
Bioremediation of Chromium-Contaminated Groundwater Using Chromate Reductase from Pseudomonas putida: An In Silico Approach

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Analysis of ChrR Sequence
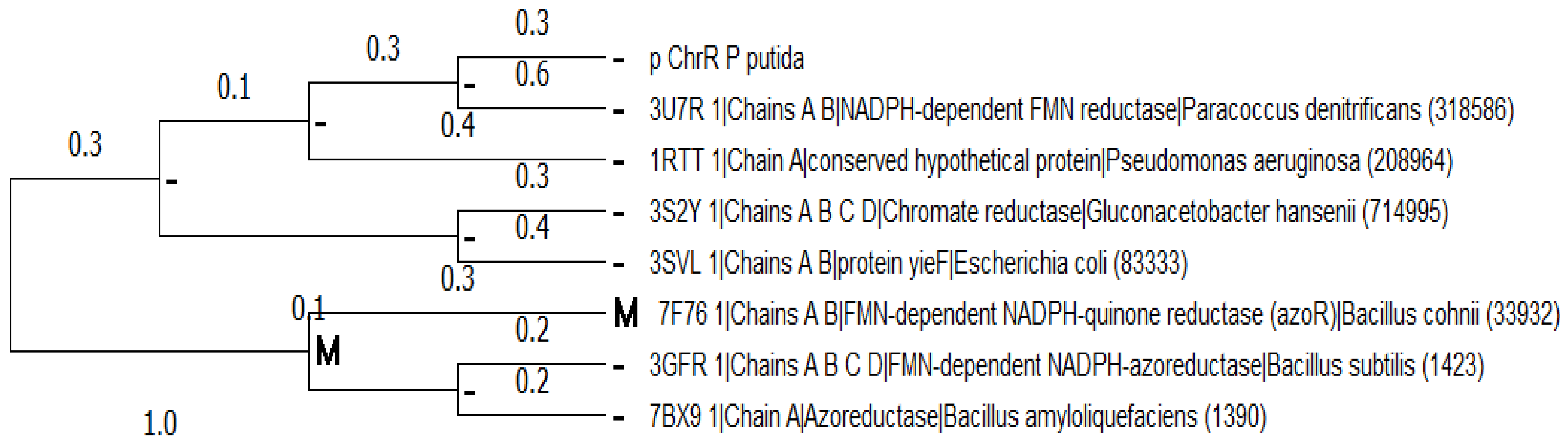
2.2. Multiple Sequence Alignment and Evolutionary Analysis
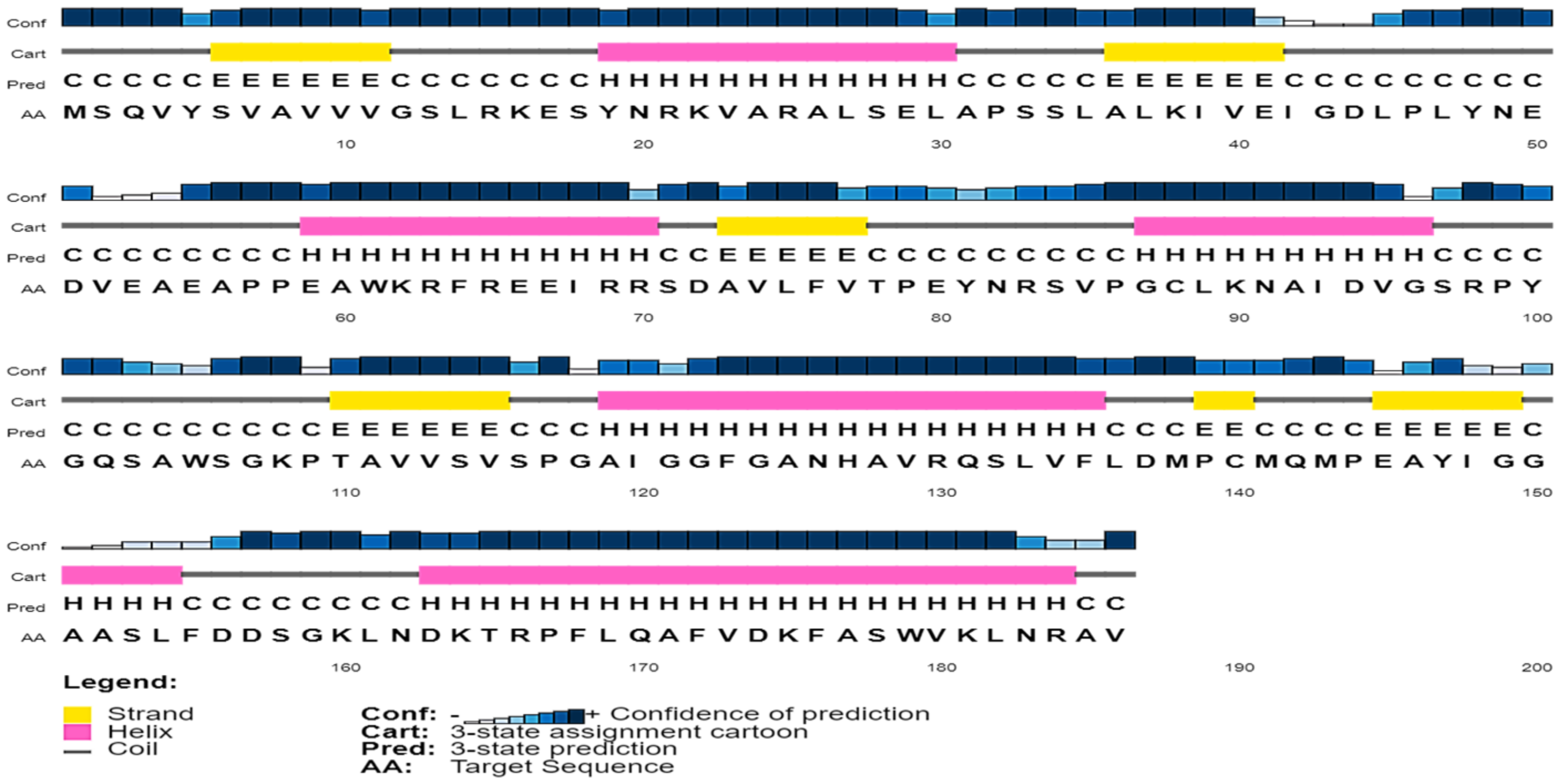
2.3. Secondary Structure Assessment
2.4. Three-Dimensional Structure Generation
2.5. Three-Dimensional Model Generation and Optimization
2.6. Validation of ChrR Three-Dimensional Structure
2.7. Molecular Docking and Interaction Studies
2.8. In Silico Mutant Generation and Interaction with Cr(VI)
3. Results
3.1. Sequence Analysis
3.2. Multiple Sequence Alignment and Evolutionary Analysis
3.3. Conserved Regions in the Sequence
3.4. Secondary Structure Content in the Sequence
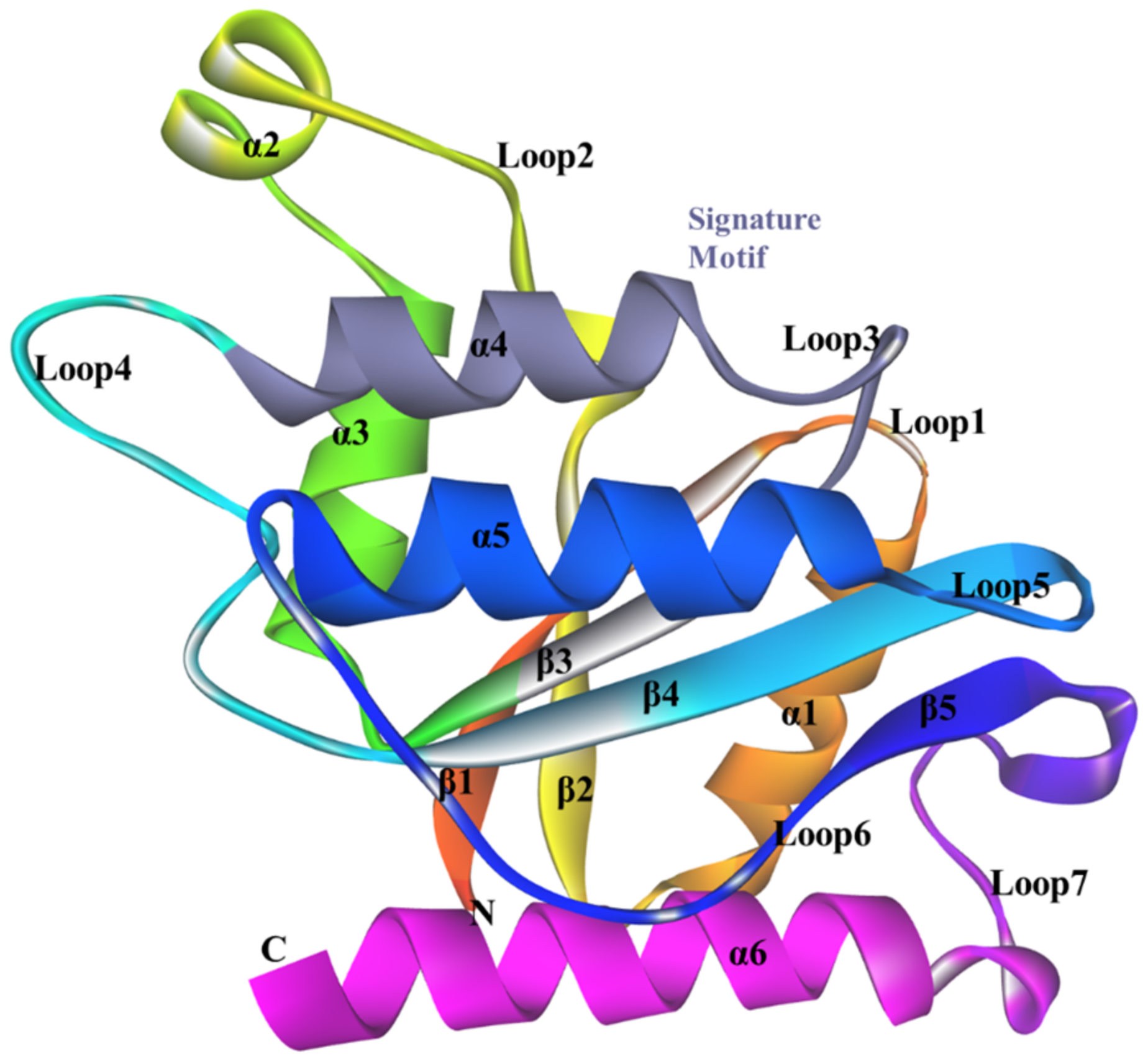
3.5. Structure of ChrR from P. putida
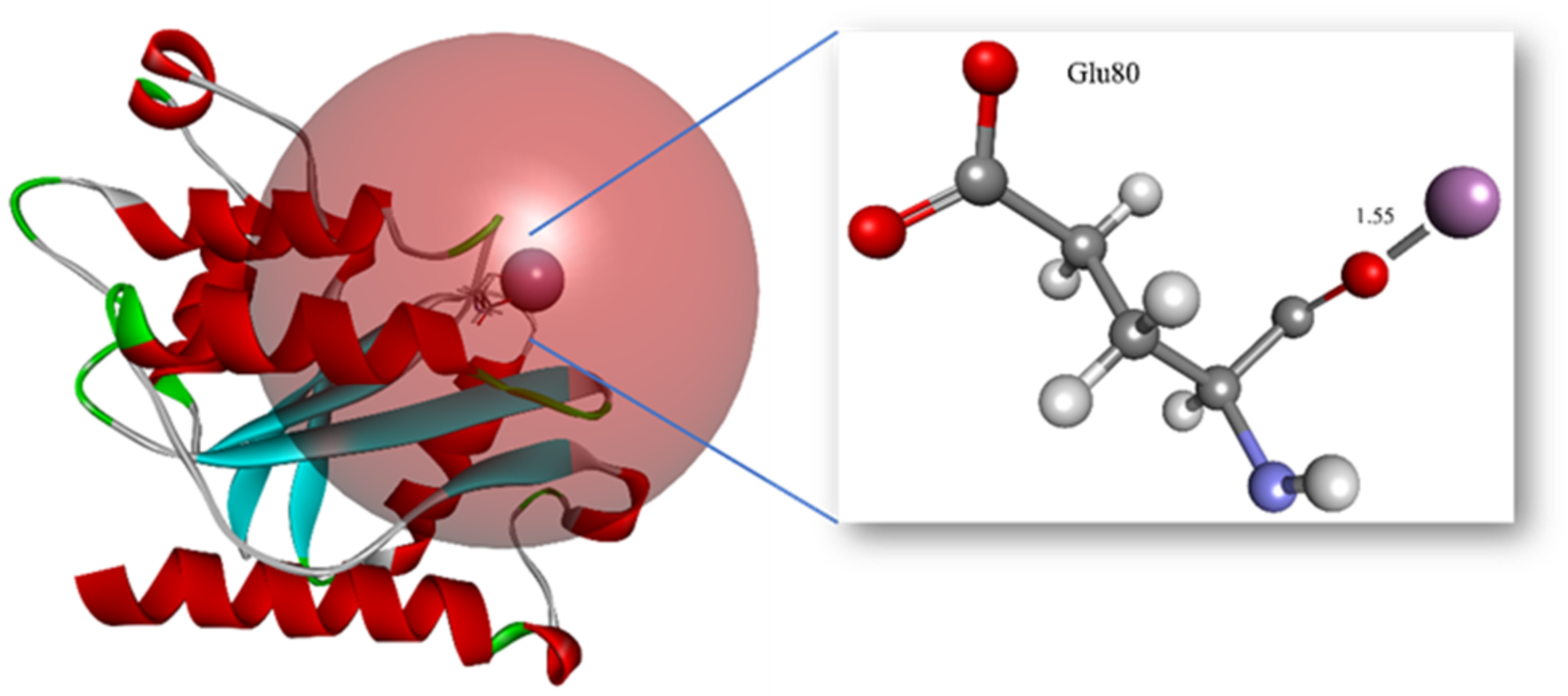
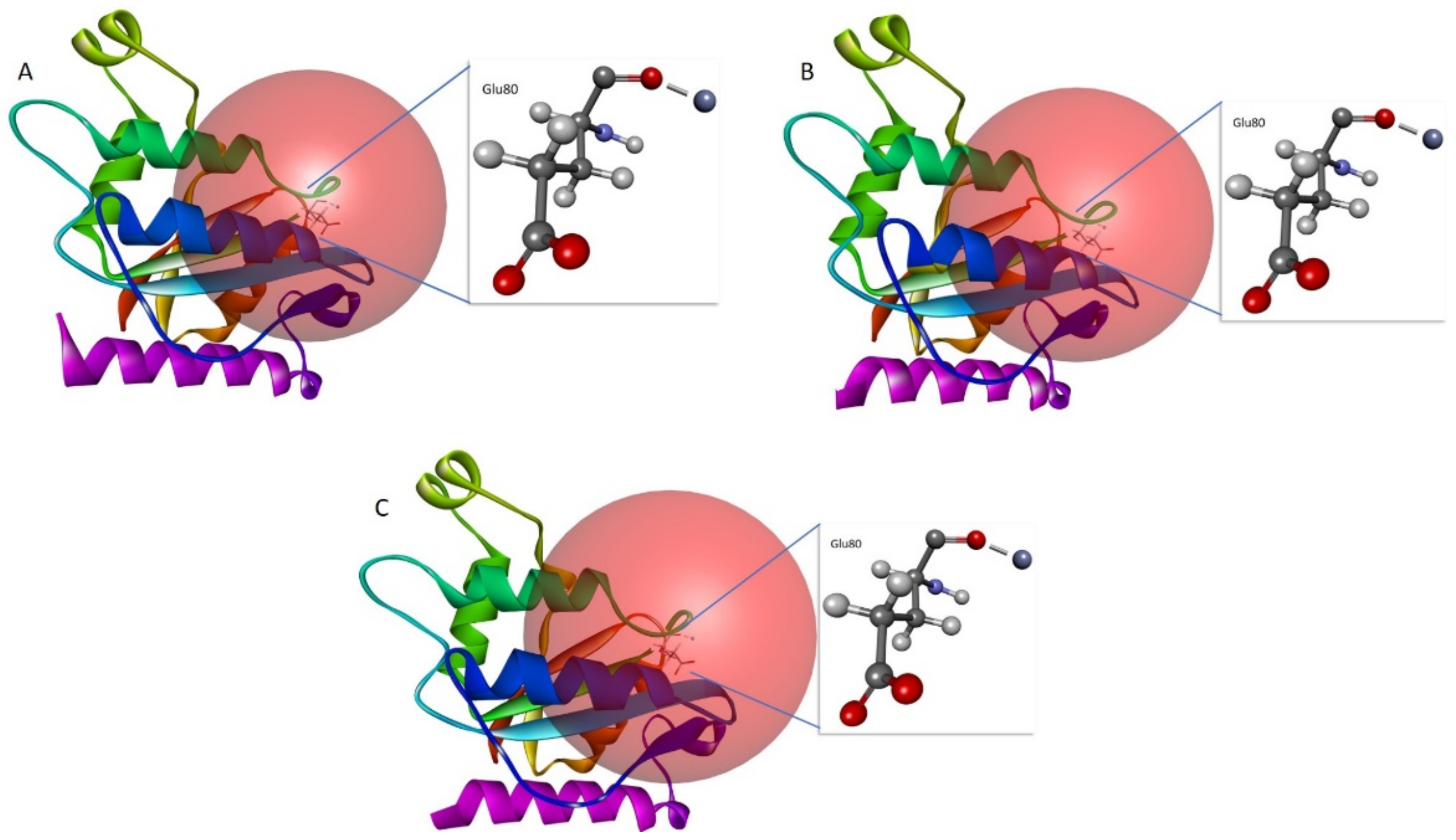
3.6. In Silico Site-Directed Mutagenesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ali, I.; Hasan, M.A.; Alharbi, O.M.L. Toxic metal ions contamination in the groundwater, Kingdom of Saudi Arabia. J. Taibah Univ. Sci. 2020, 14, 1571–1579. [Google Scholar] [CrossRef]
- Abdulrahman, M. Seawater desalination: The strategic choice for Saudi Arabia. Desalination Water Treat. 2012, 51, 1–4. [Google Scholar]
- Corteel, C.; Dini, A.; Deyhle, A. Element and isotope mobility during water–rock interaction processes. Phys. Chem. Earth Parts A/B/C 2005, 30, 993–996. [Google Scholar] [CrossRef]
- Krishnaraj, R.N.; Samanta, D.; Kumar, A.; Sani, R. Bioprospecting of Thermostable Cellulolytic Enzymes through Modeling and Virtual Screening Method. Can. J. Biotechnol. 2017, 1, 19–25. [Google Scholar] [CrossRef]
- Jan, A.T.; Azam, M.; Siddiqui, K.; Ali, A.; Choi, I.; Haq, Q.M.R. Heavy metals and human health: Mechanistic insight into toxicity and counter defense system of antioxidants. Int. J. Mol. Sci. 2015, 16, 29592–29630. [Google Scholar] [CrossRef]
- Maghraby, M.; Nasr, O.; Hamouda, M. Quality assessment of groundwater at south Al Madinah Al Munawarah area, Saudi Arabia. Environ. Earth Sci. 2013, 70, 1525–1538. [Google Scholar] [CrossRef]
- Jaishankar, M.; Tseten, T.; Anbalagan, N.; Mathew, B.B.; Beeregowda, K.N. Toxicity, mechanism and health effects of some heavy metals. Interdiscip. Toxicol. 2014, 7, 60–72. [Google Scholar] [CrossRef]
- Alharbi, B.H.; Pasha, M.J.; Al-Shamsi, M.A.S. Influence of Different Urban Structures on Metal Contamination in Two Metropolitan Cities. Sci. Rep. 2019, 9, 4920. [Google Scholar] [CrossRef]
- Bucher, J.R. NTP toxicity studies of sodium dichromate dihydrate (CAS No. 7789-12-0) administered in drinking water to male and female F344/N rats and B6C3F1 mice and male BALB/c and am3-C57BL/6 mice. Toxic. Rep. Ser. 2007, 7, g1–g4. [Google Scholar]
- Ryan, R.P.; Monchy, S.; Cardinale, M.; Taghavi, S.; Crossman, L.; Avison, M.B.; Berg, G.; Van Der Lelie, D.; Dow, J.M. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 2009, 7, 514–525. [Google Scholar] [CrossRef]
- Das, S.; Raj, R.; Mangwani, N.; Dash, H.R.; Chakraborty, J. 2-Heavy Metals and Hydrocarbons: Adverse Effects and Mechanism of Toxicity. In Microbial Biodegradation and Bioremediation; Das, S., Ed.; Elsevier: Oxford, UK, 2014; pp. 23–54. [Google Scholar]
- Zhitkovich, A. Chromium in Drinking Water: Sources, Metabolism, and Cancer Risks. Chem. Res. Toxicol. 2011, 24, 1617–1629. [Google Scholar] [CrossRef] [PubMed]
- Dmytrenko, G.M.; Ereshko, T.V.; Konovalova, V.V. Reduction of Chromium (Vi) by Bacteria Collection Strains of Different Physiological Groups. In Bioremediation of Soils Contaminated with Aromatic Compounds; Springer: Dordrecht, The Netherlands, 2007; Volume 76, pp. 125–130. [Google Scholar]
- Elkarmi, A.Z.; Abu-Elteen, K.H.; Khader, M.A. Modeling the Biodegradation Efficiency and Growth of Pseudomonas Alcaligenes Utilizing 2,4-Dichlorophenol as a Carbon Source Pre- and Post-Exposure to UV Radiation. Jordan J. Biol. Sci. 2008, 1, 7–11. [Google Scholar]
- Francis, A.J.; Spanggord, R.J.; Ouchi, G.I.; Bramhall, R.; Bohonos, N. Metabolism of DDT analogues by a Pseudomonas sp. Appl. Environ. Microbiol. 1976, 32, 213–216. [Google Scholar] [CrossRef]
- Das, S.; Dash, H.R.; Chakraborty, J. Genetic basis and importance of metal resistant genes in bacteria for bioremediation of contaminated environments with toxic metal pollutants. Appl. Microbiol. Biotechnol. 2016, 100, 2967–2984. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2010, 38, D5–D16. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Gupta, D. VirulentPred: A SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinform. 2008, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Raghava, G. VICMpred: An SVM-based Method for the Prediction of Functional Proteins of Gram-negative Bacteria Using Amino Acid Patterns and Composition. Genom. Proteom. Bioinform. 2006, 4, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Kapil, R.; Dhakan, D.B.; Sharma, V.K. MP3: A Software Tool for the Prediction of Pathogenic Proteins in Genomic and Metagenomic Data. PLoS ONE 2014, 9, e93907. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.V.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2018, 47, D309–D314. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.X.; Chen, L.; Di Costanzo, L.; Christie, C.; Duarte, J.M.; Dutta, S.; Feng, Z.K.; et al. Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019, 47, D520–D528. [Google Scholar]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Sillitoe, I.; Bordin, N.; Dawson, N.; Waman, V.P.; Ashford, P.; Scholes, H.M.; Pang, C.S.M.; Woodridge, L.; Rauer, C.; Sen, N.; et al. CATH: Increased structural coverage of functional space. Nucleic Acids Res. 2020, 49, D266–D273. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed]
- Geer, L.Y.; Domrachev, M.; Lipman, D.J.; Bryant, S.H. CDART: Protein Homology by Domain Architecture. Genome Res. 2002, 12, 1619–1623. [Google Scholar] [CrossRef]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent updates, new developments and status in 2020. Nucleic Acids Res. 2020, 49, D458–D460. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Watson, J.D.; Thornton, J. ProFunc: A server for predicting protein function from 3D structure. Nucleic Acids Res. 2005, 33, W89–W93. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Gotoh, O. Multiple sequence alignment: Algorithms and applications. Adv. Biophys. 1999, 36, 159–206. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple Sequence Alignment Using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2002, in press. [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J.; Bryson, K.; Jones, D.T.J.B. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Pieper, U.; Webb, B.M.; Dong, G.Q.; Schneidman-Duhovny, D.; Fan, H.; Kim, S.J.; Khuri, N.; Spill, Y.G.; Weinkam, P.; Hammel, M.; et al. ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 2013, 42, D336–D346. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Wallner, B.; Elofsson, A. Can correct protein models be identified? Protein Sci. 2003, 12, 1073–1086. [Google Scholar] [CrossRef]
- Morris, A.L.; MacArthur, M.W.; Hutchinson, E.G.; Thornton, J. Stereochemical quality of protein structure coordinates. Proteins Struct. Funct. Bioinform. 1992, 12, 345–364. [Google Scholar] [CrossRef]
- Shen, M.-Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar]
- Paul, M.; Pranjaya, P.P.; Thatoi, H. In silico studies on structural, functional, and evolutionary analysis of bacterial chromate reductase family responsible for high chromate bioremediation efficiency. SN Appl. Sci. 2020, 2, 1997. [Google Scholar] [CrossRef]
- Jin, H.; Zhang, Y.; Buchko, G.W.; Varnum, S.M.; Robinson, H.; Squier, T.C.; Long, P.E. Structure Determination and Functional Analysis of a Chromate Reductase from Gluconacetobacter hansenii. PLoS ONE 2012, 7, e42432. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, J.K.; Law, S.M.; Brooks, C.L., 3rd. Flexible CDOCKER: Development and application of a pseudo-explicit structure-based docking method within CHARMM. J. Comput. Chem. 2015, 37, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Eswaramoorthy, S.; Poulain, S.; Hienerwadel, R.; Bremond, N.; Sylvester, M.D.; Zhang, Y.-B.; Berthomieu, C.; Van Der Lelie, D.; Matin, A.C. Crystal Structure of ChrR—A Quinone Reductase with the Capacity to Reduce Chromate. PLoS ONE 2012, 7, e36017. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Gonzalez, C.F.; Ackerley, D.; Lynch, S.V.; Matin, A.C. ChrR, a Soluble Quinone Reductase of Pseudomonas putida That Defends against H2O2. J. Biol. Chem. 2005, 280, 22590–22595. [Google Scholar] [CrossRef]
- Sedláček, V.; Klumpler, T.; Marek, J.; Kucera, I. The Structural and Functional Basis of Catalysis Mediated by NAD(P)H:acceptor Oxidoreductase (FerB) of Paracoccus denitrificans. PLoS ONE 2014, 9, e96262. [Google Scholar] [CrossRef]
- Hanukoglu, I. Proteopedia: Rossmann fold: A beta-alpha-beta fold at dinucleotide binding sites. Biochem. Mol. Biol. Educ. 2015, 43, 206–209. [Google Scholar] [CrossRef]
- Abeln, S.; Feenstra, K.A.; Heringa, J. Protein Three-Dimensional Structure Prediction. In Encyclopedia of Bioinformatics and Computational Biology; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Mazoch, J.; Tesarík, R.; Sedlácek, V.; Kucera, I.; Turánek, J. Isolation and biochemical characterization of two soluble iron(III) reductases from Paracoccus denitrificans. JBIC J. Biol. Inorg. Chem. 2004, 271, 553–562. [Google Scholar] [CrossRef]
- Ackerley, D.F.; Gonzalez, C.F.; Park, C.H.; Blake, R.; Keyhan, M.; Matin, A. Chromate-Reducing Properties of Soluble Flavoproteins from Pseudomonas putida and Escherichia coli. Appl. Environ. Microbiol. 2004, 70, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Wilding, M.; Hong, N.; Spence, M.; Buckle, A.; Jackson, C.J. Protein engineering: The potential of remote mutations. Biochem. Soc. Trans. 2019, 47, 701–711. [Google Scholar] [CrossRef] [PubMed]
- McCarty, P.L.; Semprini, L. Ground-Water Treatment for Chlorinated Solvents. In Handbook of Bioremediation; CRC Press: Boca Raton, FL, USA, 2017; pp. 87–116. [Google Scholar]
- Matin, A. Starvation Promoters of Escherichia coli: Their Function, Regulation, and Use in Bioprocessing and Bioremediation. Ann. N. Y. Acad. Sci. 1994, 721, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.; Brugna, M.; Aubert, C.; Bernadac, A.; Bruschi, M. Enzymatic reduction of chromate: Comparative studies using sulfate-reducing bacteria. Appl. Microbiol. Biotechnol. 2001, 55, 95–100. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Measures |
|---|---|
| No. of amino acids | 186 |
| Molecular weight | 20,282.20 |
| Theoretical pI | 8.53 |
| Total number of negatively charged residues (Asp + Glu) | 20 |
| Total number of positively charged residues (Arg + Lys) | 22 |
| Ext. coefficient | 25,565 |
| Estimated half-life | 30 h (mammalian reticulocytes, in vitro) |
| Instability index (II) | 53.81 |
| Aliphatic index | 86.51 |
| Grand average of hydropathicity (GRAVY) | −0.101 |
| Tool | Prediction Approach | Prediction |
|---|---|---|
| VirulentPred | Amino Acid Composition-Based | 0.4223 (Non-virulent) |
| Dipeptide Composition-Based | −0.647 (Non-virulent) | |
| Higher-Order Dipeptide Composition-Based | −0.542 | |
| Similarity-Based using PSI-BLAST | 0 No hits obtained | |
| PSI-BLAST created PSSM Profiles | 0.6117 (Virulent) | |
| Cascade of SVMs and PSI-BLAST | 0.2162 (Non-virulent) | |
| MP3 | HMM | Non-pathogenic |
| Hybrid | Non-pathogenic | |
| SVM | Pathogenic | |
| VICMPred | Patterns + Compositions | −3.0806466 Metabolism molecule |
| Tool | Domain | Residues |
|---|---|---|
| SMART | FMN_red | 5–153 |
| Flavodxin_2 | 6–143 | |
| Pfam | FMN_red | 5–153 |
| CDD | SsuE (NAD(P)H-dependent FMN reductase) | 6–177 |
| CDART | SsuE (NAD(P)H-dependent FMN reductase) | 3–188 |
| InterProScan | FMN_rdtase-like | 7–150 |
| FMN_red | 7–150 |
| Modeling Tool | Residues | ProQ | Verify Protein | PROCHECK | |||||
|---|---|---|---|---|---|---|---|---|---|
| LG | Max Sub | MODELER | Profile 3D | Most Favored | Additionally Allowed | Generous Allowed | Dis Allowed Region | ||
| Phyre2 | 4–183 | −0.31 | 8.58 | −20,023.46 | 87.74 | 87.6 | 9.8 | 2.6 | 0.0 |
| ModWeb | 4–183 | −0.31 | 8.2 | −20,425.416 | 87.97 | 91.5 | 7.8 | 0.7 | 0.0 |
| SwissModel | 1–186 | −0.36 | 7.67 | −20,981.69 | 86.28 | 92.5 | 4.4 | 0.6 | 1.3 |
| Discovery Studio | 185 | −0.604 | 8.5 | −19,997.08 | 80.85 | 77.25 | 20.3 | 1.3 | 1.3 |
| I-Tasser | 1–186 | −0.55 | 8.28 | −21,941.75 | 84.23 | 75.5 | 22.0 | 2.5 | 0.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tasleem, M.; El-Sayed, A.-A.A.A.; Hussein, W.M.; Alrehaily, A. Bioremediation of Chromium-Contaminated Groundwater Using Chromate Reductase from Pseudomonas putida: An In Silico Approach. Water 2023, 15, 150. https://doi.org/10.3390/w15010150
Tasleem M, El-Sayed A-AAA, Hussein WM, Alrehaily A. Bioremediation of Chromium-Contaminated Groundwater Using Chromate Reductase from Pseudomonas putida: An In Silico Approach. Water. 2023; 15(1):150. https://doi.org/10.3390/w15010150
Chicago/Turabian StyleTasleem, Munazzah, Abdel-Aziz A. A. El-Sayed, Wesam Mekawy Hussein, and Abdulwahed Alrehaily. 2023. "Bioremediation of Chromium-Contaminated Groundwater Using Chromate Reductase from Pseudomonas putida: An In Silico Approach" Water 15, no. 1: 150. https://doi.org/10.3390/w15010150
APA StyleTasleem, M., El-Sayed, A.-A. A. A., Hussein, W. M., & Alrehaily, A. (2023). Bioremediation of Chromium-Contaminated Groundwater Using Chromate Reductase from Pseudomonas putida: An In Silico Approach. Water, 15(1), 150. https://doi.org/10.3390/w15010150