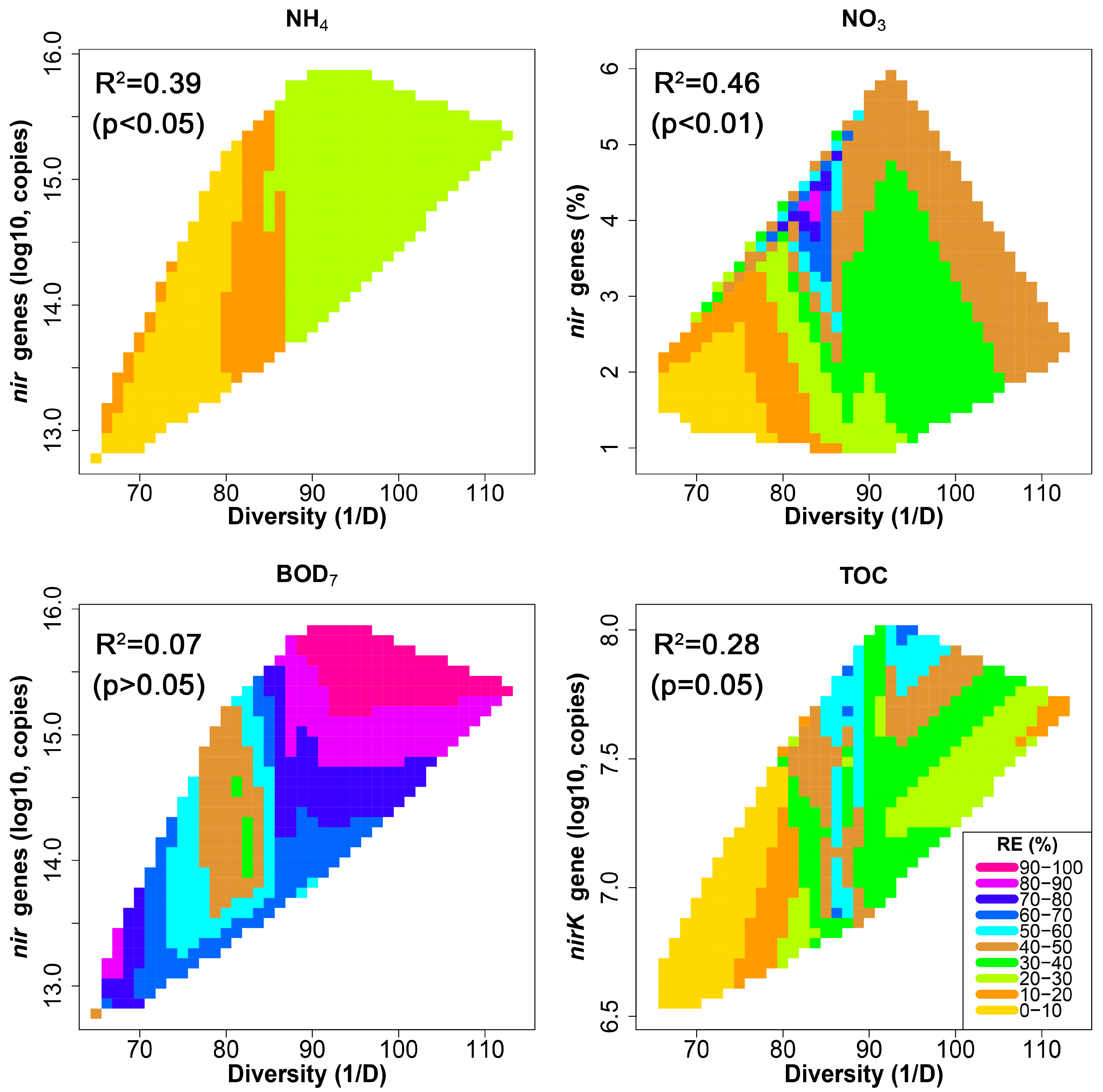
| Parameter | Ntotal | NO3–N | NO2–N | NH4–N | BOD7 | TOC | pH |
|---|---|---|---|---|---|---|---|
| Influent | 25 | 12 | 0.85 | 9.4 | 24 | 19 | 7.6 |
| n = 5 | (±5) | (±1.6) | (±0.51) | (±5.1) | (±4.9) | (±6.1) | (±0.3) |
| Effluent | 18 | 8 | 0.39 | 8.0 | 6.5 | 14 | 8.3 |
| n = 5 | (±4) | (±3) | (±0.16) | (±4.2) | (±4.4) | (±7.0) | (±0.3) |
| RE (%) | 24 | 33 | 41 | 16 | 71 | 26 | 0.7 a |
| n = 15 | (±17) | (±24) | (±34) | (±12) | (±21) | (±32) | (±0.2) |
| Samples (Days) | 1/D | Richness | Evenness | NRI | NTI |
|---|---|---|---|---|---|
| LECA | 38.1 | 662 | 0.72 | −2.84 | −10.37 |
| Influent | 29.4 | 630 | 0.69 | −0.96 | −10.07 |
| WMB (26) | 66.4 (±2.1) | 751 (±9) | 0.76 (±0.01) | −3.64 (±0.32) | −9.69 (±0.68) |
| WMB (45) | 87.1 (±1.9) | 808 (±12) | 0.79 (±0.00) | −2.67 (±0.72) | −8.98 (±0.36) |
| WMB (64) | 81.0 (±2.0) | 795 (±18) | 0.78 (±0.00) | −2.71 (±0.95) | −8.68 (±1.02) |
| WMB (94) | 98.0 (±13.9) | 803 (±28) | 0.80 (±0.01) | −3.92 (±0.77) | −9.52 (±0.91) |
| WMB (150) | 90.6 (±2.6) | 783 (±6) | 0.79 (±0.01) | −3.97 (±0.37) | −8.38 (±1.02) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oopkaup, K.; Truu, M.; Nõlvak, H.; Ligi, T.; Preem, J.-K.; Mander, Ü.; Truu, J. Dynamics of Bacterial Community Abundance and Structure in Horizontal Subsurface Flow Wetland Mesocosms Treating Municipal Wastewater. Water 2016, 8, 457. https://doi.org/10.3390/w8100457
Oopkaup K, Truu M, Nõlvak H, Ligi T, Preem J-K, Mander Ü, Truu J. Dynamics of Bacterial Community Abundance and Structure in Horizontal Subsurface Flow Wetland Mesocosms Treating Municipal Wastewater. Water. 2016; 8(10):457. https://doi.org/10.3390/w8100457
Chicago/Turabian StyleOopkaup, Kristjan, Marika Truu, Hiie Nõlvak, Teele Ligi, Jens-Konrad Preem, Ülo Mander, and Jaak Truu. 2016. "Dynamics of Bacterial Community Abundance and Structure in Horizontal Subsurface Flow Wetland Mesocosms Treating Municipal Wastewater" Water 8, no. 10: 457. https://doi.org/10.3390/w8100457
APA StyleOopkaup, K., Truu, M., Nõlvak, H., Ligi, T., Preem, J.-K., Mander, Ü., & Truu, J. (2016). Dynamics of Bacterial Community Abundance and Structure in Horizontal Subsurface Flow Wetland Mesocosms Treating Municipal Wastewater. Water, 8(10), 457. https://doi.org/10.3390/w8100457