
Transcriptomic Analysis of Ulva prolifera in Response to Salt Stress

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Culture
2.2. Experimental Processing and Sample Collection
2.3. RNA Extraction and Library Construction
2.4. Transcript Splicing and Function Annotation
2.5. Identification and Enrichment Analysis of Differentially Expressed Genes (DEGs)
2.6. Validation of mRNA Expression Patterns by Quantitative Real-Time PCR (qRT-PCR)
2.7. Photosynthetic Parameter Determination
2.8. Measurement of Photosynthetic Oxygen Release and Respiratory Oxygen Consumption
2.9. Statistical Analysis
3. Results
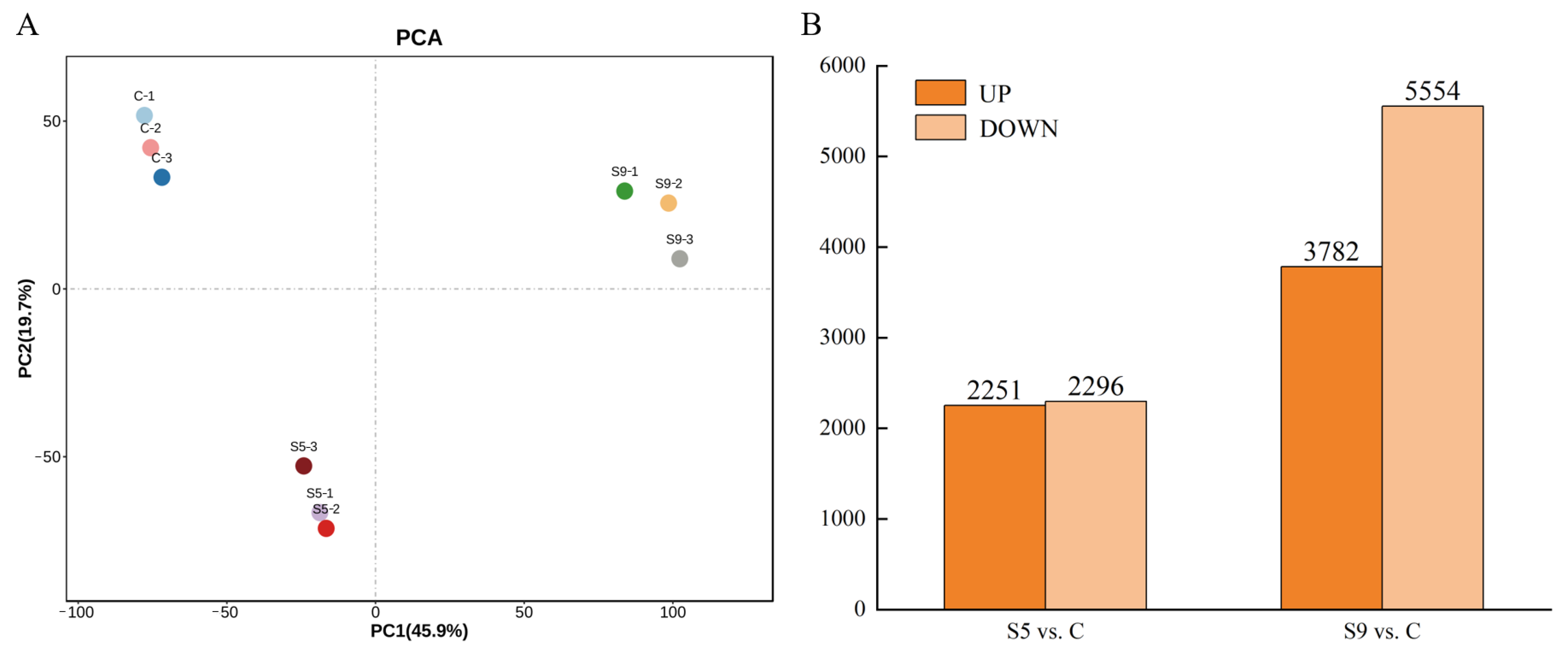
3.1. Transcriptome Sequencing Quality and Annotation Results
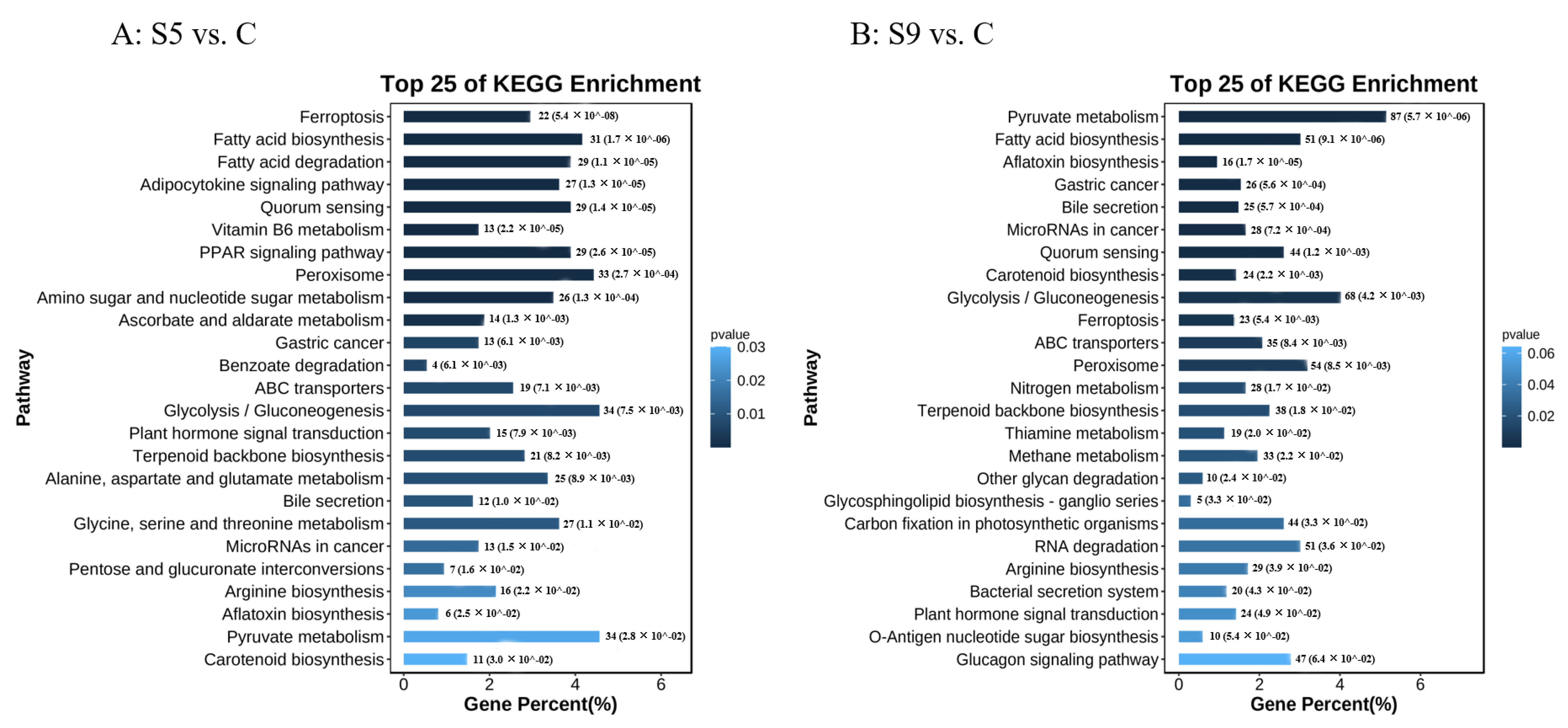
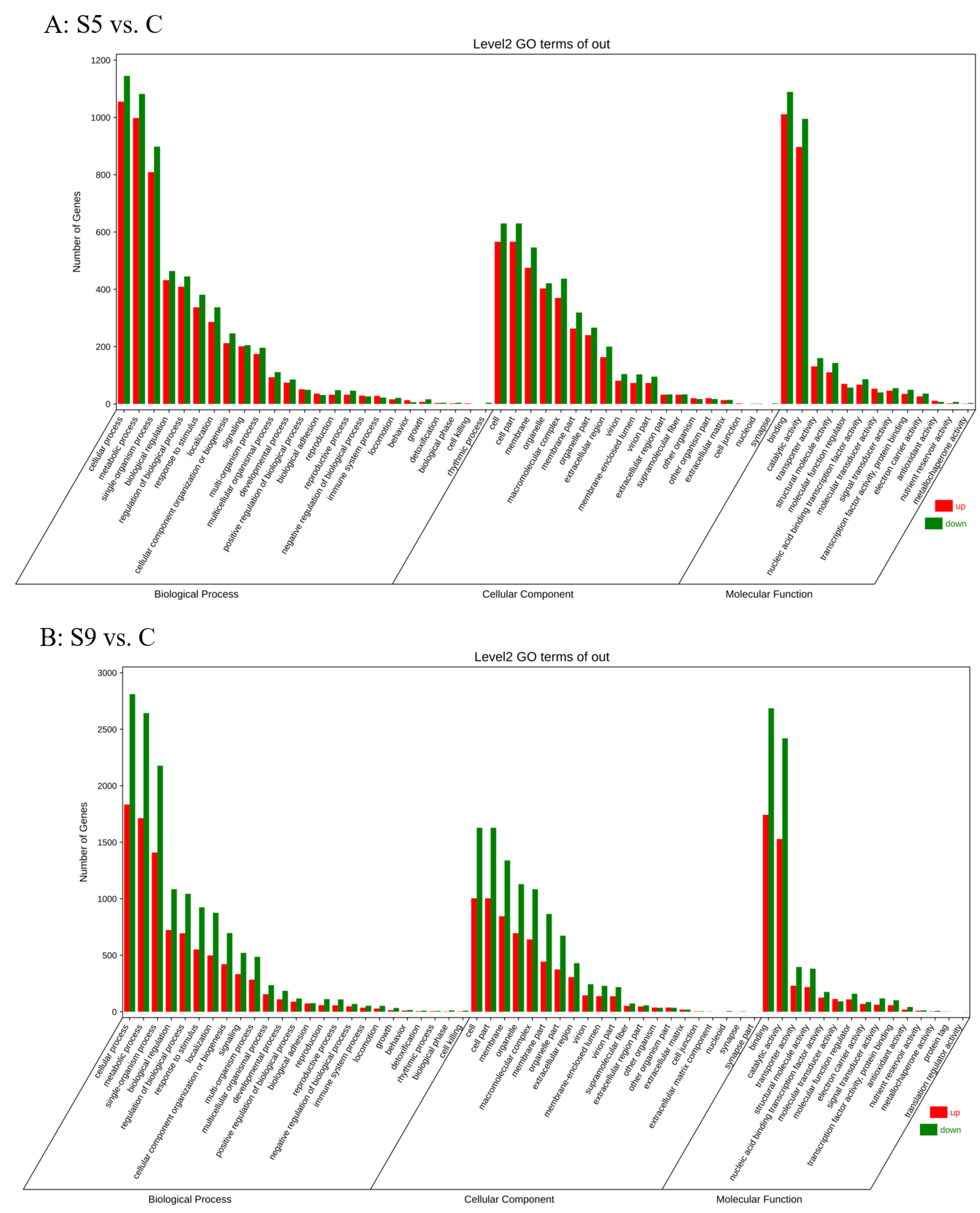
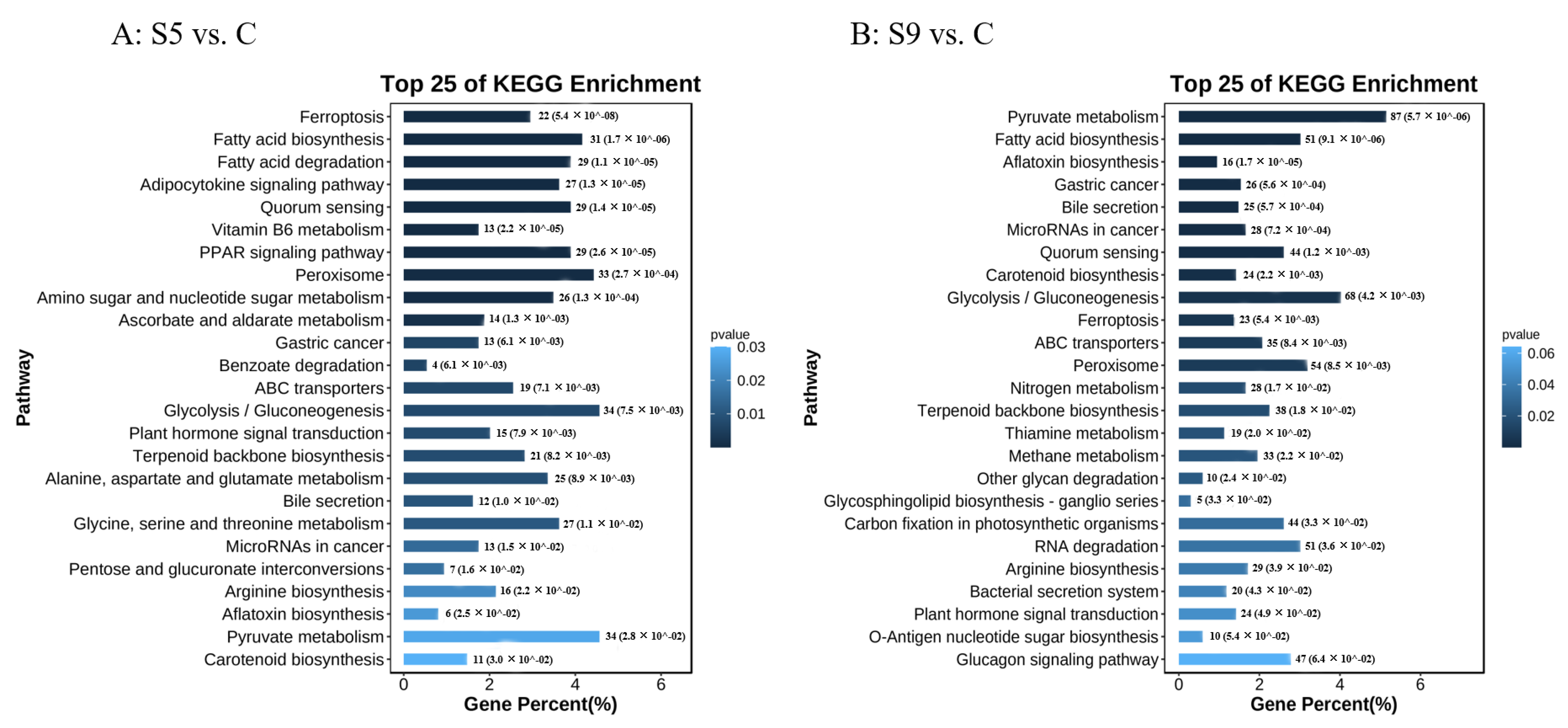
3.2. Screening and Enrichment Analysis of DEGs
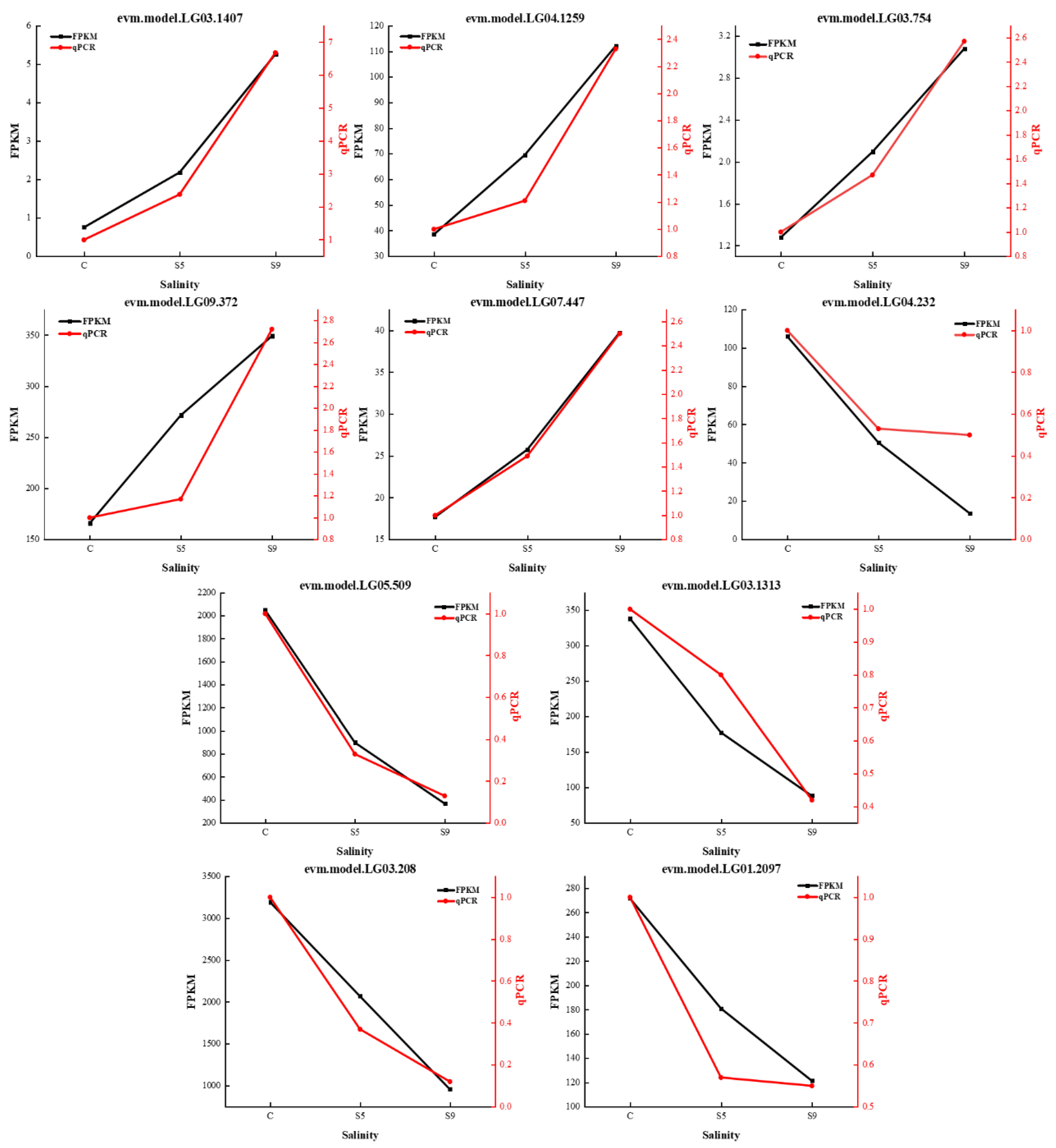
3.3. qRT-PCR Validation of Gene Expression Patterns
3.4. Resistance-Related Gene Expression
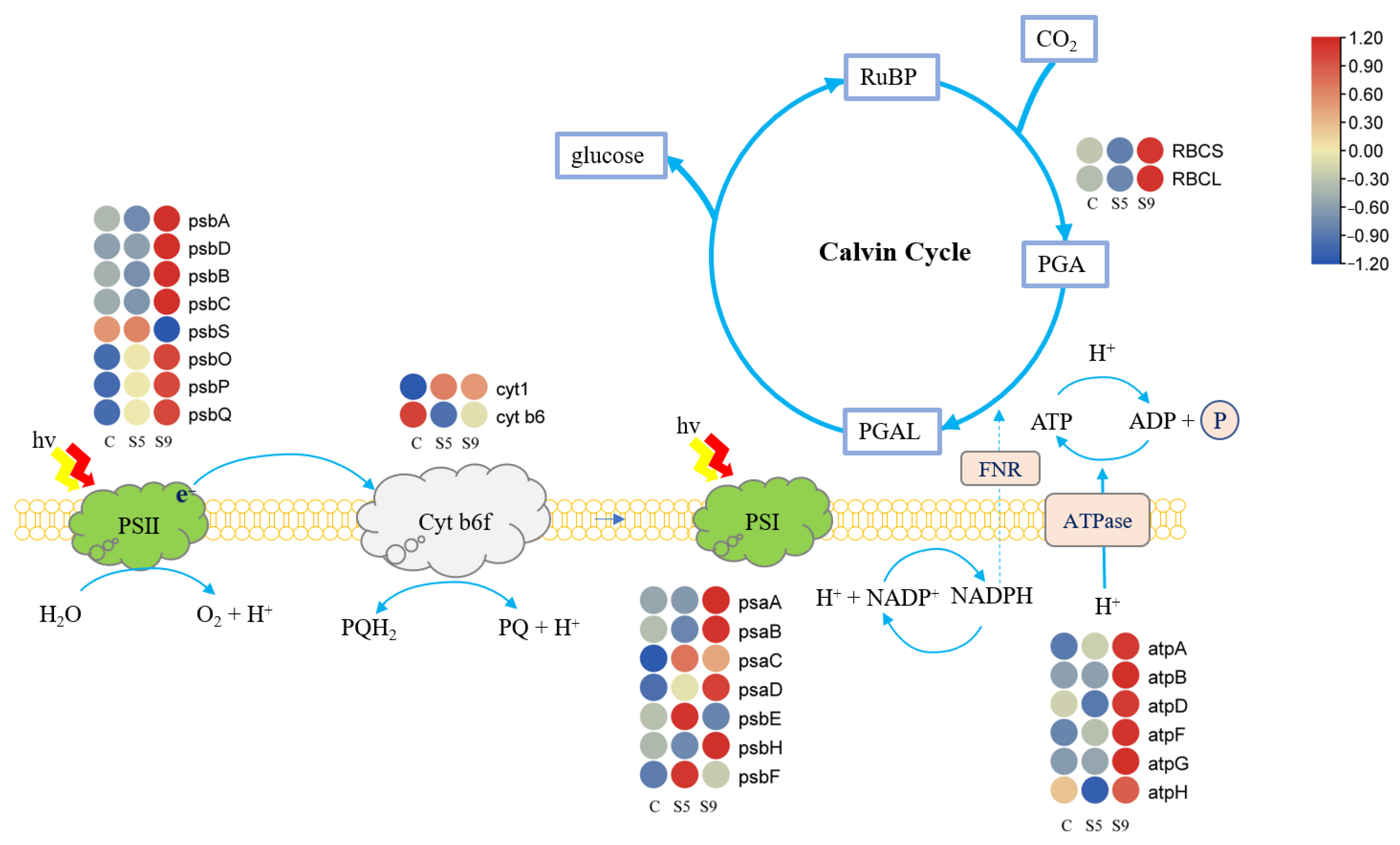
3.5. Expression of Genes Related to Photosynthesis and Carbon Fixation Pathways
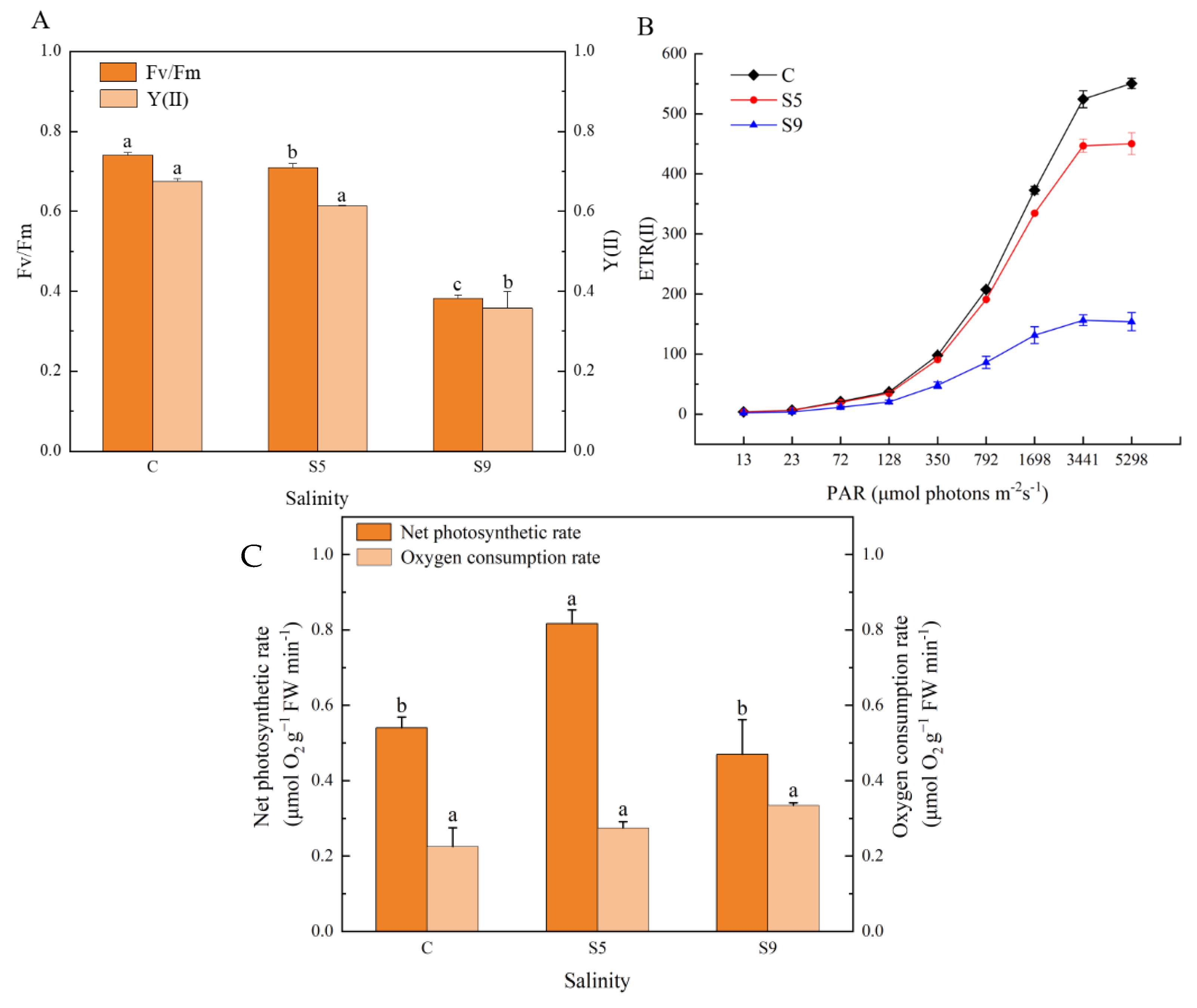
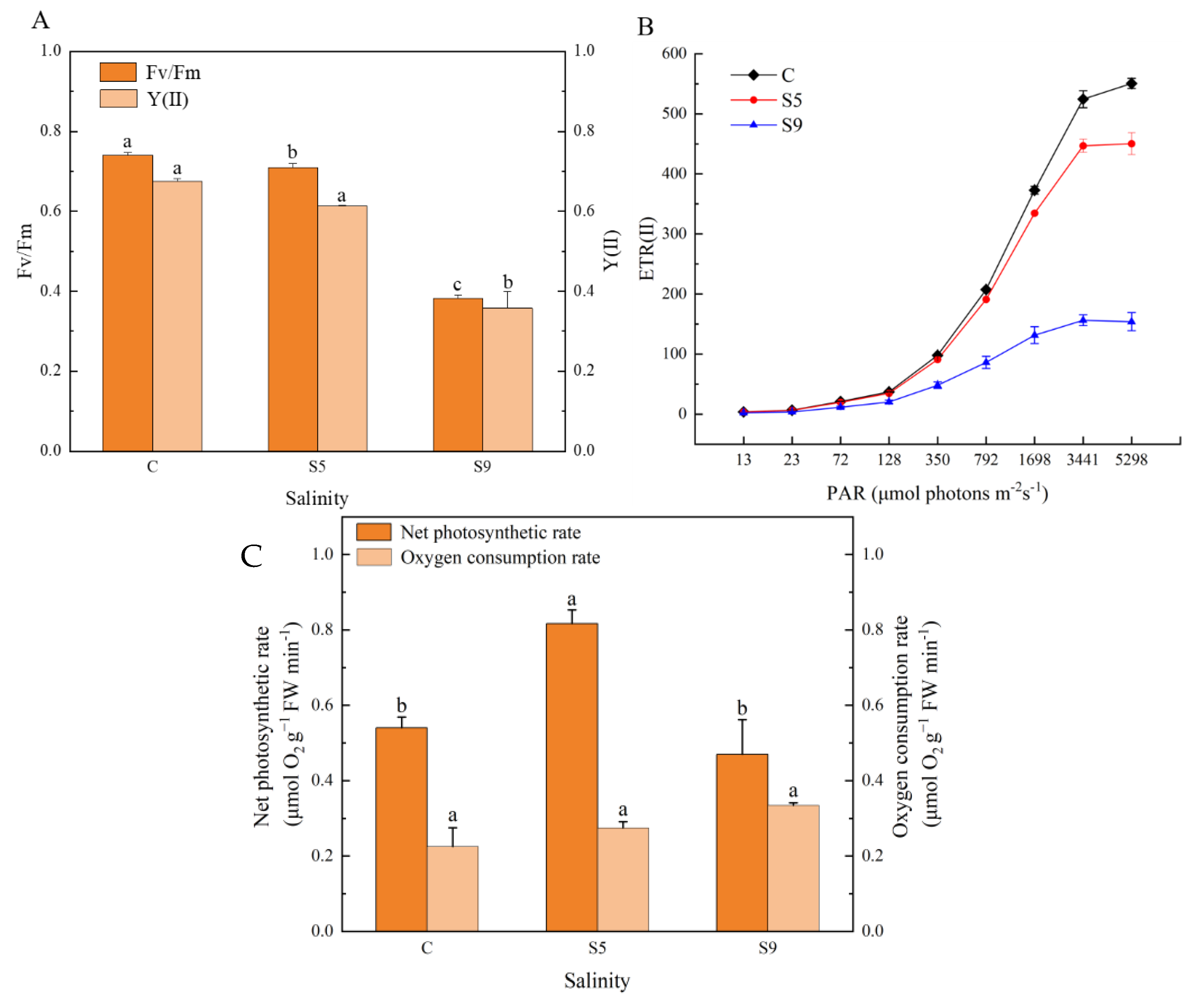
3.6. Changes in Physiological Activity, Photosynthetic Oxygen Release, and Respiratory Oxygen Consumption
4. Discussion
4.1. Downregulated Genes Increase Significant under Salt Stress
4.2. Expression of Resistance-Related Genes was Upregulated under Salt Stress
4.3. Photosynthetic Oxygen Release Rate Enhanced Significantly under Salt Stress
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Guidone, M.; Thornber, C.S. Examination of Ulva bloom species richness and relative abundance reveals two cryptically co-occurring bloom species in narragansett bay, rhode island. Harmful Algae 2013, 24, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Yu, R.; Zhou, M. Effects of the decomposing green macroalga Ulva (Enteromorpha) prolifera on the growth of four red-tide species. Harmful Algae 2012, 16, 12–19. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, X. Release and microbial degradation of dissolved organic matter (DOM) from the macroalgae Ulva prolifera. Mar. Pollut. Bull. 2017, 125, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, P.; Li, H.; Li, G.; Liu, J.; Jiao, F.; Zhang, J.; Huo, Y.; Shi, X.; Su, R.; et al. Ulva prolifera green-tide outbreaks and their environmental impact in the yellow sea, China. Natl. Sci. Rev. 2019, 6, 825–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Li, H.; Zhang, Z.; He, C.; Shi, Q.; Jiao, N.; Zhang, Y. Doc dynamics and bacterial community succession during long-term degradation of Ulva prolifera and their implications for the legacy effect of green tides on refractory DOC pool in seawater. Water Res. 2020, 185, 116268. [Google Scholar] [CrossRef]
- Liu, D.; Keesing, J.K.; Dong, Z.; Zhen, Y.; Di, B.; Shi, Y.; Fearns, P.; Shi, P. Recurrence of the world’s largest green-tide in 2009 in yellow sea, China: Porphyra yezoensis aquaculture rafts confirmed as nursery for macroalgal blooms. Mar. Pollut. Bull. 2010, 60, 1423–1432. [Google Scholar] [CrossRef]
- Cui, J.; Shi, J.; Zhang, J.; Wang, L.; Fan, S.; Xu, Z.; Huo, Y.; Zhou, Q.; Lu, Y.; He, P. Rapid expansion of Ulva blooms in the yellow sea, China through sexual reproduction and vegetative growth. Mar. Pollut. Bull. 2018, 130, 223–228. [Google Scholar] [CrossRef]
- Lin, A.; Shen, S.; Wang, G.; Yi, Q.; Qiao, H.; Niu, J.; Pan, G. Comparison of chlorophyll and photosynthesis parameters of floating and attached Ulva prolifera. J. Integr. Plant. Biol. 2011, 53, 25–34. [Google Scholar] [CrossRef]
- Choi, T.S.; Kang, E.J.; Kim, K.Y. Effect of salinity on growth and nutrient uptake of Ulva pertusa (Chlorophyta) from an eelgrass bed. Algae 2010, 25, 17–26. [Google Scholar] [CrossRef]
- Huan, L.; Xie, X.; Zheng, Z.; Sun, F.; Wu, S.; Li, M.; Gao, S.; Gu, W.; Wang, G. Positive correlation between PSI response and oxidative pentose phosphate pathway activity during salt stress in an intertidal macroalga. Plant Cell Physiol. 2014, 55, 1395–1403. [Google Scholar] [CrossRef]
- Jimenez, C.; Niell, F.X. Influence of high salinity and nitrogen limitation on package effect and C/N ratio in Dunaliella viridis. Hydrobiologia 2003, 492, 201–206. [Google Scholar] [CrossRef]
- Dan, A.; Hiraoka, M.; Ohno, M.; Critchley, A.T. Observations on the effect of salinity and photon fluence rate on the induction of sporulation and rhizoid formation in the green alga Enteromorpha prolifera (Muller) J. Agardh (Chlorophyta, Ulvales). Fish. Sci. 2002, 68, 1182–1188. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, X.; He, N.; Wang, X.; Zhu, P.; Ji, Z. Transcriptome profiling of the salt-stress response in the halophytic green alga Dunaliella salina. Plant Mol. Biol. Rep. 2019, 37, 421–435. [Google Scholar] [CrossRef]
- Wang, N.; Qian, Z.; Luo, M.; Fan, S.; Zhang, X.; Zhang, L. Identification of salt stress responding genes using transcriptome analysis in green alga Chlamydomonas reinhardtii. Int. J. Mol. Sci. 2018, 19, 3359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Q.; Bi, G.; Cao, M.; Belcour, A.; Aite, M.; Mo, Z.; Mao, Y. Comparative transcriptome analysis provides insights into response of Ulva compressa to fluctuating salinity conditions. J. Phycol. 2021, 57, 1295–1308. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillies, M.A.; Rau, A.; Aubert, J.; Hennequet-Antier, C.; Jeanmougin, M.; Servant, N.; Keime, C.; Marot, G.; Castel, D.; Estelle, J.; et al. A comprehensive evaluation of normalization methods for illumina high-throughput RNA sequencing data analysis. Brief. Bioinform. 2013, 14, 671–683. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Genty, B.; Briantais, J.M.; Baker, N.R. The relationship between the quantum yield of photosynthetic electron-transport and quenching of chlorophyll fluorescence. BBA Rev. Cancer 1989, 990, 87–92. [Google Scholar] [CrossRef]
- Deng, Y.; Srivastava, R.; Howell, S.H. Endoplasmic reticulum (ER) stress response and its physiological roles in plants. Int. J. Mol. Sci. 2013, 14, 8188–8212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Ming, T.; Zhou, J.; Lu, C.; Wang, R.; Su, X. The response and survival mechanisms of Staphylococcus aureus under high salinity stress in salted foods. Foods 2022, 11, 1503. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Saini, K.C.; Ojah, H.; Sahoo, R.; Gupta, K.; Kumar, A.; Bast, F. Abiotic stress in algae: Response, signaling and transgenic approaches. J. Appl. Phycol. 2022, 34, 1843–1869. [Google Scholar] [CrossRef]
- Perrineau, M.-M.; Zelzion, E.; Gross, J.; Price, D.C.; Boyd, J.; Bhattacharya, D. Evolution of salt tolerance in a laboratory reared population of Chlamydomonas reinhardtii. Environ. Microbiol. 2014, 16, 1755–1766. [Google Scholar] [CrossRef]
- Li, K.; Pang, C.H.; Ding, F.; Sui, N.; Feng, Z.T.; Wang, B.S. Overexpression of Suaeda salsa stroma ascorbate peroxidase in Arabidopsis chloroplasts enhances salt tolerance of plants. South Afr. J. Bot. 2012, 78, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Santangeli, M.; Capo, C.; Beninati, S.; Pietrini, F.; Forni, C. Gradual exposure to salinity improves tolerance to salt stress in rapeseed (Brassica napus L.). Water 2019, 11, 1667. [Google Scholar] [CrossRef] [Green Version]
- Ballinger, D.G.; Pardue, M.L. The control of protein-synthesis during heat-shock in Drosophila cells involves altered polypeptide elongation rates. Cell 1983, 33, 103–113. [Google Scholar] [CrossRef]
- Sugino, M.; Hibino, T.; Tanaka, Y.; Nii, N.; Takabe, T.; Takabe, T. Overexpression of Dnak from a halotolerant cyanobacterium Aphanothece halophytica acquires resistance to salt stress in transgenic tobacco plants. Plant Sci. 1999, 146, 81–88. [Google Scholar] [CrossRef]
- Klein, E.M.; Mascheroni, L.; Pompa, A.; Ragni, L.; Weimar, T.; Lilley, K.S.; Dupree, P.; Vitale, A. Plant endoplasmin supports the protein secretory pathway and has a role in proliferating tissues. Plant J. 2006, 48, 657–673. [Google Scholar] [CrossRef]
- Dittami, S.M.; Heesch, S.; Olsen, J.L.; Collen, J. Transitions between marine and freshwater environments provide new clues about the origins of multicellular plants and algae. J. Phycol. 2017, 53, 731–745. [Google Scholar] [CrossRef]
- Plaxton, W.C. The organization and regulation of plant glycolysis. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 185–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, Y.; Ho, S.H.; Vavricka, C.J.; Chang, J.S.; Hasunuma, T.; Kondo, A. Evolutionary engineering of salt-resistant Chlamydomonas sp. Strains reveals salinity stress-activated starch-to-lipid biosynthesis switching. Bioresour. Technol. 2017, 245, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Canora, D.D.; Guasch, L.L.; Zuazo, R.S. Species-specific responses of antarctic terrestrial microalgae to salinity stress. Comparative study in klebsormidium sp. and stigeoclonium sp. Czech Polar Rep. 2022, 12, 89–102. [Google Scholar]
- Guo, J.; Li, Y.; Han, G.; Song, J.; Wang, B. Nacl markedly improved the reproductive capacity of the euhalophyte Suaeda salsa. Funct. Plant Biol. 2018, 45, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lu, Y.; Jiang, J.G. Comparative analysis on the key enzymes of the glycerol cycle metabolic pathway in Dunaliella salina under osmotic stresses. PLoS ONE 2012, 7, e37578. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Forward Primer (5′–3′) | Reverse Primer (5′–3′) |
|---|---|---|
| evm.model.LG03.754 | GCGAAGGACAGAGCACAGTAAGG | CGTTGCCATTCTTGCCGTTGTTG |
| evm.model.LG07.447 | CAGCCGACGCAGGTCTCAAAG | CCTCGTGATGTGCCAGCCAAC |
| evm.model.LG09.372 | CCGCAGACATCATTGACCAGGAG | TAGCAGAACCGCCGCAACATC |
| evm.model.LG03.1407 | CGTGAGGCGTTCCTGGCTTTC | TGGCTGTCTGCTGCTGTCATTG |
| evm.model.LG04.1259 | GACGATCACGACAAGCAGGG | CATCTCATCGGCCTTGTCGG |
| evm.model.LG04.232 | CCTCATCGCCTTCATCGCCTTC | CGGTGGGAGCAACAGTAACGAC |
| evm.model.LG05.509 | GGAGGAGCCAGAGGAGGATGAC | TCCTCACGGGTTGTCTCCACTTC |
| evm.model.LG03.1313 | GCGAGGCGGTACGAAGTCAAAC | CGGTCGGAGGATACAGGGTGAG |
| evm.model.LG01.2097 | ACAGTTATTACGACGCCGATGACC | ACTCATCCGCCTCCTCATCCAC |
| evm.model.LG03.208 | GCAGGAACTCCTTCAGCACGAC | GATGACGGAGGACGAGAAGTTTGAC |
| evm.model.LG05.203 (β-actin) | AGGATGCATACGTTGGTGAA | TTGTGGTGCCAAATCTTCTC |
| Sample | Total Reads | Total Bases | Clean Reads | Clean Bases | Q20 (%) | Q30 (%) | GC (%) | Uniquely Mapped | Total Mapped |
|---|---|---|---|---|---|---|---|---|---|
| C-1 | 48,022,138 | 7,203,320,700 | 47,717,318 | 7,146,882,951 | 97.63 | 93.51 | 61.23 | 89.07% | 91.64% |
| C-2 | 55,044,484 | 8,256,672,600 | 54,388,580 | 8,139,529,615 | 96.81 | 91.78 | 61.36 | 87.09% | 89.62% |
| C-3 | 59,523,928 | 8,928,589,200 | 58,841,366 | 8,810,555,804 | 96.79 | 91.76 | 61.63 | 87.70% | 90.24% |
| S5-1 | 56,877,658 | 8,531,648,700 | 56,247,326 | 8,421,268,782 | 96.80 | 91.76 | 61.82 | 87.75% | 90.17% |
| S5-2 | 58,866,622 | 8,829,993,300 | 58,041,962 | 8,685,862,641 | 96.25 | 90.60 | 61.55 | 86.98% | 89.41% |
| S5-3 | 53,921,832 | 8,088,274,800 | 53,250,108 | 7,967,943,016 | 94.69 | 86.74 | 61.74 | 86.22% | 88.51% |
| S9-1 | 53,983,414 | 8,097,512,100 | 53,259,220 | 7,973,724,974 | 96.47 | 91.12 | 61.66 | 86.23% | 88.84% |
| S9-2 | 56,064,434 | 8,409,665,100 | 55,380,048 | 8,288,100,283 | 96.68 | 91.55 | 61.41 | 87.27% | 90.04% |
| S9-3 | 61,928,180 | 9,289,227,000 | 61,146,286 | 9,152,944,506 | 96.68 | 91.57 | 61.61 | 83.99% | 86.51% |
| Gene ID | KEGG Ortholog | Relative Change Multiple | ||
|---|---|---|---|---|
| C | S5 | S9 | ||
| evm.model.LG04.1209 | K09562 HSPBP1; hsp70-interacting protein | 1.00 | 0.92 | 1.40 |
| evm.model.LG08.120 | K13993 HSP20; hsp20 family protein | 1.00 | 1.53 | 2.60 |
| evm.model.LG02.2352 | K09553 STIP1; stress-induced-phosphoprotein 1 | 1.00 | 1.18 | 1.38 |
| evm.model.LG04.1314 | K09490 HSPA5, BIP; heat shock 70kDa protein 5 | 1.00 | 1.08 | 1.43 |
| evm.model.LG01.496 | K07374 TUBA; tubulin alpha | 1.00 | 0.97 | 1.22 |
| evm.model.LG04.1142 | K14498 SNRK2; serine/threonine-protein kinase | 1.00 | 1.01 | 1.50 |
| evm.model.LG02.1115 | K10588 UBE3B; ubiquitin-protein ligase E3 B | 1.00 | 1.36 | 1.17 |
| evm.model.LG01.457 | K00432 E1.11.1.9; glutathione peroxidase | 1.00 | 1.37 | 1.32 |
| evm.model.LG03.168 | K00026 MDH2; malate dehydrogenase | 1.00 | 1.10 | 1.17 |
| evm.model.LG02.856 | K04564 SOD2; superoxide dismutase, Fe-Mn family | 1.00 | 1.24 | 1.64 |
| evm.model.LG02.1983 | K00383 GSR, gor; glutathione reductase (NADPH) | 1.00 | 1.22 | 1.48 |
| evm.model.LG03.1372 | K01919 gshA; glutamate-cysteine ligase | 1.00 | 1.56 | 3.54 |
| evm.model.LG01.1003 | K05928 E2.1.1.95; tocopherol O-methyltransferase | 1.00 | 0.90 | 1.02 |
| evm.model.LG04.832 | K00434 E1.11.1.11; L-ascorbate peroxidase | 1.00 | 1.37 | 1.80 |
| evm.model.LG01.1168 | K03781 katE, CAT, catB, srpA; catalase | 1.00 | 1.22 | 1.40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuo, J.; Wang, H.; Du, Y.; Shi, M.; Huan, L.; Wang, G. Transcriptomic Analysis of Ulva prolifera in Response to Salt Stress. Water 2023, 15, 63. https://doi.org/10.3390/w15010063
Zhuo J, Wang H, Du Y, Shi M, Huan L, Wang G. Transcriptomic Analysis of Ulva prolifera in Response to Salt Stress. Water. 2023; 15(1):63. https://doi.org/10.3390/w15010063
Chicago/Turabian StyleZhuo, Jintao, Hong Wang, Yifei Du, Mengmeng Shi, Li Huan, and Guangce Wang. 2023. "Transcriptomic Analysis of Ulva prolifera in Response to Salt Stress" Water 15, no. 1: 63. https://doi.org/10.3390/w15010063