
Geochemical Equilibrium Modelling of the Aqueous Speciation of Select Trace Elements in the Great Lakes


Abstract
:1. Introduction
2. Methods and Materials
2.1. Water Quality Data Selection
2.2. Geochemical Equilibrium Modelling Approach
2.3. Sensitivity Analysis and Extrapolations
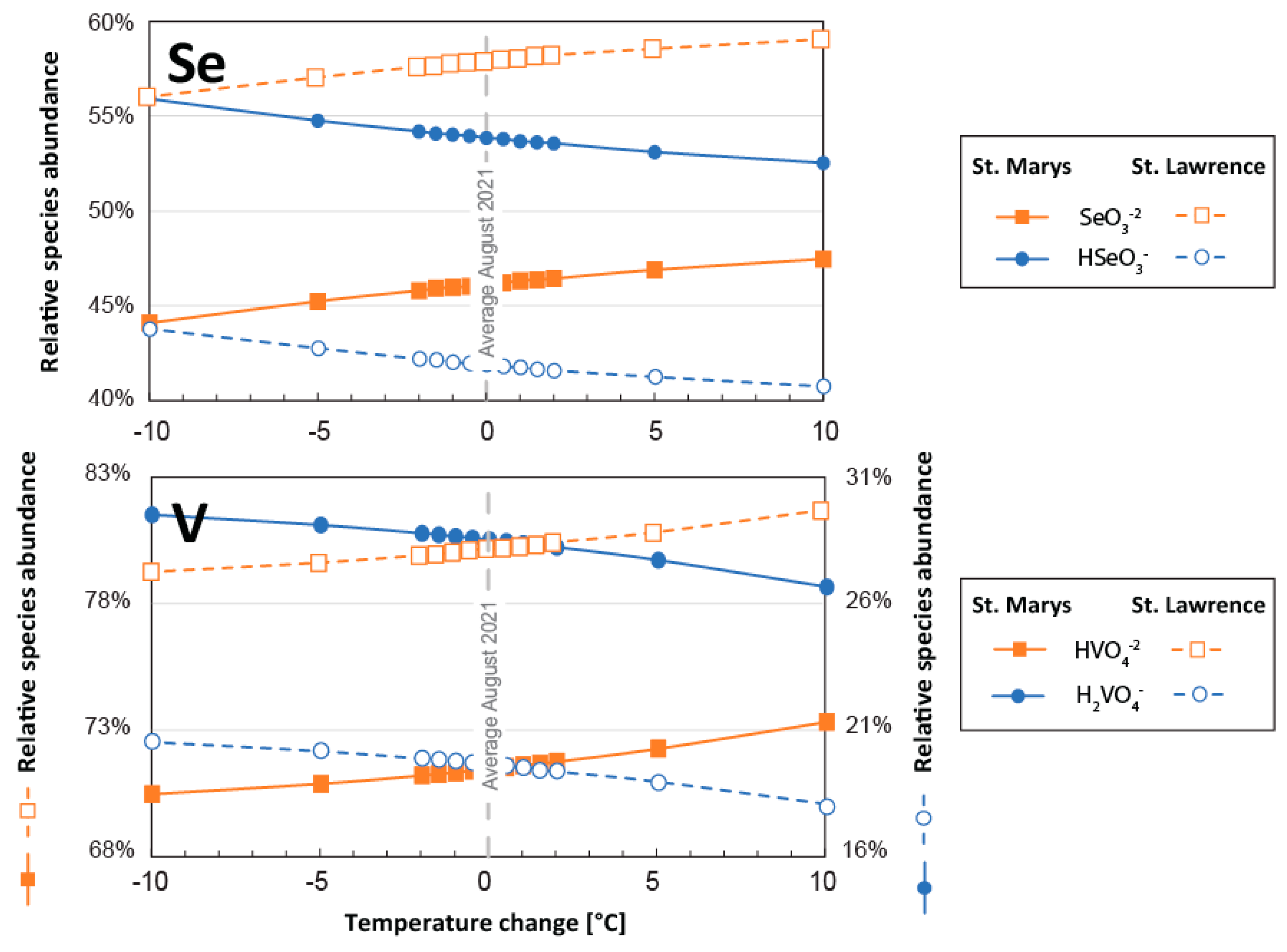
- Temperature: Long-term average seasonal variability in surface water temperatures in the Great Lakes ranges from 0 °C to 16 °C in Lake Superior and from 0 °C to 24 °C in Lake Erie (https://coastwatch.glerl.noaa.gov/statistic/ (accessed on 1 December 2022). Surface waters in the Great Lakes are further expected to warm at a rate of ~0.5 °C per decade [39]. To reflect both seasonal and long-term variability, simulations were performed with connecting channel water temperatures up to 10 degrees below and above the average water temperatures recorded in 2021 (Table S1; [18]).
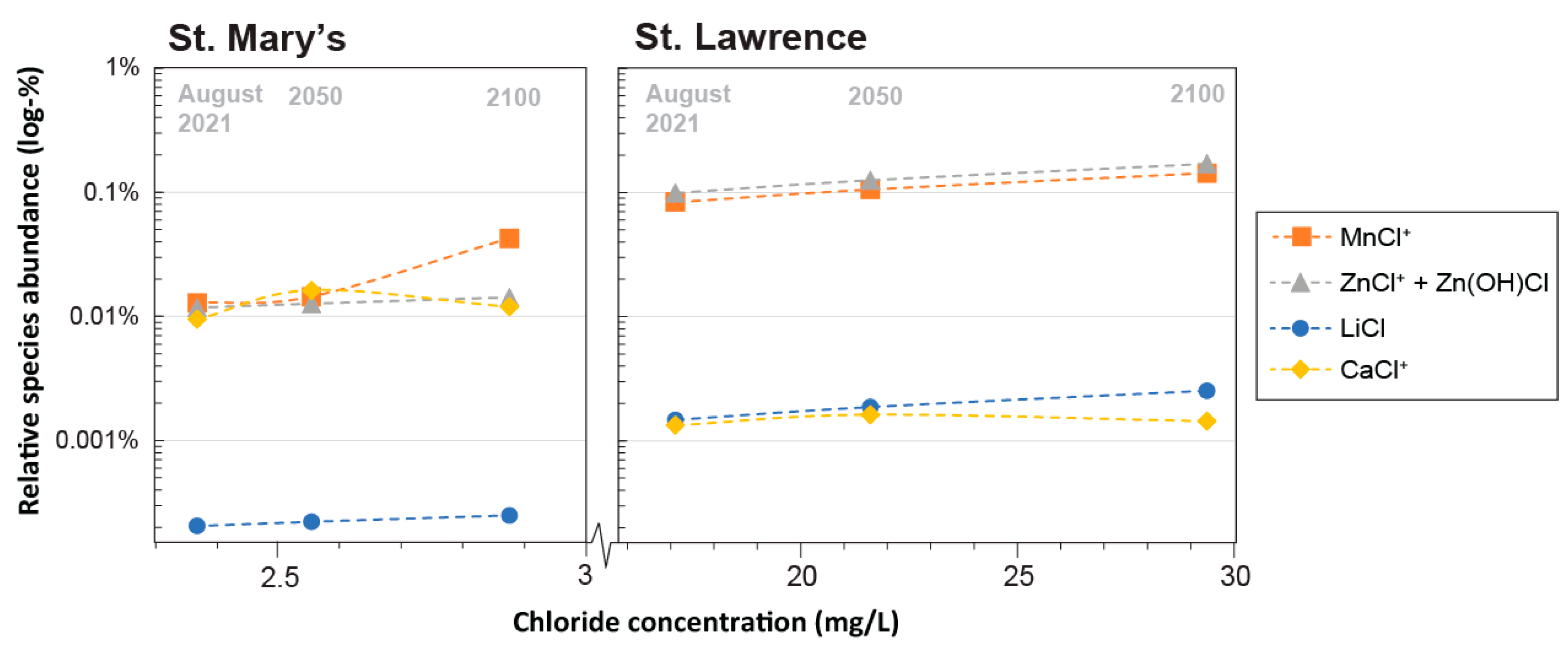
- Salinity: All the Great Lakes are experiencing increasing salinification [3,40]. Previously assessed regression analysis on a long-term Cl concentration time-series for each of the Great Lakes presented in [41] were extrapolated to 2100 for their downstream connecting channels examined here, and the predicted Cl concentrations were adopted directly (Table S2).
- Alkalinity: Previous longitudinal analysis of a carbonate alkalinity time-series for each of the Great Lakes between 1965–2005 revealed increasing levels in Lakes Superior and Huron yet decreasing levels in Lakes Erie and Ontario [3]. The average (linear) rates of change over that time period were extrapolated to 2100 and predicted alkalinity concentrations were adopted directly (Table S2).
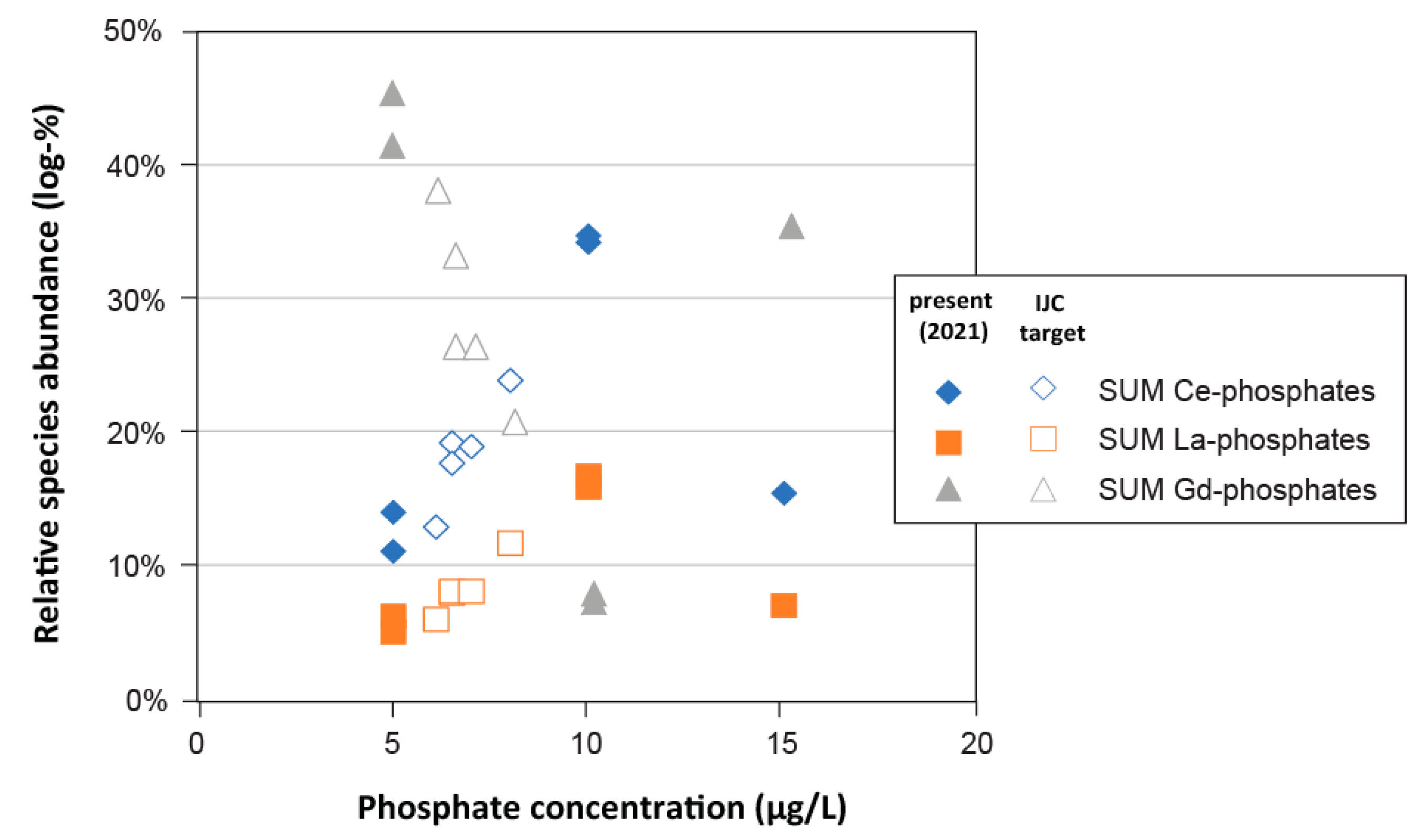
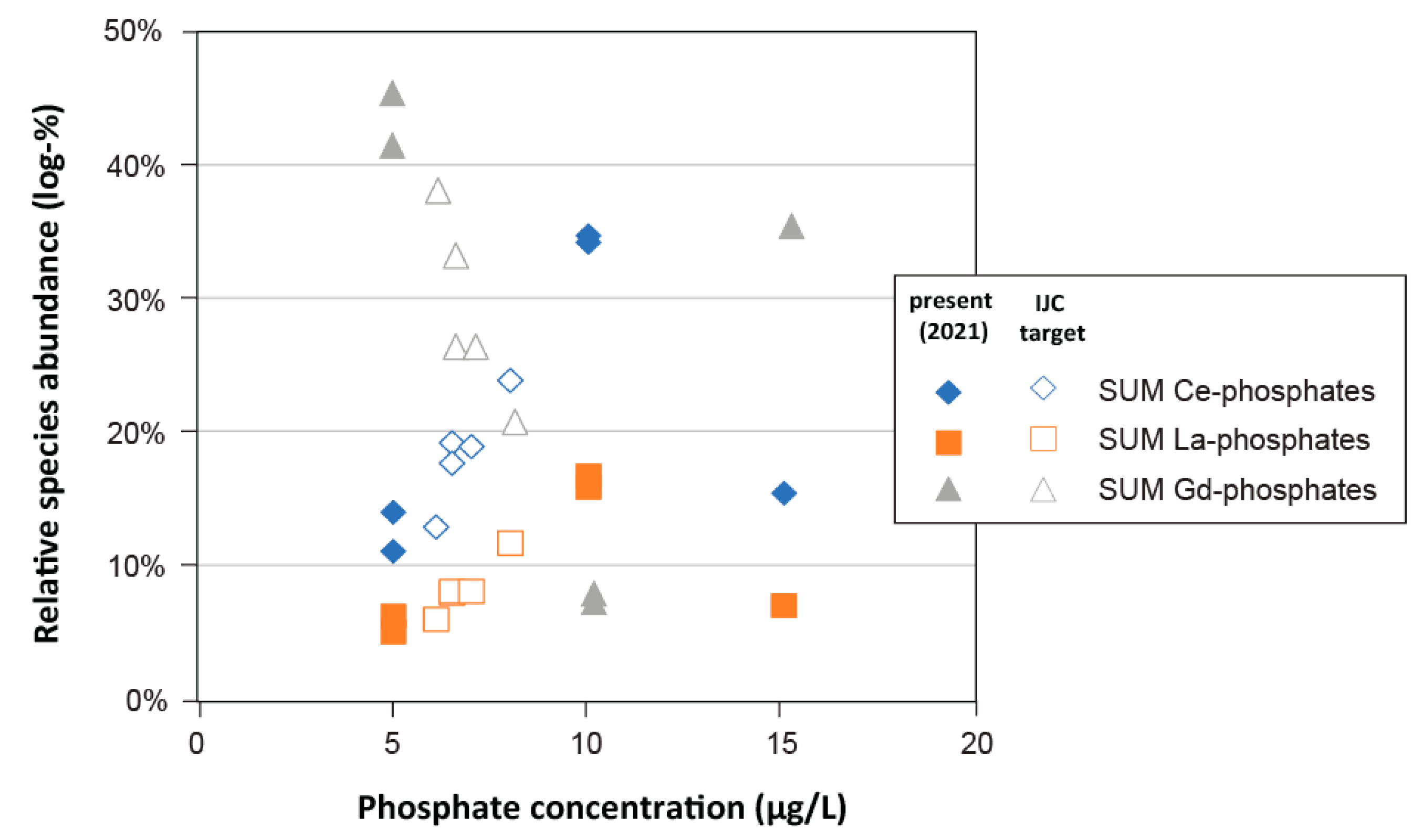
- Phosphate: Analysis of phosphorus (P) dynamics in the Great Lakes surface waters has revealed a large spatiotemporal variation of phosphate levels [19], well-beyond what was recorded in the connecting channel waters in 2021 (Table S1; [10]). In addition to performing simulations with the maximum dissolved phosphate concentrations measured in the Great Lakes (i.e., 15 μg L−1 in Western Lake Erie; [42]), additional simulations were performed with phosphate solution levels that were adjusted to phosphate targets set by the Great Lakes Water Quality Agreement (GLWQA; International Joint Commission (IJC); Table S2; [42,43]), which ranged between 5 and 15 μg L−1 for the upper Great Lakes versus Western Lake Erie, respectively.
3. Results and Discussion
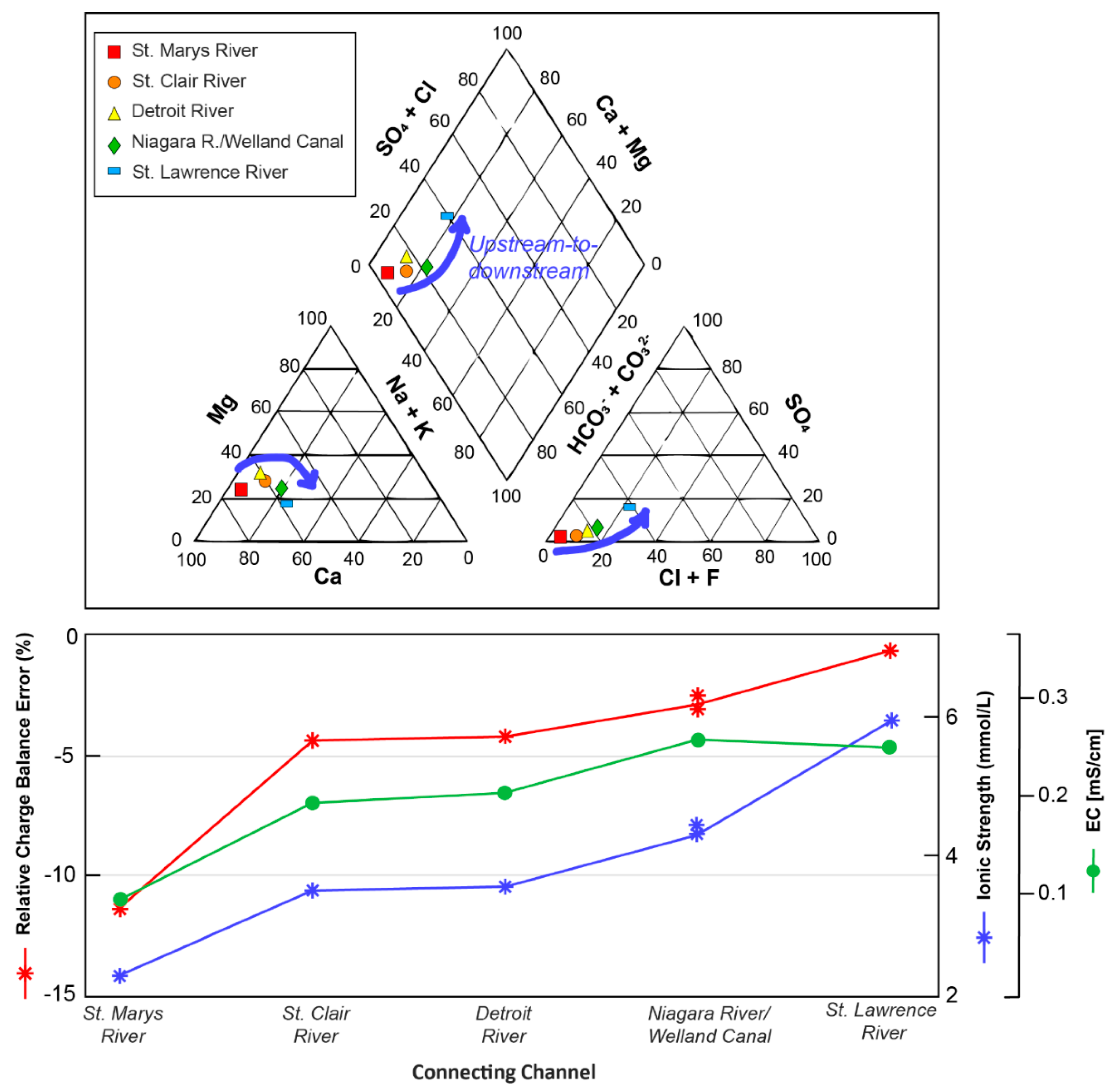
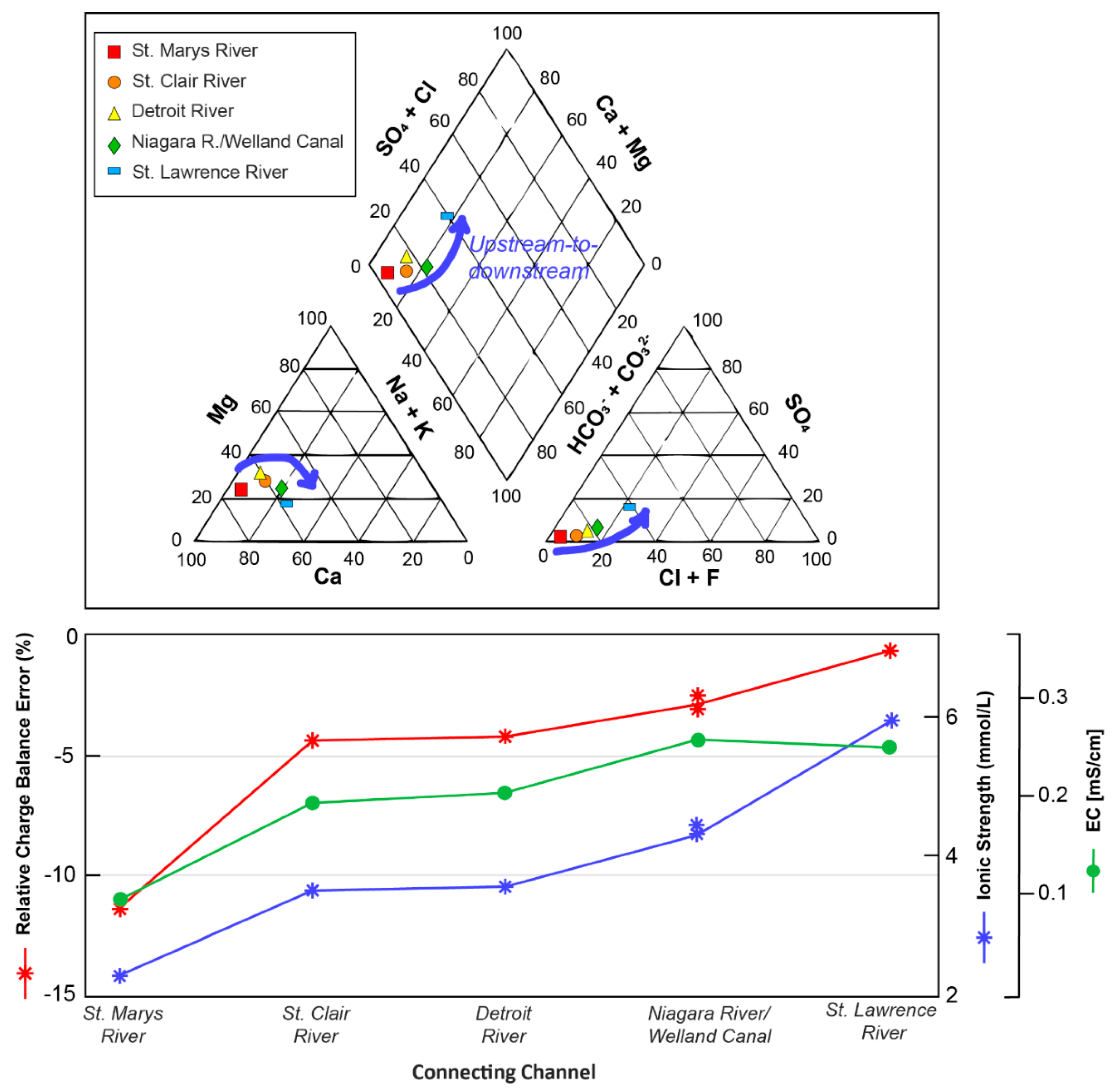
3.1. General Solution Chemistries and Model Charge Balance
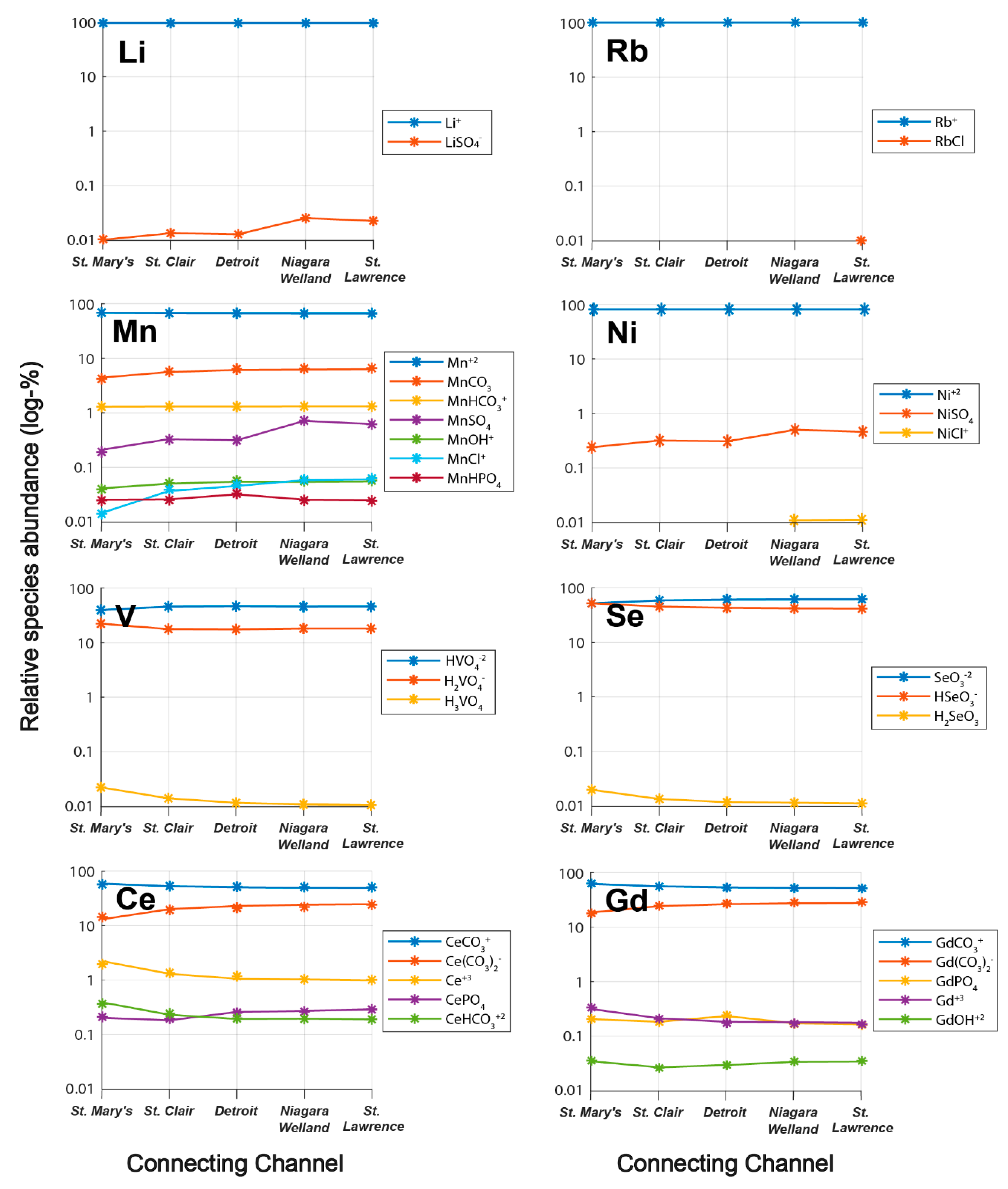
3.2. Trends in Aqueous Trace Element Speciation across the Great Lakes
3.3. Stable Trace Element Speciation with Changing Great Lakes Water Quality
3.3.1. Temperature
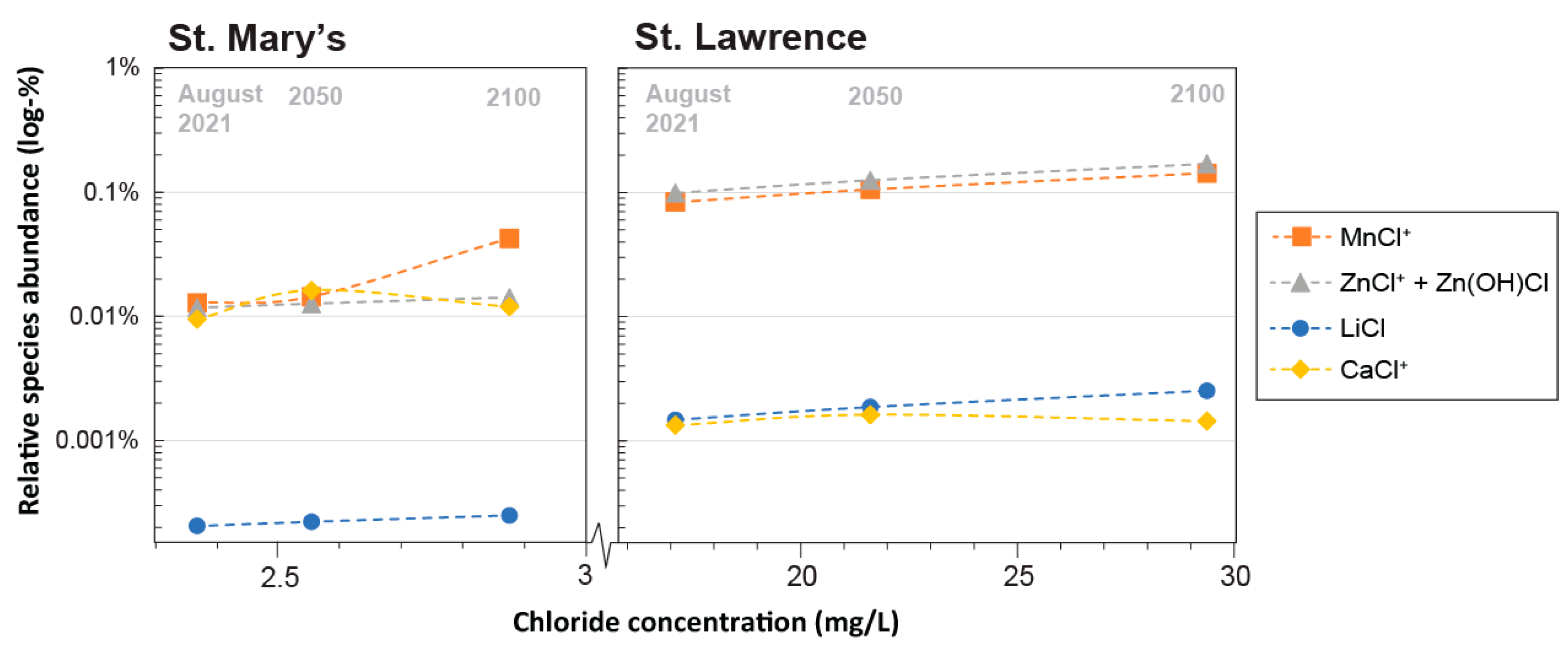
3.3.2. Salinity
3.3.3. Alkalinity
3.3.4. Phosphate
4. Conclusions
- The aqueous speciation of most trace elements appears relatively consistent across the basin, in line with a comparatively stable water quality (pH, alkalinity) upstream-to-downstream across the Great Lakes. Trace element complexation with ligands such as phosphate or chloride generally followed basin-wide trends in salinity or nutrient levels;
- Alkali trace metals (Li, Rb) are dominantly speciated as free monovalent cations, oxyanion-forming elements (Se, V) as oxoacids, rare earth elements (La, Ce, Gd) as carbonates, and various other trace elements as complexes with sulfate or phosphate;
- Simulations of aqueous trace element speciation under extrapolated future water quality scenarios (e.g., increased temperature, salinity, varying alkalinity, and phosphate) suggest that the speciation of most trace elements is robust, not only spatially, but temporally as well.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Behmel, S.; Damour, M.; Ludwig, R.; Rodriguez, M.J. Water quality monitoring strategies—A review and future perspectives. Sci. Total Environ. 2016, 571, 1312–1329. [Google Scholar] [CrossRef] [PubMed]
- Mahdiyan, O.; Filazzola, A.; Molot, L.A.; Gray, D.; Sharma, S. Drivers of water quality changes within the Laurentian Great Lakes region over the past 40 years. Limnol. Oceanogr. 2020, 66, 237–254. [Google Scholar] [CrossRef]
- Chapra, S.C.; Dove, A.; Warren, G.J. Long-term trends of Great Lakes major ion chemistry. J. Great Lakes Res. 2012, 38, 550–560. [Google Scholar] [CrossRef]
- Sterner, R.W.; Ostrom, P.; Ostrom, N.E.; Klump, J.V.; Steinman, A.D.; Dreelin, E.A.; Zanden, M.J.V.; Fisk, A.T. Grand challenges for research in the Laurentian Great Lakes. Limnol. Oceanogr. 2017, 62, 2510–2523. [Google Scholar] [CrossRef] [Green Version]
- Hartig, J.H.; Krantzberg, G.; Alsip, P. Thirty-five years of restoring Great Lakes Areas of Concern: Gradual progress, hopeful future. J. Great Lakes Res. 2020, 46, 429–442. [Google Scholar] [CrossRef]
- Hill, B.; Dove, A. Concentrations and loads of nutrients and major ions in the Niagara River, 1975–2018. J. Great Lakes Res. 2021, 47, 844–861. [Google Scholar] [CrossRef]
- Elliott, S.M.; Brigham, M.E.; Lee, K.E.; Banda, J.A.; Choy, S.J.; Gefell, D.J.; Minarik, T.A.; Moore, J.N.; Jorgenson, Z.G. Contaminants of emerging concern in tributaries to the Laurentian Great Lakes: I. Patterns of occurrence. PLoS ONE 2017, 12, e0182868. [Google Scholar] [CrossRef] [Green Version]
- Sterner, R.W. The Laurentian Great Lakes: A Biogeochemical Test Bed. Annu. Rev. Earth Planet. Sci. 2021, 49, 201–229. [Google Scholar] [CrossRef]
- Ozersky, T.; Bramburger, A.J.; Elgin, A.K.; Vanderploeg, H.A.; Wang, J.; Austin, J.A.; Carrick, H.J.; Chavarie, L.; Depew, D.C.; Fisk, A.T.; et al. The changing face of winter: Lessons and questions from the Laurentian great lakes. J. Geophys. Res. Biogeosci. 2021, 126, e2021JG006247. [Google Scholar] [CrossRef]
- Bentley, C.; Fitzgerald; Dove, A.; Richardson, V.; Bradley, L.; Vriens, B. Trace Element Loads in the Great Lakes Basin: A Reconnaissance. J. Great Lakes Res. 2023; in press. [Google Scholar] [CrossRef]
- Baehr, M.M.; McManus, J. The measurement of phosphorus and its spatial and temporal variability in the western arm of Lake Superior. J. Great Lakes Res. 2003, 29, 479–487. [Google Scholar] [CrossRef]
- Chen, C.; Kamman, N.; Williams, J.; Bugge, D.; Taylor, V.; Jackson, B.; Miller, E. Spatial and temporal variation in mercury bioaccumulation by zooplankton in Lake Champlain (North America). Environ. Pollut. 2012, 161, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Coale, K.H.; Flegal, A.R. Copper, zinc, cadmium and lead in surface waters of Lakes Erie and Ontario. Sci. Total Environ. 1989, 87–88, 297–304. [Google Scholar] [CrossRef]
- Rossman, R.; Barres, J. Trace Element Concentrations in Near-Surface Waters of the Great Lakes and Methods of Collection, Storage, and Analysis. J. Great Lakes Res. 1988, 14, 188–204. [Google Scholar] [CrossRef]
- Nriagu, J.O.; Lawson, G.; Wong, H.K.T.; Cheam, V. Dissolved Trace Metals in Lakes Superior, Erie, and Ontario. Environ. Sci. Technol. 1995, 30, 178–187. [Google Scholar] [CrossRef]
- Yuan, F.; Chaffin, J.; Xue, B.; Wattrus, N.; Zhu, Y.; Sun, Y. Contrasting sources and mobility of trace metals in recent sediments of western Lake Erie. J. Great Lakes Res. 2018, 44, 1026–1034. [Google Scholar] [CrossRef]
- Aliff, M.N.; Reavie, E.D.; Post, S.P.; Zanko, L.M. Metallic elements and oxides and their relevance to Laurentian Great Lakes geochemistry. PeerJ 2020, 8, e9053. [Google Scholar] [CrossRef]
- Bentley, C.; Junqueira, T.; Dove, A.; Vriens, B. Mass-balance Modeling of Metal Loading Rates in the Great Lakes. Environ. Res. 2022, 205, 112557. [Google Scholar] [CrossRef]
- Dove, A.; Chapra, S.C. Long-term trends of nutrients and trophic response variables for the Great Lakes: Great Lakes nutrient trends. Limnol. Oceanogr. 2015, 60, 696–721. [Google Scholar] [CrossRef]
- Bazzi, A.; Lehman, J.T.; Nriagu, J.O.; Hollandsworth, D.; Irish, N.; Nosher, T. Chemical Speciation of Dissolved Copper in Saginaw Bay, Lake Huron, with Square Wave Anodic Stripping Voltammetry. J. Great Lakes Res. 2002, 28, 466–478. [Google Scholar] [CrossRef]
- Twiss, M.R.; Campbell, P.G.C. Scavenging of 137Cs, 109Cd, 65Zn, and 153Gd by plankton of the microbial food web in pelagic Lake Erie surface waters. J. Great Lakes Res. 1998, 24, 776–790. [Google Scholar] [CrossRef]
- Twiss, M.R.; Campbell, P.G.C. Trace metal cycling in the surface waters of Lake Erie: Linking ecological and geochemical fates. J. Great Lakes Res. 1998, 24, 791–807. [Google Scholar] [CrossRef]
- Leal, A.M.M.; Kulik, D.A.; Smith, W.R.; Saar, M.O. An overview of computational methods for chemical equilibrium and kinetic calculations for geochemical and reactive transport modeling. Pure Appl. Chem. 2017, 89, 597–643. [Google Scholar] [CrossRef]
- Seigneur, N.; Mayer, K.U.; Steefel, C.I. Reactive transport in evolving porous media. Rev. Mineral. Geochem. 2019, 85, 197–238. [Google Scholar] [CrossRef] [Green Version]
- Caruso, B.S.; Cox, T.J.; Runkel, R.L.; Velleux, M.L.; Bencala, K.E.; Nordstrom, D.K.; Julien, P.Y.; Butler, B.; Alpers, C.; Marion, A.; et al. Metals fate and transport modelling in streams and watersheds: State of the science and USEPA workshop review. Hydrol. Process. 2008, 22, 4011–4021. [Google Scholar] [CrossRef] [Green Version]
- Schecher, W.D.; McAvoy, D.C. MINEQL+, A Software Environment for Chemical Equilibrium Modeling. Comput. Environ. Urban Syst. 1992, 16, 65–76. [Google Scholar] [CrossRef]
- Parkhurst, D.L. User’s Guide to PHREEQC, a Computer Model for Speciation, Reaction-Path, Advective-Transport and Inverse Geochemical Calculations; US Geological Survey Water-Resources Investigations Report 95-4227; U.S. Geological Survey: Reston, VA, USA, 1995.
- Haase, C.; Ebert, M.; Dethlefsen, F. Uncertainties of geochemical codes and thermodynamic databases for predicting the impact of carbon dioxide on geologic formations. Appl. Geochem. 2016, 67, 81–92. [Google Scholar] [CrossRef]
- Zhu, C.; Nordstrom, D.K. Flying Blind: Geochemical Modeling and Thermodynamic Data Files. Groundwater 2022, 60, 699–700. [Google Scholar] [CrossRef]
- Mühr-Ebert, E.L.; Wagner, F.; Walther, C. Speciation of uranium: Compilation of a thermodynamic database and its experimental evaluation using different analytical techniques. Appl. Geochem. 2019, 100, 213–222. [Google Scholar] [CrossRef]
- Ball, J.W.; Nordstrom, D.K. User’s Manual for WATEQ4F, with Revised Thermodynamic Database and Test Cases for Calculating Speciation of Major, Trace, and Redox Elements in Natural Waters; US Geological Survey Open File Report 91-183; U.S. Geological Survey: Reston, VA, USA, 1991.
- Zhu, C.; Anderson, G. Environmental Applications of Geochemical Modeling; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Quinn, F.H. Hydraulic residence times for the Laurentian great lakes. J. Great Lakes Res. 1992, 18, 22–28. [Google Scholar] [CrossRef]
- EGLE, State of Michigan, Department of the Environment, Great Lakes and Energy. Great Lakes Connecting Channel—Water Quality Data, 2020 Integrated Report. 2020. Available online: https://www.michigan.gov/-/media/Project/Websites/egle/Documents/Programs/WRD/SWAS/ir2020-all.pdf?rev=2983e2380a7a438a86a0d636077ceb51 (accessed on 1 October 2022).
- Smith, R.E.H.; Allen, C.D.; Charlton, M.N. Dissolved Organic Matter and Ultraviolet Radiation Penetration in the Laurentian Great Lakes and Tributary Waters. J. Great Lakes Res. 2004, 30, 367–380. [Google Scholar] [CrossRef]
- Zhou, Z.; Guo, L.; Minor, E.C. Characterization of bulk and chromophoric dissolved organic matter in the Laurentian Great Lakes during summer 2013. J. Great Lakes Res. 2016, 42, 789–801. [Google Scholar] [CrossRef] [Green Version]
- Allison, J.D.; Brown, D.S.; Novo-Gradac, K.J. MINTEQA2/PRODEFA2—A Geochemical Assessment Model for Environmental Systems. Version 3.0 User’s Manual; Environmental Research Laboratory, Office of Research and Development U.S. Environmental Protection Agency: Athens, GA, USA, 1998.
- Blanc, P.; Lassin, A.; Piantone, P.; Azaroual, M.; Jacquemet, N.; Fabbri, A.; Gaucher, E. Thermoddem: A geochemical database focused on low temperature water/rock interactions and waste materials. Appl. Geochem. 2012, 27, 2107–2116. [Google Scholar] [CrossRef]
- Trumpickas, J.; Shuter, B.J.; Minns, C.K. Forecasting impacts of climate change on Great Lakes surface water temperatures. J. Great Lakes Res. 2009, 35, 454–463. [Google Scholar] [CrossRef]
- Dugan, H.A.; Bartlett, S.L.; Burke, S.M.; Doubek, J.P.; Krivak-Tetley, F.E.; Skaff, N.K.; Summers, J.C.; Farrell, K.J.; McCullough, I.M.; Morales-Williams, A.M.; et al. Salting our freshwater lakes. Proc. Natl. Acad. Sci. USA 2017, 114, 4453–4458. [Google Scholar] [CrossRef] [Green Version]
- Chapra, S.C.; Dove, A.; Rockwell, D.C. Great Lakes chloride trends: Long-term mass balance and loading analysis. J. Great Lakes Res. 2009, 35, 272–284. [Google Scholar] [CrossRef]
- Chapra, S.C.; Dolan, D.M. Great Lakes total phosphorus revisited: 2. Mass balance modeling. J. Great Lakes Res. 2012, 38, 741–754. [Google Scholar] [CrossRef]
- ECCC, Environment and Climate Change Canada, Canadian Environmental Sustainability Indicators: Phosphorus levels in the offshore waters of the Great Lakes. 2020. Available online: www.canada.ca/en/environment-climate-change/services/environmental-indicators/phosphoruslevels-off-shore-great-lakes.html (accessed on 12 December 2022).
- McKay, R.M.L.; Bullerjahn, G.S.; Porta, D.; Brown, E.T.; Sherrell, R.M.; Smutka, T.M.; Sterner, R.W.; Twiss, M.R.; Wilhelm, S.W. Consideration of the bioavailability of iron in the North American Great Lakes: Development of novel approaches toward understanding iron biogeochemistry. Aquat. Ecosyst. Health Manag. 2004, 7, 475–490. [Google Scholar] [CrossRef]
- Stoffyn-Egli, P. Conservative Behaviour of Dissolved Lithium in Estuarine Waters. Estuar. Coast. Shelf Sci. 1982, 14, 577–587. [Google Scholar] [CrossRef]
- Kszos, L.A.; Stewart, A.J. Review of lithium in the aquatic environment: Distribution in the United States, toxicity and case example of groundwater contamination. Ecotoxicology 2003, 12, 439–447. [Google Scholar] [CrossRef]
- Smith, J.P.; Boyd, T.J.; Cragan, J.; Ward, M.C. Dissolved rubidium to strontium ratio as a conservative tracer for wastewater effluent-sourced contaminant inputs near a major urban wastewater treatment plant. Water Res. 2021, 205, 117691. [Google Scholar] [CrossRef]
- Johannesson, K.H.; Lyons, W.B.; Stetzenbach, K.J.; Byrne, R.H. The solubility control of rare earth elements in natural terrestrial waters and the significance of PO43− and CO32− in limiting dissolved rare earth concentrations: A review of recent information. Aquat. Geochem. 1995, 1, 157–173. [Google Scholar] [CrossRef]
- Byrne, R.H.; Kim, K.-H. Rare earth element scavenging in seawater. Geochim. Et Cosmochim. Acta 1990, 54, 2645–2656. [Google Scholar] [CrossRef]
- Hayhoe, K.; VanDorn, J.; Croley, T.; Schlegal, N.; Wuebbles, D. Regional climate change projections for Chicago and the US Great Lakes. J. Great Lakes Res. 2010, 36, 7–21. [Google Scholar] [CrossRef]
- Byun, K.; Hamlet, A.F. Projected changes in future climate over the Midwest and Great Lakes region using downscaled CMIP5 ensembles. Int. J. Climatol. 2018, 38, e5310553. [Google Scholar] [CrossRef]
- Correll, D.L. The Role of Phosphorus in the Eutrophication of Receiving Waters: A Review. J. Environ. Qual. 1998, 27, 261–266. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Redox States | Carbonate Complexes | Halogen Complexes | Sulfate Complexes | Phosphate Complexes | Major Cations | Minor Cations | |
|---|---|---|---|---|---|---|---|
| Li | {1} | N/A | Cl | Included | N/A | N/A | N/A |
| Rb | {1} | N/A | Cl, Br, I | Included | N/A | N/A | N/A |
| Mn | {0, 2, 3, 6} | Included | F, Cl | Included | Included | N/A | N/A |
| Ni | {2} | N/A | Cl, Br | Included | Included | N/A | N/A |
| V | {2, 3, 4, 5} | N/A | F | Included | Included | Fe, K, Ca, Mg | Mn |
| Se | {−2, 4, 6} | N/A | N/A | N/A | N/A | Ca, Cu, Fe, K, Mg, Na | Cd, Co, Mn, Ni, Zn, Ba, Cd, Pb, Li, Mn, Ag, Sn, Sr, Th |
| Gd | {2, 3, 5} | Included | F, Cl | Included | Included | N/A | N/A |
| Ce | {2, 3, 4} | Included | F, Cl, Br, I | Included | Included | N/A | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fitzgerald, J.; Bentley, C.; Vriens, B. Geochemical Equilibrium Modelling of the Aqueous Speciation of Select Trace Elements in the Great Lakes. Water 2023, 15, 1483. https://doi.org/10.3390/w15081483
Fitzgerald J, Bentley C, Vriens B. Geochemical Equilibrium Modelling of the Aqueous Speciation of Select Trace Elements in the Great Lakes. Water. 2023; 15(8):1483. https://doi.org/10.3390/w15081483
Chicago/Turabian StyleFitzgerald, John, Colton Bentley, and Bas Vriens. 2023. "Geochemical Equilibrium Modelling of the Aqueous Speciation of Select Trace Elements in the Great Lakes" Water 15, no. 8: 1483. https://doi.org/10.3390/w15081483