

Providencia alcalifaciens—Assisted Bioremediation of Chromium-Contaminated Groundwater: A Computational Study
 , , and
, , and
Abstract
:1. Introduction
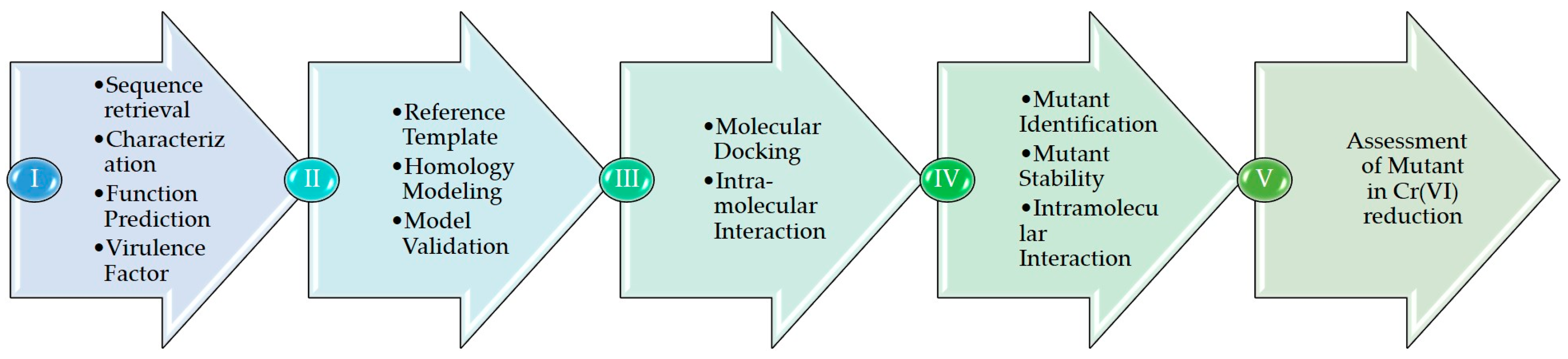
2. Materials and Methods
2.1. ChrR Sequence Annotation
2.2. Homology Model and Validation of ChrR
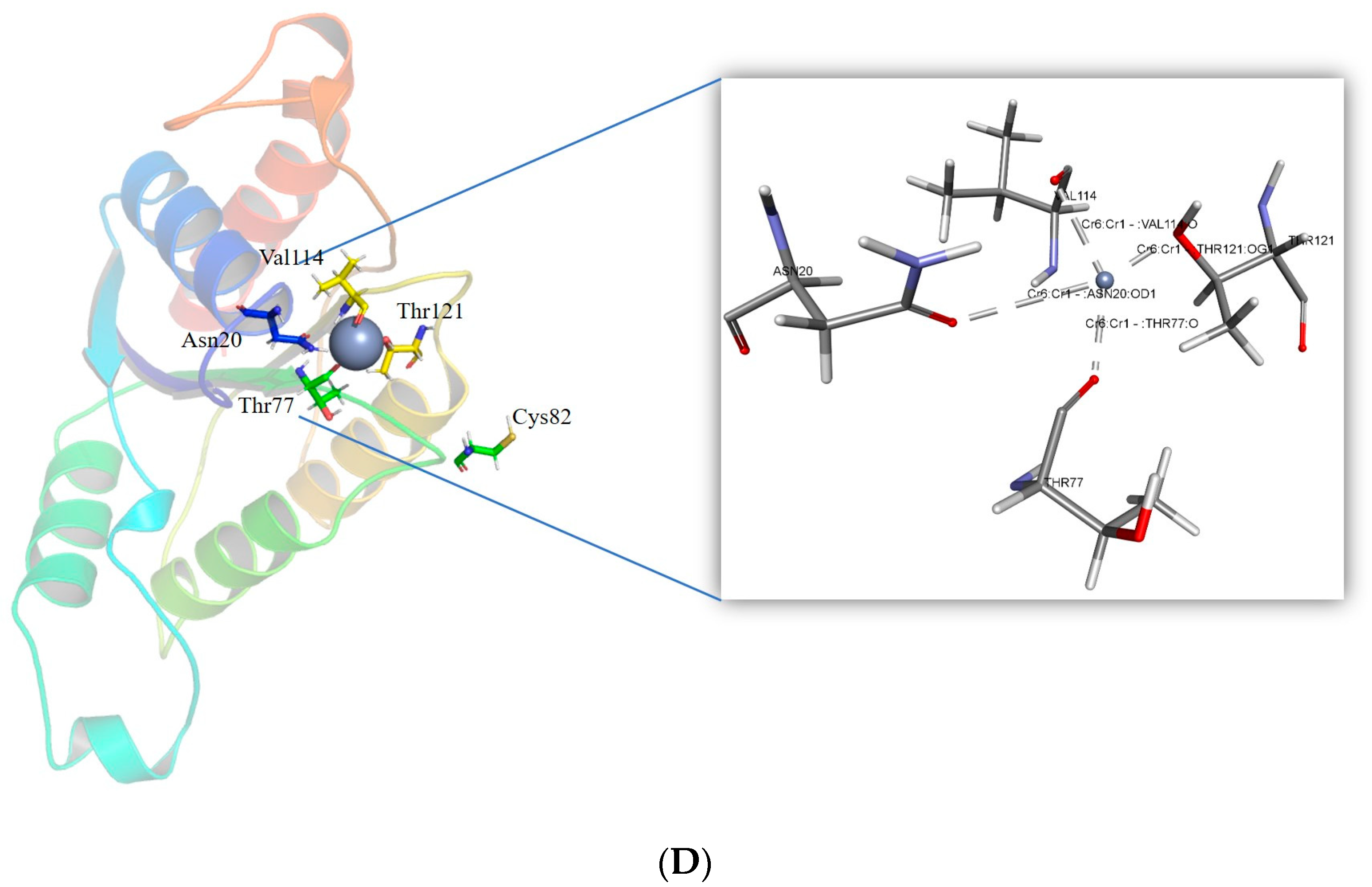
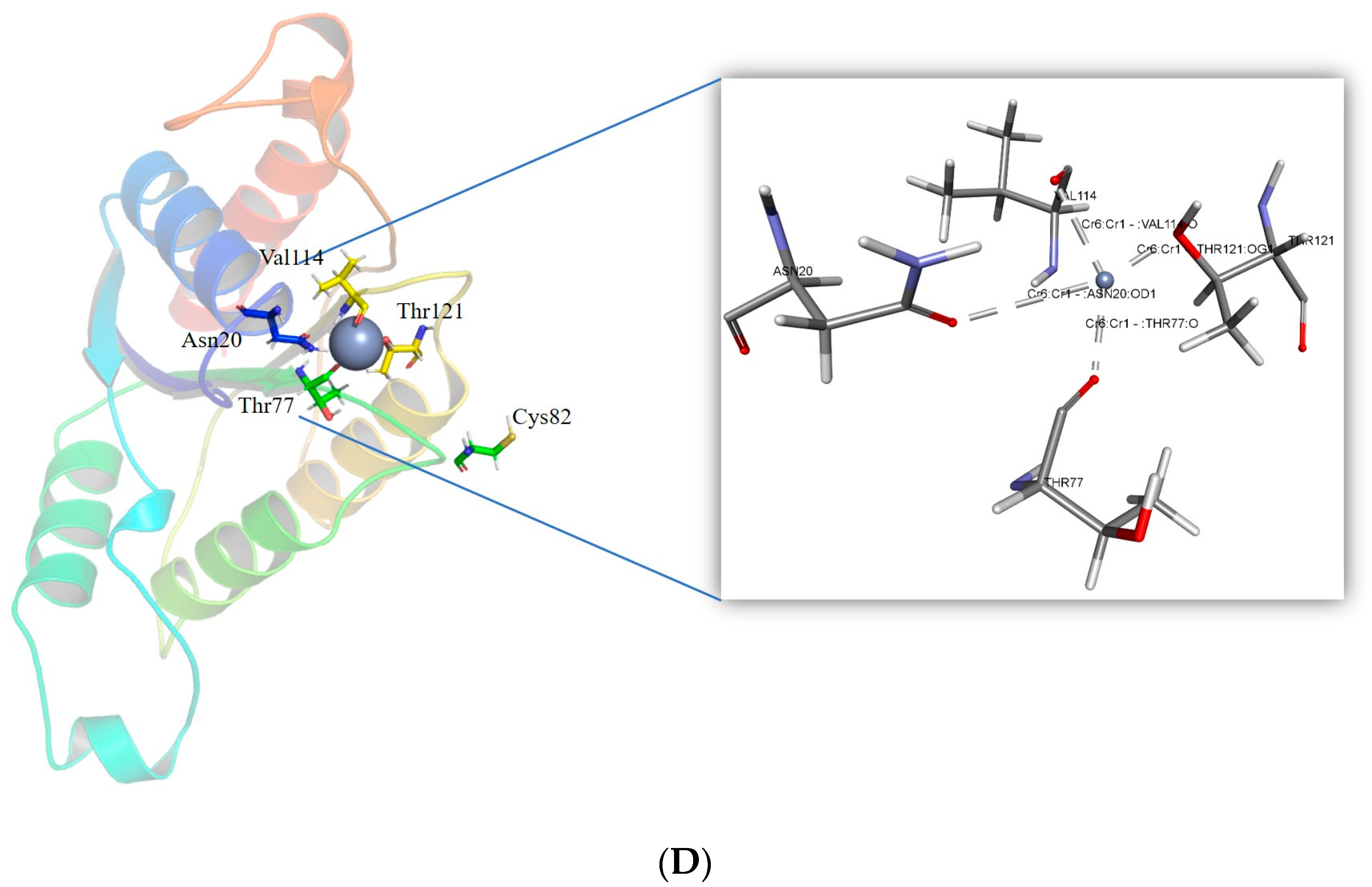
2.3. Cr(VI) Molecular Docking and Intramolecular Interaction Studies
2.4. Generation and Molecular Docking of In Silico Mutant of ChrR to Analyze Intramolecular Interaction with Cr(VI)
3. Results
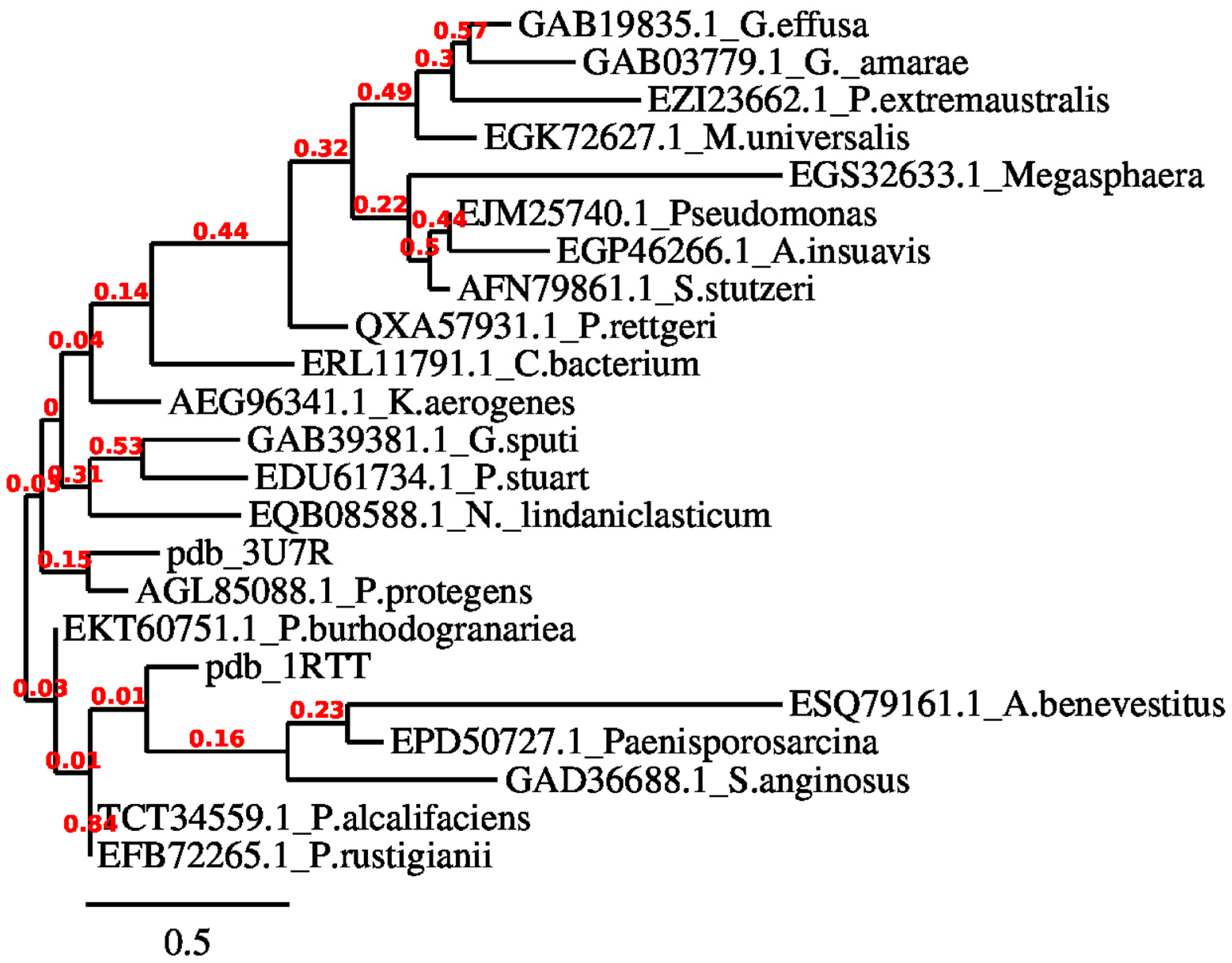
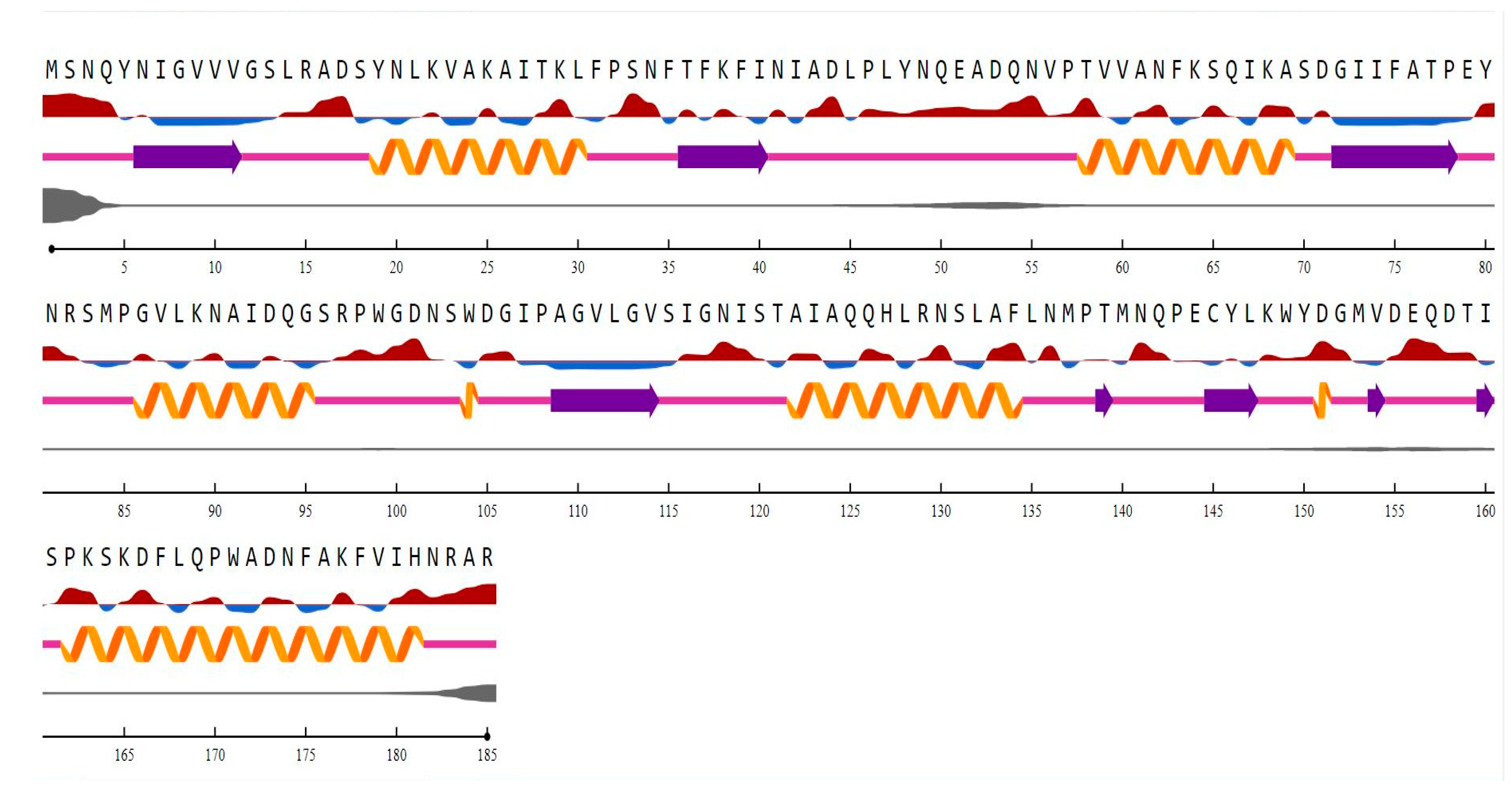
3.1. ChrR Sequence Annotation
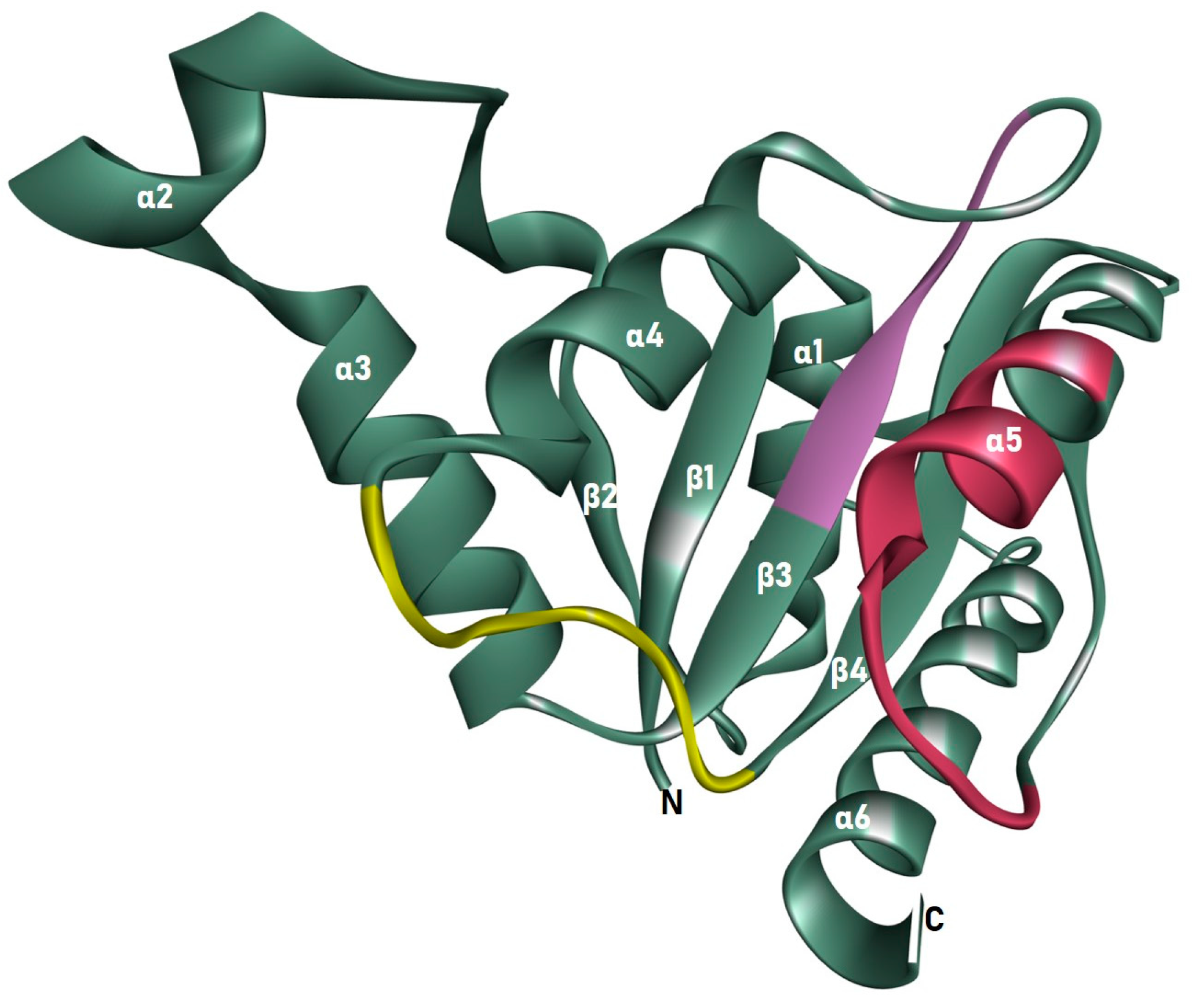
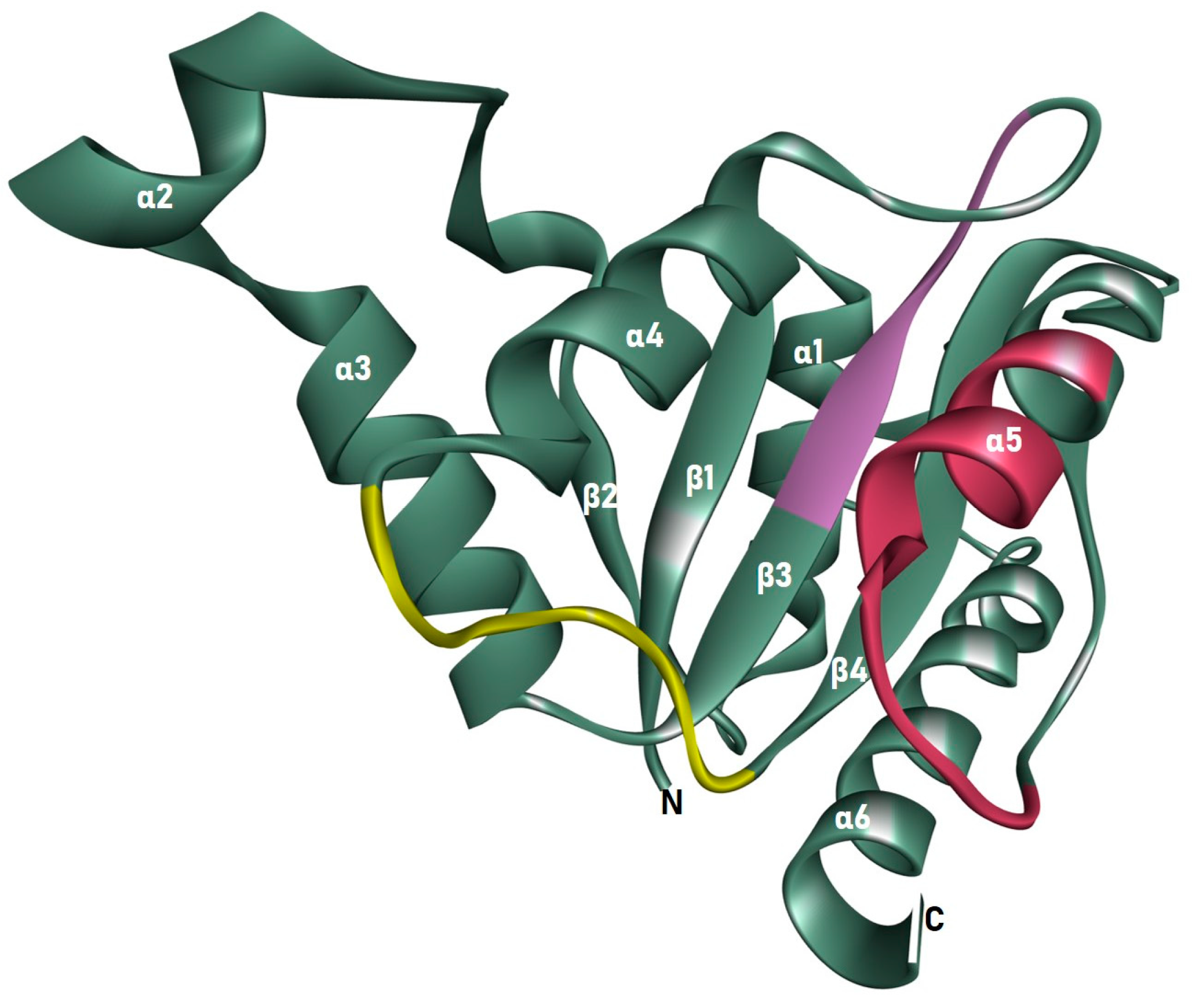
3.2. Homology Model and Validation of ChrR
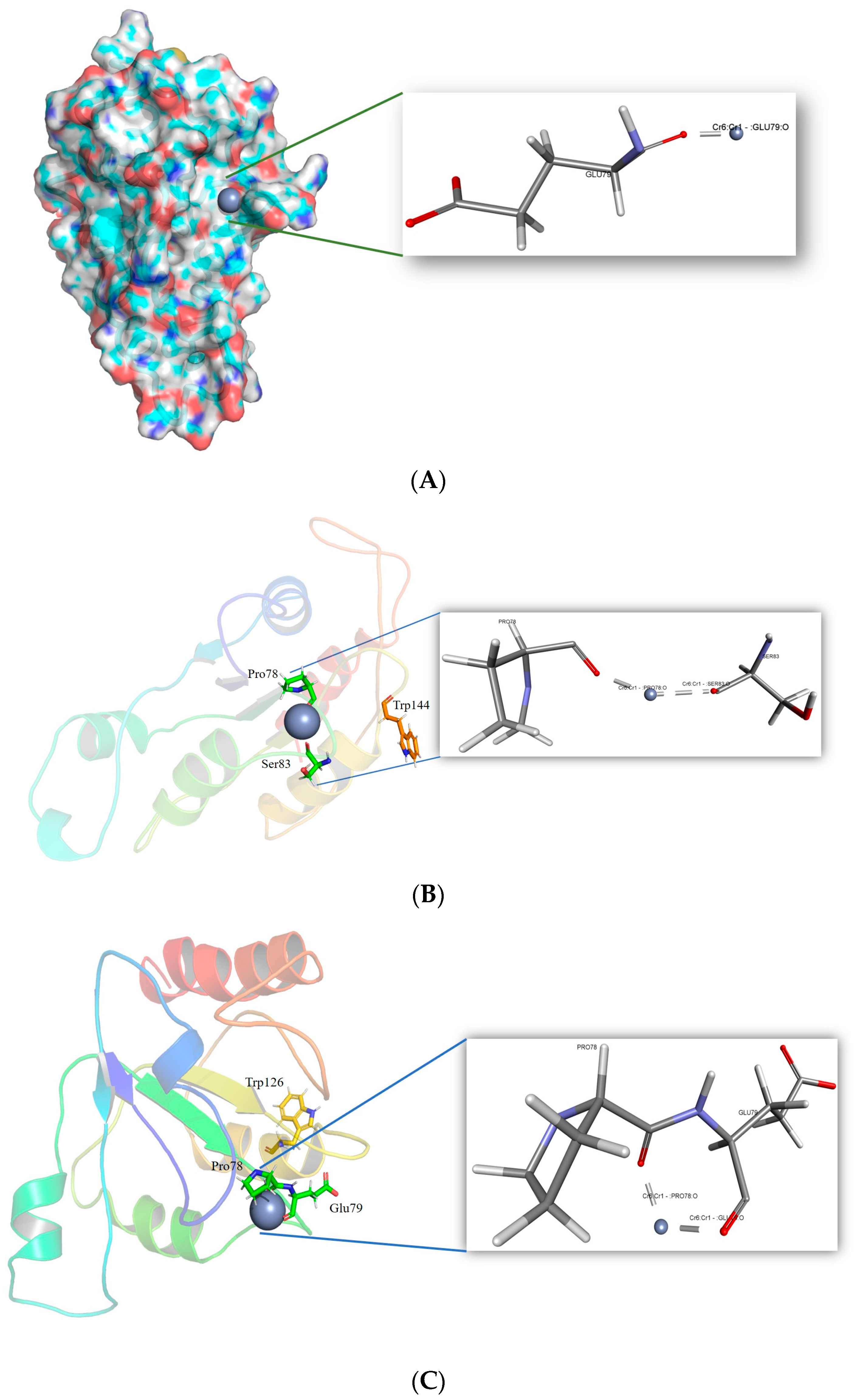
3.3. Generation and Molecular Docking of In Silico Mutant of ChrR to Analyse Intramolecular Interaction with Cr(VI)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Secretary-General, U. Progress towards the Sustainable Development Goals: Report of the Secretary-General; UN: New York, NY, USA, 2021. [Google Scholar]
- Abdulrahman, M. Seawater desalination: The strategic choice for Saudi Arabia. Desalination Water Treat. 2012, 51, 1–4. [Google Scholar]
- Ghaffar, A.; Sehgal, S.A.; Fatima, R.; Batool, R.; Aimen, U.; Awan, S.; Batool, S.; Ahmad, F.; Nurulain, S.M. Molecular docking analyses of CYP450 monooxygenases of Tribolium castaneum (Herbst) reveal synergism of quercetin with paraoxon and tetraethyl pyrophosphate: In vivo and in silico studies. Toxicol. Res. 2020, 9, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- WHO. Guidelines for Drinking-Water Quality, 4th Edition, Incorporating the 1st Addendum. 24 April 2017. Available online: https://www.who.int/publications/i/item/9789241549950 (accessed on 24 April 2022).
- Hotta, N. Clinical aspect of chronic arsenic poisoning due to environmental and occupational pollution in and around a small refining spot. JPN J. Const. Med. 1989, 53, 49–70. [Google Scholar]
- Maghraby, M.; Nasr, O.; Hamouda, M. Quality assessment of groundwater at south Al Madinah Al Munawarah area, Saudi Arabia. Environ. Earth Sci. 2013, 70, 1525–1538. [Google Scholar] [CrossRef]
- Ali, I.; Hasan, M.A.; Alharbi, O.M.L. Toxic metal ions contamination in the groundwater, Kingdom of Saudi Arabia. J. Taibah Univ. Sci. 2020, 14, 1571–1579. [Google Scholar] [CrossRef]
- Mohanty, M.; Patra, H.K. Attenuation of Chromium Toxicity by Bioremediation Technology. Rev. Environ. Contam. Toxicol. 2011, 210, 1–34. [Google Scholar]
- Liang, J.; Huang, X.; Yan, J.; Li, Y.; Zhao, Z.; Liu, Y.; Ye, J.; Wei, Y. A review of the formation of Cr(VI) via Cr(III) oxidation in soils and groundwater. Sci. Total Environ. 2021, 774, 145762. [Google Scholar]
- Zhitkovich, A. Chromium in drinking water: Sources, metabolism, and cancer risks. Chem. Res. Toxicol. 2011, 24, 1617–1629. [Google Scholar] [CrossRef]
- Chen, Z.; Song, S.; Wen, Y. Reduction of Cr(VI) into Cr [10] by organelles of Chlorella vulgaris in aqueous solution: An organelle-level attempt. Sci. Total Environ. 2016, 572, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Fernández, P.M.; Viñarta, S.C.; Bernal, A.R.; Cruz, E.L.; Figueroa, L.I. Bioremediation strategies for chromium removal: Current research, scale-up approach and future perspectives. Chemosphere 2018, 208, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Jaishankar, M.; Tseten, T.; Anbalagan, N.; Mathew, B.B.; Beeregowda, K.N. Toxicity, mechanism and health effects of some heavy metals. Interdiscip. Toxicol. 2014, 7, 60–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradhan, D.; Sukla, L.B.; Sawyer, M.; Rahman, P.K. Recent bioreduction of hexavalent chromium in wastewater treatment: A review. J. Ind. Eng. Chem. 2017, 55, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Daneshvar, E.; Zarrinmehr, M.J.; Kousha, M.; Hashtjin, A.M.; Saratale, G.D.; Maiti, A.; Vithanage, M.; Bhatnagar, A. Hexavalent chromium removal from water by microalgal-based materials: Adsorption, desorption and recovery studies. Bioresour. Technol. 2019, 293, 122064. [Google Scholar] [CrossRef]
- Wołowiec, M.; Komorowska-Kaufman, M.; Pruss, A.; Rzepa, G.; Bajda, T. Removal of Heavy Metals and Metalloids from Water Using Drinking Water Treatment Residuals as Adsorbents: A Review. Minerals 2019, 9, 487. [Google Scholar] [CrossRef] [Green Version]
- Kanmani, P.L.; Aravind, J.; Preston, D. Remediation of chromium contaminants using bacteria. Int. J. Environ. Sci. Technol. 2012, 9, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Spain, O.; Plöhn, M.; Funk, C. The cell wall of green microalgae and its role in heavy metal removal. Physiol. Plant. 2021, 173, 526–535. [Google Scholar] [CrossRef]
- Asha, L.P.; Sandeep, R.S. Review on bioremediation–potential tool for removing environmental pollution. Int. J. Basic Appl. Chem. Sci. 2013, 3, 21–33. [Google Scholar]
- Sharma, J.; Shamim, K.; Dubey, S.K.; Meena, R.M. Metallothionein assisted periplasmic lead sequestration as lead sulfite by Providencia vermicola strain SJ2A. Sci. Total Environ. 2017, 579, 359–365. [Google Scholar] [CrossRef]
- Naik, M.M.; Khanolkar, D.; Dubey, S.K. Lead-resistant Providencia alcalifaciens strain 2EA bioprecipitates Pb+2 as lead phosphate. Lett. Appl. Microbiol. 2013, 56, 99–104. [Google Scholar] [CrossRef]
- Abo-Amer, A.E.; Ramadan, A.B.; Abo-State, M.; Abu-Gharbia, M.A.; Ahmed, H.E. Biosorption of aluminum, cobalt, and copper ions by Providencia rettgeri isolated from wastewater. J. Basic Microbiol. 2013, 53, 477–488. [Google Scholar] [CrossRef]
- Thacker, U.; Parikh, R.; Shouche, Y.; Madamwar, D. Hexavalent chromium reduction by Providencia sp. Process Biochem. 2006, 41, 1332–1337. [Google Scholar] [CrossRef]
- Zimmer, A.L.; Thoden, J.B.; Holden, H.M. Three-dimensional structure of a sugar N-formyltransferase from Francisella tularensis. Protein Sci. 2014, 23, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bolton, E.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2010, 38, D5–D16. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.; Gupta, D. VirulentPred: A SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinform. 2008, 9, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Raghava, G.P. VICMpred: An SVM-based method for the prediction of functional proteins of Gram-negative bacteria using amino acid patterns and composition. Genom. Proteom. Bioinform. 2006, 4, 42–47. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Kapil, R.; Dhakan, D.B.; Sharma, V.K. MP3: A Software Tool for the Prediction of Pathogenic Proteins in Genomic and Metagenomic Data. PLoS ONE 2014, 9, e93907. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.V.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar]
- Sillitoe, I.; Bordin, N.; Dawson, N.; Waman, V.P.; Ashford, P.; Scholes, H.M.; Pang, C.S.M.; Woodridge, L.; Rauer, C.; Sen, N.; et al. CATH: Increased structural coverage of functional space. Nucleic Acids Res. 2021, 49, D266–D273. [Google Scholar] [CrossRef]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar]
- Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019, 47, D520–D528. [CrossRef] [Green Version]
- Combet, C.; Jambon, M.; Deléage, G.; Geourjon, C. Geno3D: Automatic comparative molecular modelling of protein. Bioinformatics 2002, 18, 213–214. [Google Scholar] [CrossRef] [Green Version]
- McGuffin, L.J.; Adiyaman, R.; Maghrabi, A.H.A.; Shuid, A.N.; Brackenridge, D.A.; Nealon, J.O.; Philomina, L.S. IntFOLD: An integrated web resource for high performance protein structure and function prediction. Nucleic Acids Res. 2019, 47, W408–W413. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Wallner, B.; Elofsson, A. Can correct protein models be identified? Protein Sci. 2003, 12, 1073–1086. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.; Pranjaya, P.P.; Thatoi, H. In silico studies on structural, functional, and evolutionary analysis of bacterial chromate reductase family responsible for high chromate bioremediation efficiency. SN Appl. Sci. 2020, 2, 1997. [Google Scholar] [CrossRef]
- Jin, H.; Zhang, Y.; Buchko, G.W.; Varnum, S.M.; Robinson, H.; Squier, T.C.; Long, P.E. Structure determination and functional analysis of a chromate reductase from Gluconacetobacter hansenii. PLoS ONE 2012, 7, e42432. [Google Scholar] [CrossRef] [Green Version]
- Gagnon, J.K.; Law, S.M.; Brooks, C.L., 3rd. Flexible CDOCKER: Development and application of a pseudo-explicit structure-based docking method within CHARMM. J. Comput. Chem. 2016, 37, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Eswaramoorthy, S.; Poulain, S.; Hienerwadel, R.; Bremond, N.; Sylvester, M.D.; Zhang, Y.B.; Berthomieu, C.; Van Der Lelie, D.; Matin, A. Crystal Structure of ChrR—A Quinone Reductase with the Capacity to Reduce Chromate. PLoS ONE 2012, 7, e36017. [Google Scholar] [CrossRef]
- Cristobal, S.; Zemla, A.; Fischer, D.; Rychlewski, L.; Elofsson, A. A study of quality measures for protein threading models. BMC Bioinform. 2001, 2, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siew, N.; Elofsson, A.; Rychlewski, L.; Fischer, D. MaxSub: An automated measure for the assessment of protein structure prediction quality. Bioinformatics 2000, 16, 776–785. [Google Scholar] [CrossRef] [Green Version]
- Luthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35 (Suppl. S2), W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., III; de Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Calpha geometry: Phi, psi and Cbeta deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Sedláček, V.; Klumpler, T.; Marek, J.; Kucera, I. The structural and functional basis of catalysis mediated by NAD(P)H:acceptor Oxidoreductase (FerB) of Paracoccus denitrificans. PLoS ONE 2014, 9, e96262. [Google Scholar] [CrossRef]
- Hanukoglu, I. Proteopedia: Rossmann fold: A beta-alpha-beta fold at dinucleotide binding sites. Biochem. Mol. Biol. Educ. 2015, 43, 206–209. [Google Scholar] [CrossRef]
- Abeln, S.; Feenstra, K.A.; Heringa, J. Protein Three-Dimensional Structure Prediction. In Encyclopedia of Bioinformatics and Computational Biology; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Dor, O.; Zhou, Y. Achieving 80% ten-fold cross-validated accuracy for secondary structure prediction by large-scale training. Proteins Struct. Funct. Bioinform. 2007, 66, 838–845. [Google Scholar] [CrossRef]
- Pollastri, G.; McLysaght, A. Porter: A new, accurate server for protein secondary structure prediction. Bioinformatics 2005, 21, 1719–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooney, C.; Vullo, A.; Pollastri, G. Protein structural motif prediction in multidimensional phi-psi space leads to improved secondary structure prediction. J. Comput. Biol. 2006, 13, 1489–1502. [Google Scholar] [CrossRef] [PubMed]
- Ackerley, D.F.; Gonzalez, C.F.; Park, C.H.; Blake, R.; Keyhan, M.; Matin, A. Chromate-reducing properties of soluble flavoproteins from Pseudomonas putida and Escherichia coli. Appl. Environ. Microbiol. 2004, 70, 873–882. [Google Scholar] [CrossRef] [Green Version]
- Wilding, M.; Hong, N.; Spence, M.; Buckle, A.; Jackson, C.J. Protein engineering: The potential of remote mutations. Biochem. Soc. Trans. 2019, 47, 701–711. [Google Scholar] [CrossRef] [PubMed]
- McCarty, P.L.; Semprini, L. Ground-Water Treatment for Chlorinated Solvents. In Handbook of Bioremediation; CRC Press: Boca Raton, FL, USA, 1994. [Google Scholar]
- Matin, A. Starvation Promoters of Escherichia coli: Their Function, Regulation, and Use in Bioprocessing and Bioremediation. Ann. N. Y. Acad. Sci. 1994, 721, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.; Brugna, M.; Aubert, C.; Bernadac, A.; Bruschi, M. Enzymatic reduction of chromate: Comparative studies using sulfate-reducing bacteria. Key role of polyheme cytochromes c and hydrogenases. Appl. Microbiol. Biotechnol. 2001, 55, 95–100. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Measures |
|---|---|
| No. of amino acids | 185 |
| Mw | 20,604.39 |
| Theoretical pI | 7.81 |
| Negatively charged residues | 16 |
| Positively charged residues | 17 |
| Ext. coefficient | 30,940 |
| Estimated half-life | 30 h (mammalian reticulocytes, in vitro) |
| IA | 41.23 |
| AI | 82.27 |
| GRAVY | −0.262 |
| Webserver | Method | Result |
|---|---|---|
| MP3 (Prediction of Pathogenic Proteins in Metagenomic Datasets) | HMM | Non-pathogenic |
| Hybrid | Non-pathogenic | |
| SVM | Non-pathogenic | |
| VirulentPred | Based on Amino acid Composition | 0.8062 (Virulent) |
| Based on Dipeptide Composition | 0.0167 (Virulent) | |
| Based on Higher Order Dipeptide Composition | −0.186 (Non-virulent) | |
| Similarity-Based using PSI-BLAST | 0 No Hits obtained | |
| PSI-BLAST created PSSM Profiles | −0.017 (Non-Virulent) | |
| Cascade of SVMs and PSI-BLAST | 0.6832 (Virulent) | |
| VICMPred | Patterns + Compositions | −1.7423155 |
| Modeling Tool | Residues | ProQ | Global Quality Score | PSVS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| LG | Max Sub | Verify3D | ProsaII | MolProbity Clashscore | Most Favoured | Additionally Allowed | Generous Allowed | RMSD (Bond Angle) | RMSD (Bond Length) | ||
| Phyre2 | 178 | 8.45 | −0.35 | 0.20 | 0.62 | 88.47 | 92.6 | 4 | 3.4 | 1.4 | 0.011 |
| Geno3D | 174 | 7.96 | −0.35 | 0.19 | 0.49 | 27.86 | 87.1 | 7.6 | 5.3 | 0.9 | 0.004 |
| Robetta | 185 | 8.36 | −0.58 | 0.22 | 0.70 | 2.43 | 89.4 | 9.3 | 1.2 | 1.7 | 0.018 |
| IntFold | 185 | 7.79 | −0.37 | 0.20 | 0.66 | 96.53 | 93.4 | 2.7 | 3.8 | 4.0 | 0.030 |
| I-Tasser | 185 | 8.15 | −0.58 | 0.19 | 0.65 | 8.68 | 86.3 | 11.5 | 2.2 | 2.4 | 0.014 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tasleem, M.; Hussein, W.M.; El-Sayed, A.-A.A.A.; Alrehaily, A. Providencia alcalifaciens—Assisted Bioremediation of Chromium-Contaminated Groundwater: A Computational Study. Water 2023, 15, 1142. https://doi.org/10.3390/w15061142
Tasleem M, Hussein WM, El-Sayed A-AAA, Alrehaily A. Providencia alcalifaciens—Assisted Bioremediation of Chromium-Contaminated Groundwater: A Computational Study. Water. 2023; 15(6):1142. https://doi.org/10.3390/w15061142
Chicago/Turabian StyleTasleem, Munazzah, Wesam M. Hussein, Abdel-Aziz A. A. El-Sayed, and Abdulwahed Alrehaily. 2023. "Providencia alcalifaciens—Assisted Bioremediation of Chromium-Contaminated Groundwater: A Computational Study" Water 15, no. 6: 1142. https://doi.org/10.3390/w15061142