
Effects of Dry and Wet Negev Soil–Dust Deposition on the Induction of Autoxidation of Soil–Dust Lipid Components
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and Chemical Characterization of the Dust Analogue Material
2.2. Major and Trace Metal Composition of the Loessic Soil
2.3. Incubations in Fresh Water
2.4. Treatment
2.5. Silylation
2.6. Gas Chromatography–Electron Ionization Quadrupole Time-of-Flight Mass Spectrometry (GC-QTOF)
2.7. Gas Chromatography/Tandem Mass Spectrometry
2.8. Metal Analyses
3. Results and Discussion
3.1. Composition of Dust Analogue
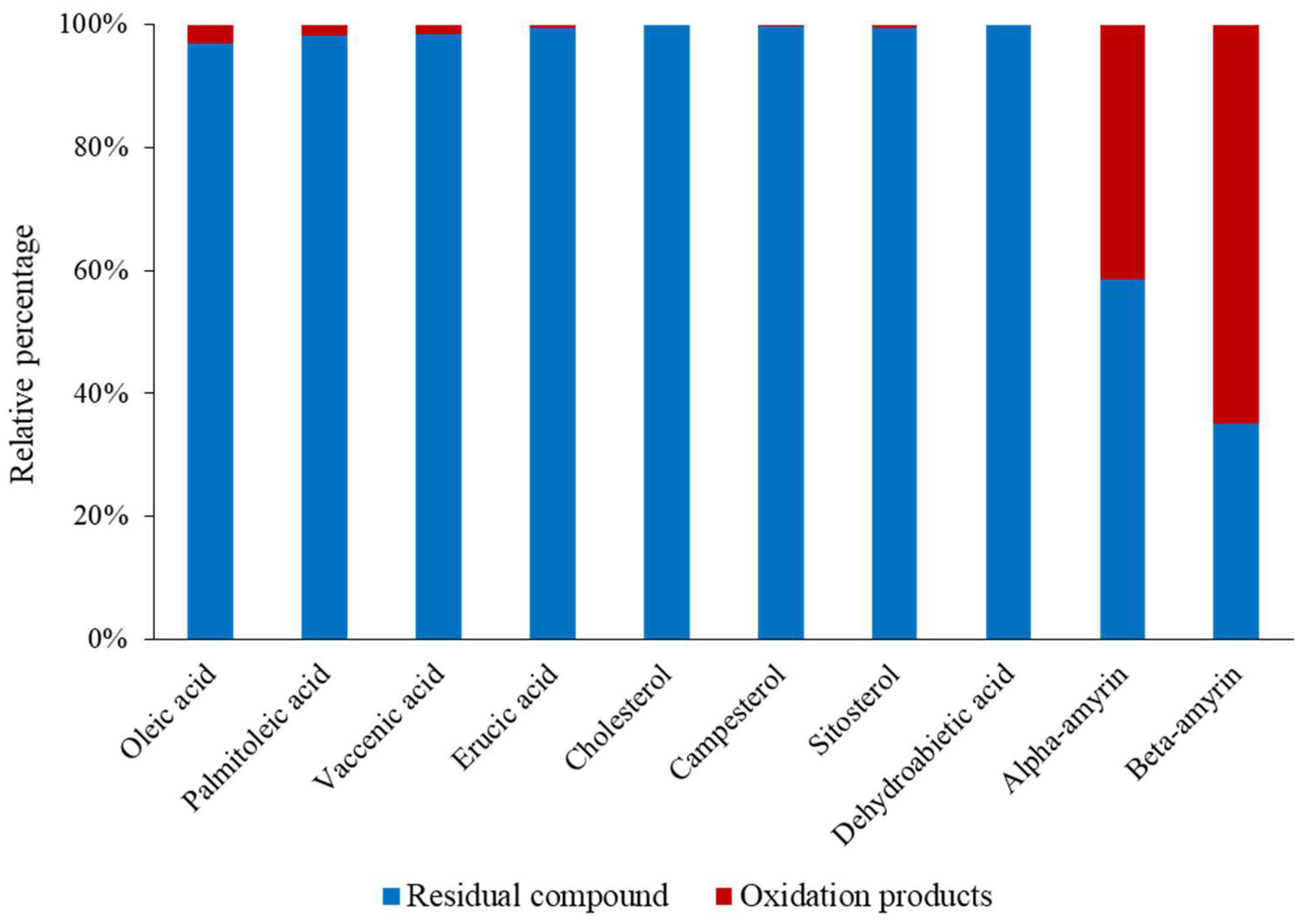
3.1.1. Organic Composition
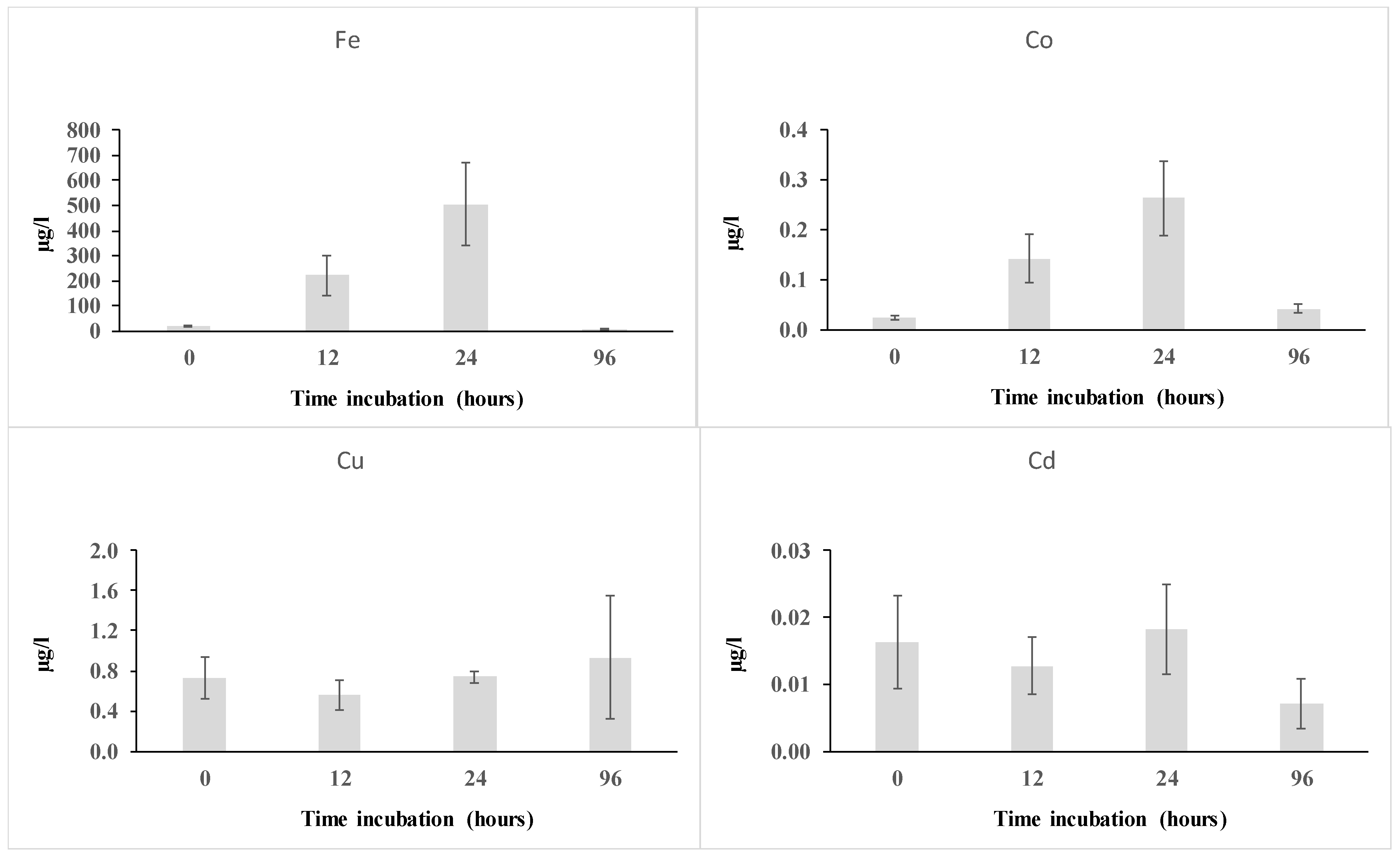
3.1.2. Metal Concentrations
3.2. Incubations of Dust Analogue in Fresh Water
3.2.1. Metal Dissolution
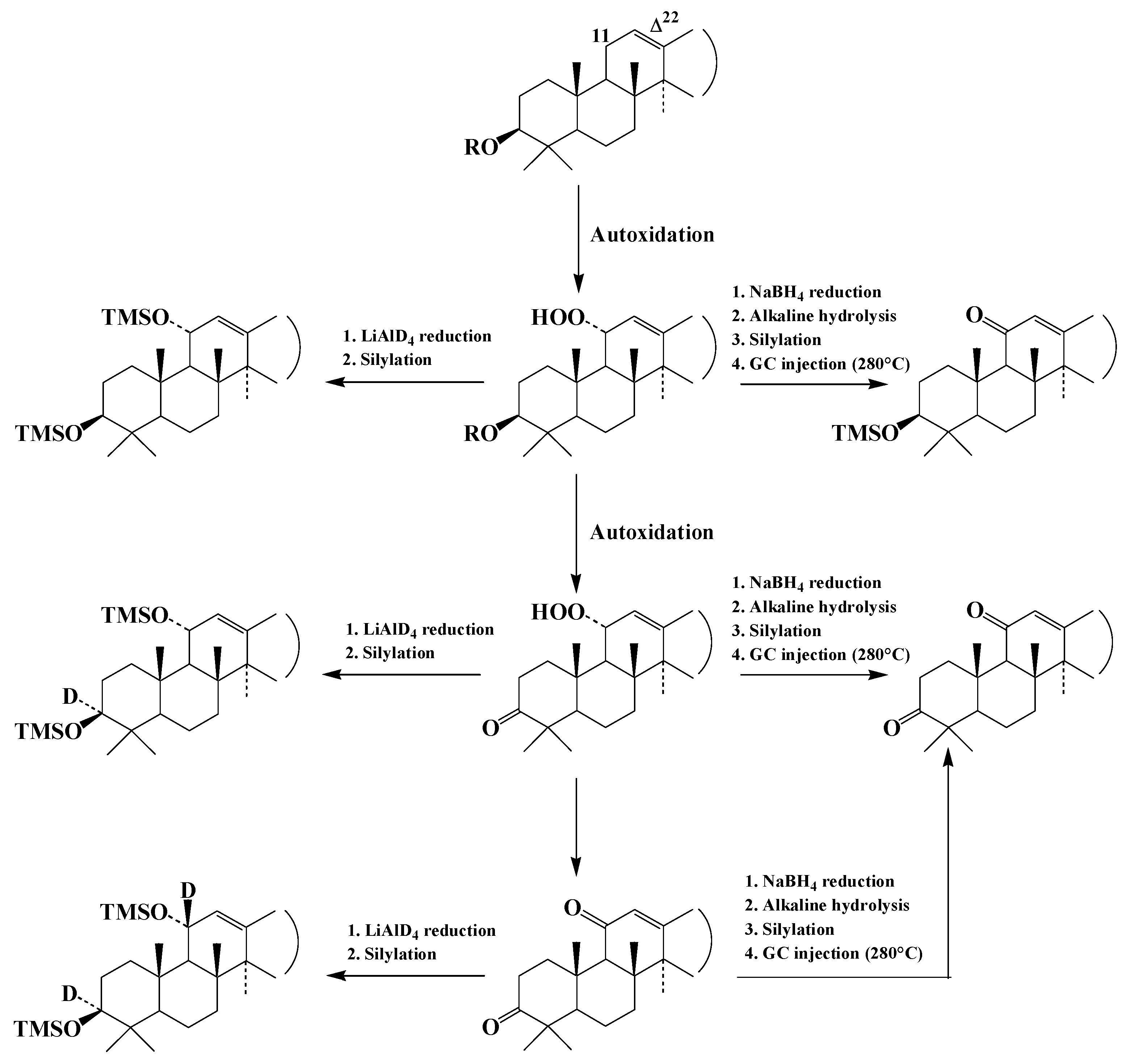
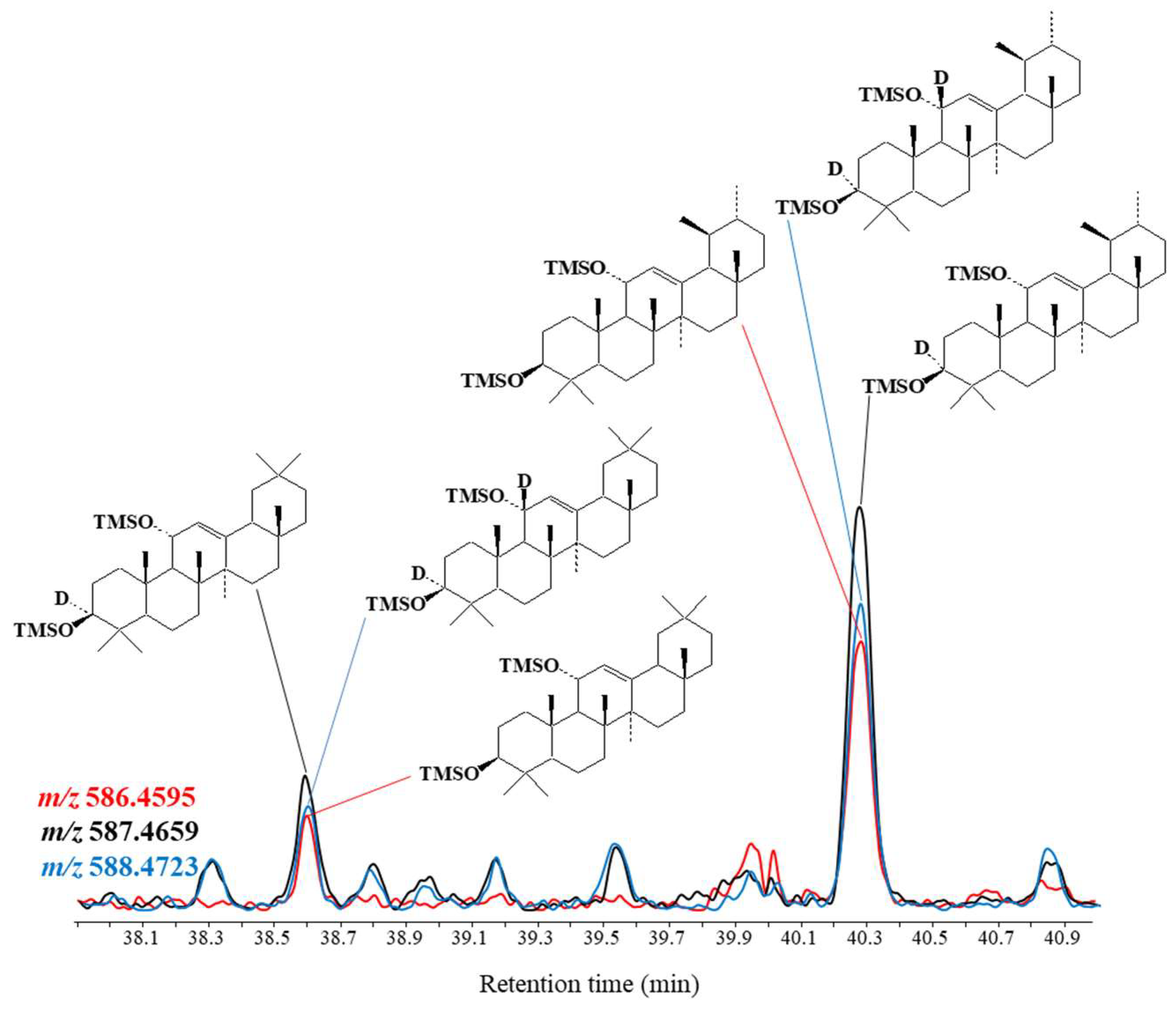
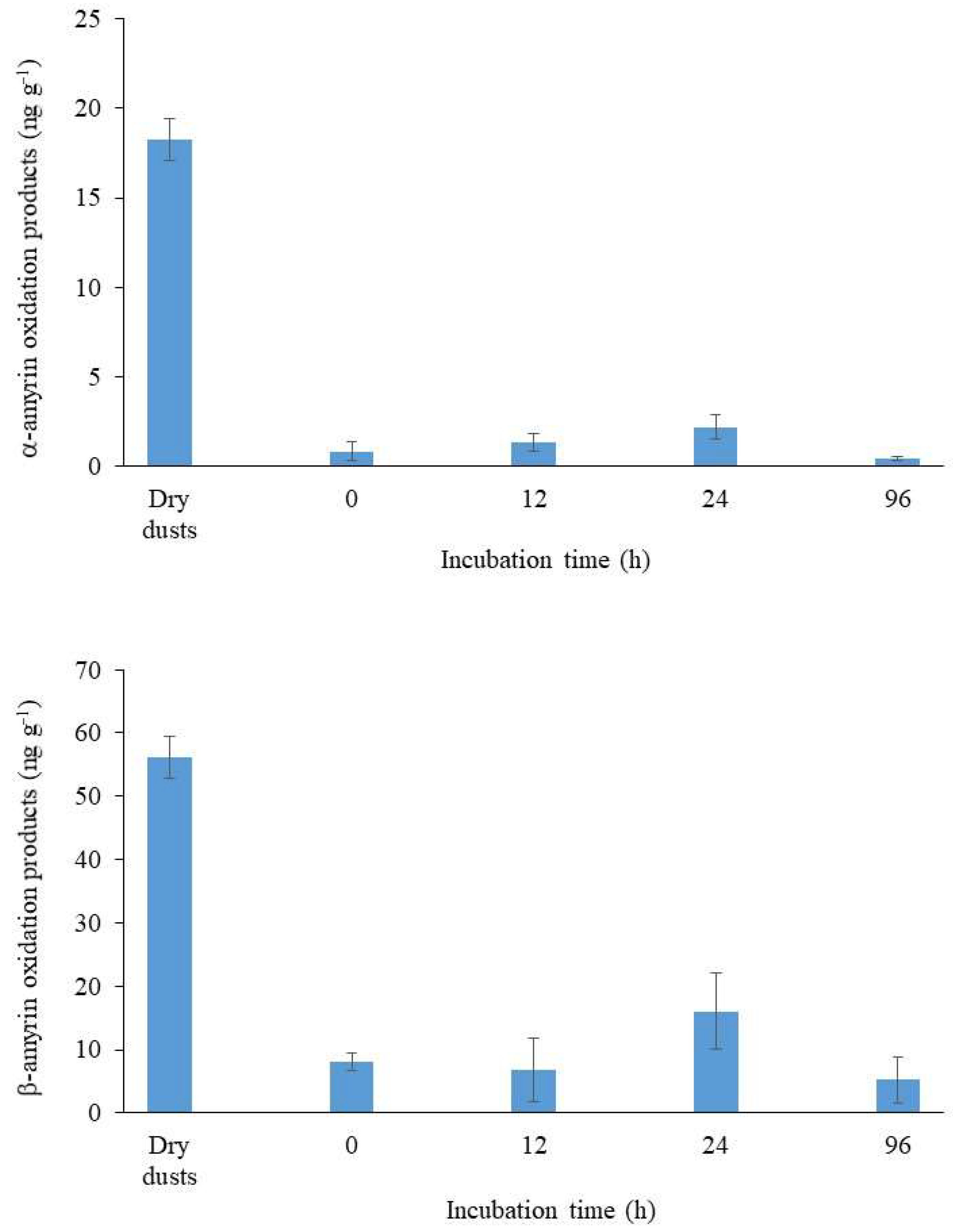
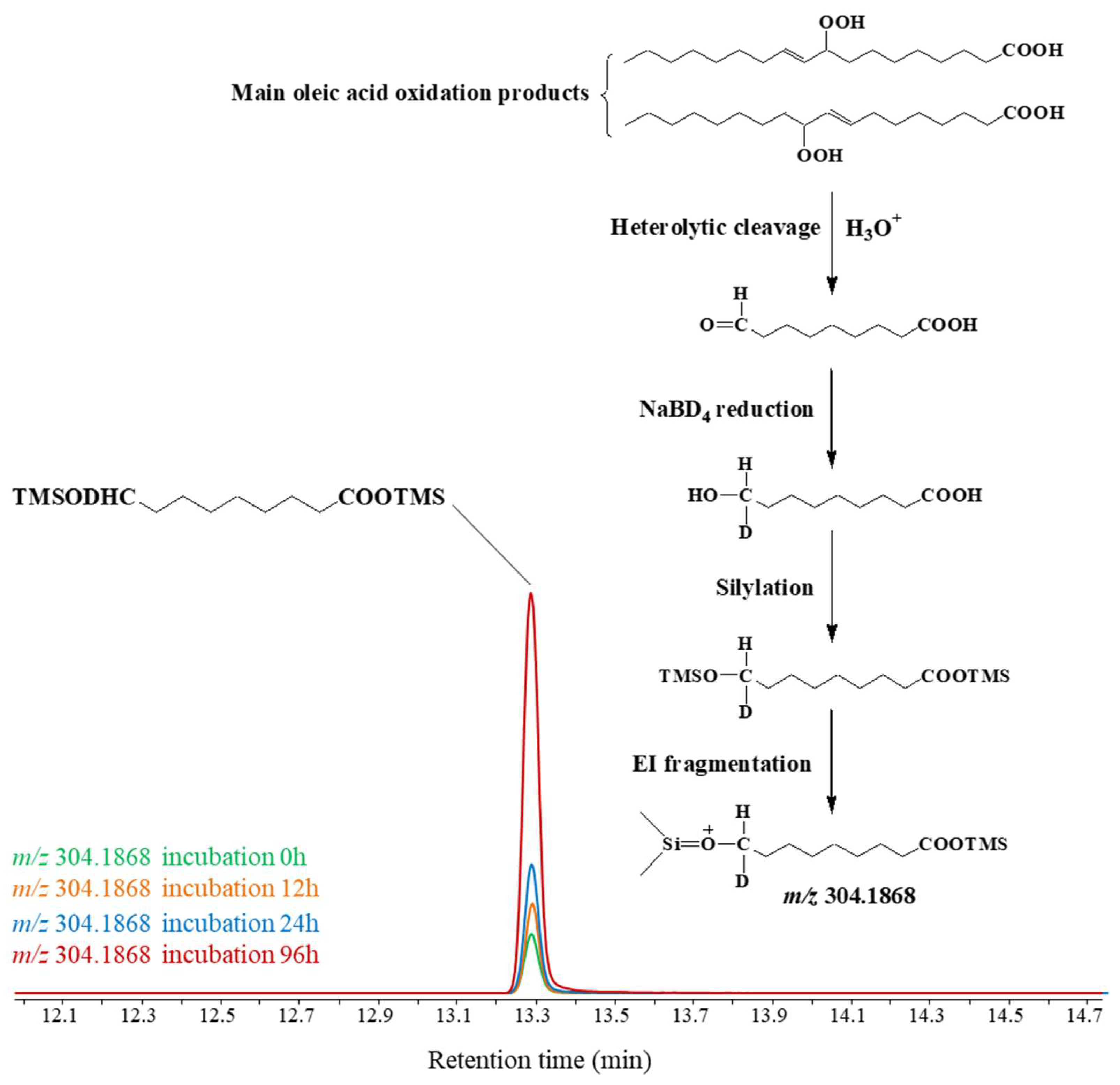
3.2.2. Impact on Lipid Oxidation Products
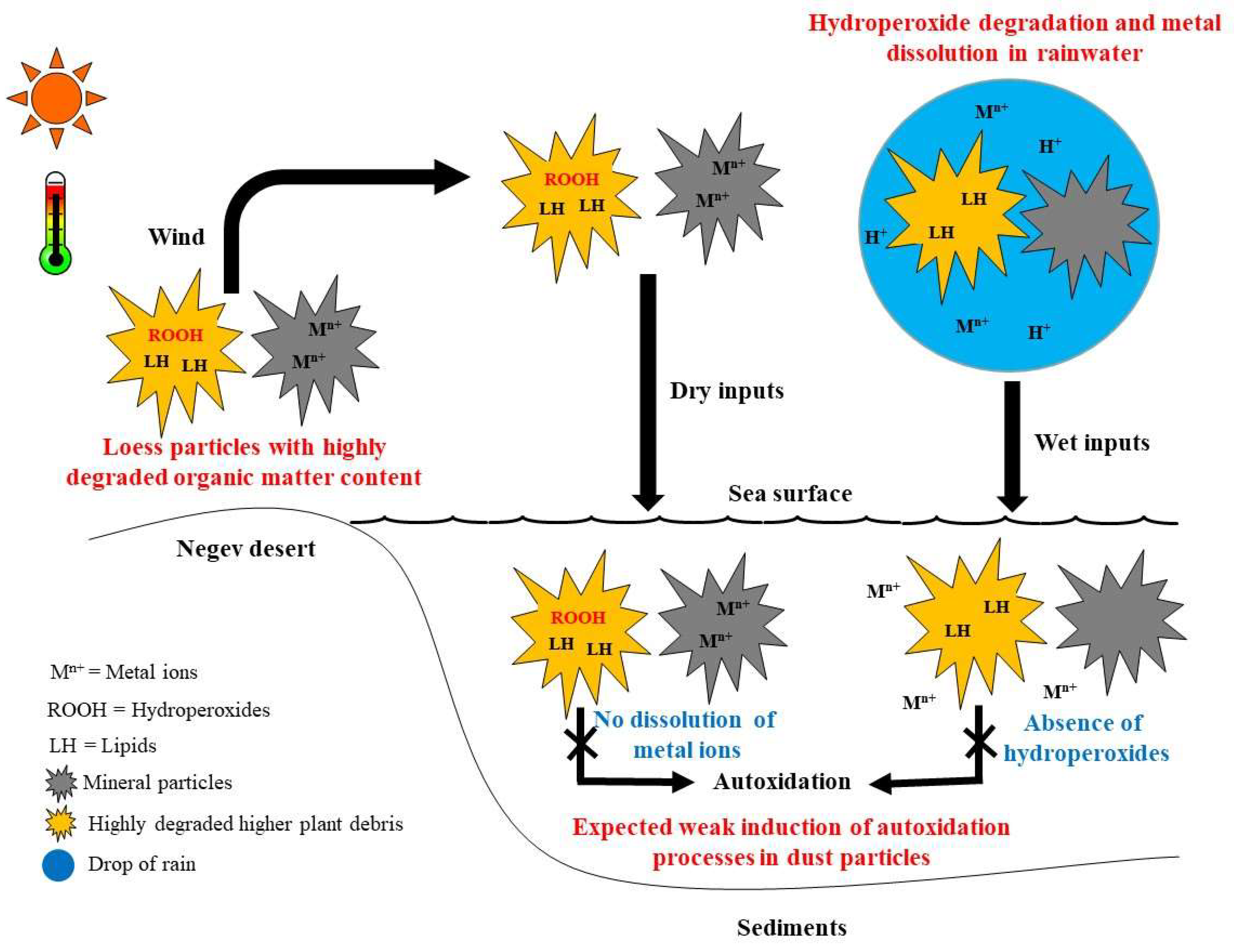
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- De Leeuw, J.W.; Largeau, C. A Review of Macromolecular Organic Compounds That Comprise Living Organisms and Their Role in Kerogen, Coal, and Petroleum Formation. In Organic Geochemistry; Engel, M.H., Macko, S.A., Eds.; Topics in Geobiology; Springer US: Boston, MA, USA, 1993; Volume 11, pp. 23–72. ISBN 978-1-4613-6252-4. [Google Scholar]
- Wakeham, S.G.; Canuel, E.A. Degradation and Preservation of Organic Matter in Marine Sediments. In Marine Organic Matter: Biomarkers, Isotopes and DNA; Volkman, J.K., Ed.; The Handbook of Environmental Chemistry; Springer-Verlag: Berlin/Heidelberg, Germany, 2006; Volume 2N, pp. 295–321. ISBN 978-3-540-28401-7. [Google Scholar]
- Hedges, J.I.; Keil, R.G. Sedimentary Organic Matter Preservation: An Assessment and Speculative Synthesis. Mar. Chem. 1995, 49, 81–115. [Google Scholar] [CrossRef]
- Vonk, J.E.; Sánchez-García, L.; Semiletov, I.; Dudarev, O.; Eglinton, T.; Andersson, A.; Gustafsson, Ö. Molecular and Radiocarbon Constraints on Sources and Degradation of Terrestrial Organic Carbon along the Kolyma Paleoriver Transect, East Siberian Sea. Biogeosciences 2010, 7, 3153–3166. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, E.S.; Charkin, A.; Dudarev, O.; Semiletov, I.; Vonk, J.E.; Sánchez-García, L.; Andersson, A.; Gustafsson, Ö. Carbon Isotopes and Lipid Biomarker Investigation of Sources, Transport and Degradation of Terrestrial Organic Matter in the Buor-Khaya Bay, SE Laptev Sea. Biogeosciences 2011, 8, 1865–1879. [Google Scholar] [CrossRef] [Green Version]
- Rontani, J.-F. Photo- and Free Radical-Mediated Oxidation of Lipid Components During the Senescence of Phototrophic Organisms. In Senescence; Nagata, T., Ed.; InTech: Saitama, Japan, 2012; ISBN 978-953-51-0144-4. [Google Scholar]
- Rontani, J.-F.; Charrière, B.; Sempéré, R.; Doxaran, D.; Vaultier, F.; Vonk, J.E.; Volkman, J.K. Degradation of Sterols and Terrigenous Organic Matter in Waters of the Mackenzie Shelf, Canadian Arctic. Org. Geochem. 2014, 75, 61–73. [Google Scholar] [CrossRef]
- Rontani, J.-F.; Galeron, M.-A.; Amiraux, R.; Artigue, L.; Belt, S.T. Identification of Di- and Triterpenoid Lipid Tracers Confirms the Significant Role of Autoxidation in the Degradation of Terrestrial Vascular Plant Material in the Canadian Arctic. Org. Geochem. 2017, 108, 43–50. [Google Scholar] [CrossRef]
- Galeron, M.-A.; Radakovitch, O.; Charrière, B.; Vaultier, F.; Volkman, J.K.; Bianchi, T.S.; Ward, N.D.; Medeiros, P.M.; Sawakuchi, H.O.; Tank, S.; et al. Lipoxygenase-Induced Autoxidative Degradation of Terrestrial Particulate Organic Matter in Estuaries: A Widespread Process Enhanced at High and Low Latitude. Org. Geochem. 2018, 115, 78–92. [Google Scholar] [CrossRef] [Green Version]
- Bonin, P.; Prime, A.; Galeron, M.; Guasco, S.; Rontani, J. Enhanced Biotic Degradation of Terrestrial POM in an Estuarine Salinity Gradient: Interactive Effects of Organic Matter Pools and Changes of Bacterial Communities. Aquat. Microb. Ecol. 2019, 83, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Rontani, J.-F.; Belt, S.T. Photo- and Autoxidation of Unsaturated Algal Lipids in the Marine Environment: An Overview of Processes, Their Potential Tracers, and Limitations. Org. Geochem. 2020, 139, 103941. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Levine, R.L. Chemical Modification of Proteins by Reactive Oxygen Species. In Redox Proteomics; Dalle-Donne, I., Scaloni, A., Butterfield, D.A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; pp. 1–23. ISBN 978-0-471-97312-6. [Google Scholar]
- Barreca, A.M.; Fabbrini, M.; Galli, C.; Gentili, P.; Ljunggren, S. Laccase/Mediated Oxidation of a Lignin Model for Improved Delignification Procedures. J. Mol. Catal. B Enzym. 2003, 26, 105–110. [Google Scholar] [CrossRef]
- Bianchi, T.S. The Role of Terrestrially Derived Organic Carbon in the Coastal Ocean: A Changing Paradigm and the Priming Effect. Proc. Natl. Acad. Sci. USA 2011, 108, 19473–19481. [Google Scholar] [CrossRef]
- d’Almeida, G.A. A Model for Saharan Dust Transport. J. Clim. Appl. Meteor. 1986, 25, 903–916. [Google Scholar] [CrossRef]
- Marticorena, B.; Bergametti, G.; Aumont, B.; Callot, Y.; N’Doumé, C.; Legrand, M. Modeling the Atmospheric Dust Cycle: 2. Simulation of Saharan Dust Sources. J. Geophys. Res. 1997, 102, 4387–4404. [Google Scholar] [CrossRef]
- Schefuß, E.; Ratmeyer, V.; Stuut, J.-B.W.; Jansen, J.H.F.; Damsté, J.S.S. Carbon Isotope Analyses of N-Alkanes in Dust from the Lower Atmosphere over the Central Eastern Atlantic. Geochim. Et Cosmochim. Acta 2003, 67, 1757–1767. [Google Scholar] [CrossRef]
- Korte, L.F.; Pausch, F.; Trimborn, S.; Brussaard, C.P.D.; Brummer, G.-J.A.; van der Does, M.; Guerreiro, C.V.; Schreuder, L.T.; Munday, C.I.; Stuut, J.-B.W. Effects of Dry and Wet Saharan Dust Deposition in the Tropical North Atlantic Ocean. Biogeosci. Discuss. 2018, 1–20. [Google Scholar]
- Schaich, K.M. Lipid Oxidation: Theoretical Aspects. In Bailey’s Industrial Oil and Fat Products; Shahidi, F., Ed.; Wiley: Hoboken, NJ, USA, 2005; ISBN 978-0-471-38460-1. [Google Scholar]
- Kouyoumdjian, H.; Saliba, N.A. Ion Concentrations of PM 10-2.5 and PM 2.5 Aerosols Over the Eastern Mediterranean Region: Seasonal Variation and Source Identification. Atmos. Chem. Phys. Discuss. 2005, 5, 13053–13073. [Google Scholar]
- Swet, N.; Katra, I. Reduction in Soil Aggregation in Response to Dust Emission Processes. Geomorphology 2016, 268, 177–183. [Google Scholar] [CrossRef]
- Katra, I.; Gross, A.; Swet, N.; Tanner, S.; Krasnov, H.; Angert, A. Substantial Dust Loss of Bioavailable Phosphorus from Agricultural Soils. Sci. Rep. 2016, 6, 24736. [Google Scholar] [CrossRef] [Green Version]
- Pardo, M.; Katra, I.; Schaeur, J.J.; Rudich, Y. Mitochondria-mediated Oxidative Stress Induced by Desert Dust in Rat Alveolar Macrophages. GeoHealth 2017, 1, 4–16. [Google Scholar] [CrossRef]
- Elad, D.; Zaretsky, U.; Avraham, S.; Gotlieb, R.; Wolf, M.; Katra, I.; Sarig, S.; Zaady, E. In Vitro Exposure of Nasal Epithelial Cells to Atmospheric Dust. Biomech. Model. Mechanobiol. 2018, 17, 891–901. [Google Scholar] [CrossRef]
- Zhan, Y.; Ginder-Vogel, M.; Shafer, M.M.; Rudich, Y.; Pardo, M.; Katra, I.; Katoshevski, D.; Schauer, J.J. Changes in Oxidative Potential of Soil and Fly Ash after Reaction with Gaseous Nitric Acid. Atmos. Environ. 2018, 173, 306–315. [Google Scholar] [CrossRef]
- Gross, A.; Tiwari, S.; Shtein, I.; Erel, R. Direct Foliar Uptake of Phosphorus from Desert Dust. New Phytol. 2021, 230, 2213–2225. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y. Integrated Wind-Erosion Modelling. In Physics and Modelling of Wind Erosion; Shao, Y., Ed.; Atmospheric and Oceanographic Sciences Library; Springer Netherlands: Dordrecht, The Netherlands, 2009; Volume 37, pp. 303–360. ISBN 978-1-4020-8894-0. [Google Scholar]
- Herut, B.; Nimmo, M.; Medway, A.; Chester, R.; Krom, M.D. Dry Atmospheric Inputs of Trace Metals at the Mediterranean Coast of Israel (SE Mediterranean): Sources and Fluxes. Atmos. Environ. 2001, 35, 803–813. [Google Scholar] [CrossRef]
- Ganor, E.; Foner, H.A. Mineral Dust Concentrations, Deposition Fluxes and Deposition Velocities in Dust Episodes over Israel. J. Geophys. Res. 2001, 106, 18431–18437. [Google Scholar] [CrossRef] [Green Version]
- Katra, I.; Arotsker, L.; Krasnov, H.; Zaritsky, A.; Kushmaro, A.; Ben-Dov, E. Richness and Diversity in Dust Stormborne Biomes at the Southeast Mediterranean. Sci. Rep. 2015, 4, 5265. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.; Palchan, D.; Krom, M.D.; Angert, A. Elemental and Isotopic Composition of Surface Soils from Key Saharan Dust Sources. Chem. Geol. 2016, 442, 54–61. [Google Scholar] [CrossRef]
- Uni, D.; Katra, I. Airborne Dust Absorption by Semi-Arid Forests Reduces PM Pollution in Nearby Urban Environments. Sci. Total Environ. 2017, 598, 984–992. [Google Scholar] [CrossRef]
- Dumas, C.; Aubert, D.; de Madron, X.D.; Ludwig, W.; Heussner, S.; Delsaut, N.; Menniti, C.; Sotin, C.; Buscail, R. Storm-Induced Transfer of Particulate Trace Metals to the Deep-Sea in the Gulf of Lion (NW Mediterranean Sea). Environ. Geochem. Health 2014, 36, 995–1014. [Google Scholar] [CrossRef]
- Maring, H.B.; Duce, R.A. The Impact of Atmospheric Aerosols on Trace Metal Chemistry in Open Ocean Surface Seawater, 1. Aluminum. Earth Planet. Sci. Lett. 1987, 84, 381–392. [Google Scholar] [CrossRef]
- Chester, R.; Nimmo, M.; Corcoran, P.A. Rain Water-Aerosol Trace Metal Relationships at Cap Ferrat: A Coastal Site in the Western Mediterranean. Mar. Chem. 1997, 58, 293–312. [Google Scholar] [CrossRef]
- Desboeufs, K.V.; Losno, R.; Vimeux, F.; Cholbi, S. The PH-Dependent Dissolution of Wind-Transported Saharan Dust. J. Geophys. Res. 1999, 104, 21287–21299. [Google Scholar] [CrossRef]
- Longo, A.F.; Feng, Y.; Lai, B.; Landing, W.M.; Shelley, R.U.; Nenes, A.; Mihalopoulos, N.; Violaki, K.; Ingall, E.D. Influence of Atmospheric Processes on the Solubility and Composition of Iron in Saharan Dust. Environ. Sci. Technol. 2016, 50, 6912–6920. [Google Scholar] [CrossRef] [PubMed]
- Meskhidze, N. Dust and Pollution: A Recipe for Enhanced Ocean Fertilization? J. Geophys. Res. 2005, 110, D03301. [Google Scholar] [CrossRef]
- Hettiarachchi, E.; Reynolds, R.L.; Goldstein, H.L.; Moskowitz, B.; Rubasinghege, G. Bioavailable Iron Production in Airborne Mineral Dust: Controls by Chemical Composition and Solar Flux. Atmos. Environ. 2019, 205, 90–102. [Google Scholar] [CrossRef]
- Desboeufs, K.V.; Sofikitis, A.; Losno, R.; Colin, J.L.; Ausset, P. Dissolution and Solubility of Trace Metals from Natural and Anthropogenic Aerosol Particulate Matter. Chemosphere 2005, 58, 195–203. [Google Scholar] [CrossRef]
- Herut, B.; Starinsky, A.; Katz, A.; Rosenfeld, D. Relationship between the Acidity and Chemical Composition of Rainwater and Climatological Conditions along a Transition Zone between Large Deserts and Mediterranean Climate, Israel. Atmos. Environ. 2000, 34, 1281–1292. [Google Scholar] [CrossRef]
- Ridame, C.; Guieu, C. Saharan Input of Phosphate to the Oligotrophic Water of the Open Western Mediterranean Sea. Limnol. Oceanogr. 2002, 47, 856–869. [Google Scholar] [CrossRef]
- Rontani, J.F.; Charrière, B.; Menniti, C.; Aubert, D.; Aubert, C. Electron Ionization Mass Spectrometry Fragmentation and Multiple Reaction Monitoring Quantification of Autoxidation Products of α- and β-Amyrins in Natural Samples. Rapid Commun. Mass Spectrom. 2018, 32, 1599–1607. [Google Scholar] [CrossRef]
- Mihara, S.; Tateba, H. Photosensitized Oxygenation Reactions of Phytol and Its Derivatives. J. Org. Chem. 1986, 51, 1142–1144. [Google Scholar] [CrossRef]
- Rontani, J.-F. Lipid Oxidation Products: Useful Tools for Monitoring Photo- and Autoxidation in Phototrophs; Cambridge Scholars Publishing: Newcastle upon Tyne, UK, 2021; ISBN 978-1-5275-7359-8. [Google Scholar]
- Shalom, O.; Crouvi, O.; Enzel, Y.; Rosenfeld, D. Locally Recycled Late Pleistocene Loess Feeds Modern Dust Storms at the Desert Margins of the Eastern Mediterranean, Israel. Aeolian Res. 2020, 46, 100612. [Google Scholar] [CrossRef]
- Sicre, M.-A.; Paillasseur, J.-L.; Marty, J.-C.; Saliot, A. Characterization of Seawater Samples Using Chemometric Methods Applied to Biomarker Fatty Acids. Org. Geochem. 1988, 12, 281–288. [Google Scholar] [CrossRef]
- McGrath, C.F.; Moss, C.W.; Burchard, R.P. Effect of Temperature Shifts on Gliding Motility, Adhesion, and Fatty Acid Composition of Cytophaga Sp. Strain U67. J. Bacteriol. 1990, 172, 1978–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deas, A.H.B.; Holloway, P.J. The Intermolecular Structure of Some Plant Cutins. In Lipids and Lipid Polymers in Higher Plants; Tevini, M., Lichtenthaler, H.K., Eds.; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 1977; pp. 293–299. ISBN 978-3-642-66634-6. [Google Scholar]
- Kolattukudy, P.E. Biopolyester Membranes of Plants: Cutin and Suberin. Science 1980, 208, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Bichot, A.; Delgenes, J.P.; Radoiu, M.; Garcia Bernet, D. Microwave Pretreatment of Lignocellulosic Biomass to Release Maximum Phenolic Acids. In Proceedings of the 17th International Conference on Microwave and High Frequency Heating, Barcelona, Spain, 9 September 2019. [Google Scholar]
- Volkman, J. Sterols in Microorganisms. Appl. Microbiol. Biotechnol. 2003, 60, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Aziz, M.; Sturtevant, D.; Chapman, K.D.; Guo, L. Heterogeneous Distribution of Erucic Acid in Brassica Napus Seeds. Front. Plant Sci. 2020, 10, 1744. [Google Scholar] [CrossRef] [Green Version]
- Brassell, S.C.; Eglinton, G.; Maxwell, J.R. The Geochemistry of Terpenoids and Steroids. Biochem. Soc. Trans. 1983, 11, 575–586. [Google Scholar] [CrossRef]
- Otto, A.; Simpson, M.J. Degradation and Preservation of Vascular Plant-Derived Biomarkers in Grassland and Forest Soils from Western Canada. Biogeochemistry 2005, 74, 377–409. [Google Scholar] [CrossRef]
- Jäger, S.; Trojan, H.; Kopp, T.; Laszczyk, M.; Scheffler, A. Pentacyclic Triterpene Distribution in Various Plants—Rich Sources for a New Group of Multi-Potent Plant Extracts. Molecules 2009, 14, 2016–2031. [Google Scholar] [CrossRef] [Green Version]
- Knoche, H.W. A Study on the Biosynthesis Ofcis-9,10-Epoxyoctadecanoic Acid. Lipids 1968, 3, 163–169. [Google Scholar] [CrossRef]
- Marchand, D.; Rontani, J.-F. Characterisation of Photo-Oxidation and Autoxidation Products of Phytoplanktonic Monounsaturated Fatty Acids in Marine Particulate Matter and Recent Sediments. Org. Geochem. 2001, 32, 287–304. [Google Scholar] [CrossRef]
- Rontani, J.-F.; Aubert, C.; Belt, S.T. EIMS Fragmentation Pathways and MRM Quantification of 7α/β-Hydroxy-Dehydroabietic Acid TMS Derivatives. J. Am. Soc. Mass Spectrom. 2015, 26, 1606–1616. [Google Scholar] [CrossRef]
- Galeron, M.-A.; Vaultier, F.; Rontani, J.-F. Oxidation Products of α- and β-Amyrins: Potential Tracers of Abiotic Degradation of Vascular-Plant Organic Matter in Aquatic Environments. Environ. Chem. 2016, 13, 732. [Google Scholar] [CrossRef]
- Buanafina, M.M.D.O. Feruloylation in Grasses: Current and Future Perspectives. Mol. Plant 2009, 2, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Minatel, I.O.; Borges, C.V.; Ferreira, M.I.; Gomez, H.A.G.; Chen, C.-Y.O.; Lima, G.P.P. Phenolic Compounds: Functional Properties, Impact of Processing and Bioavailability. In Phenolic Compounds—Biological Activity; Soto-Hernndez, M., Palma-Tenango, M., Garcia-Mateos, M.D.R., Eds.; InTech: Saitama, Japan, 2017; ISBN 978-953-51-2959-2. [Google Scholar]
- Katra, I. Soil Erosion by Wind and Dust Emission in Semi-Arid Soils Due to Agricultural Activities. Agronomy 2020, 10, 89. [Google Scholar] [CrossRef] [Green Version]
- Crouvi, O.; Amit, R.; Ben Israel, M.; Enzel, Y. Loess in the Negev Desert: Sources, Loessial Soils, Palaeosols, and Palaeoclimatic Implications. In Quaternary of the Levant; Enzel, Y., Bar-Yosef, O., Eds.; Cambridge University Press: Cambridge, UK, 2017; pp. 471–482. ISBN 978-1-316-10675-4. [Google Scholar]
- Wedepohl, K.H. The Composition of the Continental Crust. Geochim. Et Cosmochim. Acta 1995, 59, 1217–1232. [Google Scholar] [CrossRef]
- Lucke, B.; Sandler, A.; Vanselow, K.A.; Bruins, H.J.; Abu-Jaber, N.; Bäumler, R.; Porat, N.; Kouki, P. Composition of Modern Dust and Holocene Aeolian Sediments in Archaeological Structures of the Southern Levant. Atmosphere 2019, 10, 762. [Google Scholar] [CrossRef] [Green Version]
- Mahowald, N.M.; Hamilton, D.S.; Mackey, K.R.M.; Moore, J.K.; Baker, A.R.; Scanza, R.A.; Zhang, Y. Aerosol Trace Metal Leaching and Impacts on Marine Microorganisms. Nat. Commun. 2018, 9, 2614. [Google Scholar] [CrossRef] [Green Version]
- Frimer, A.A. The Reaction of Singlet Oxygen with Olefins: The Question of Mechanism. Chem. Rev. 1979, 79, 359–387. [Google Scholar] [CrossRef]
- Turner, J.A.; Herz, W. Iron(II)-Induced Decomposition of Unsaturated Cyclic Peroxides Derived from Butadienes. A Simple Procedure for Synthesis of 3-Alkylfurans. J. Org. Chem. 1977, 42, 1900–1904. [Google Scholar] [CrossRef]
- Rontani, J.-F. Photodegradation of Unsaturated Fatty Acids in Senescent Cells of Phytoplankton: Photoproduct Structural Identification and Mechanistic Aspects. J. Photochem. Photobiol. A Chem. 1998, 114, 37–44. [Google Scholar] [CrossRef]
- Vashchenko, E.V.; Knyazeva, I.V.; Krivoshey, A.I.; Vashchenko, V.V. Retro-Aldol Reactions in Micellar Media. Mon. Chem. 2012, 143, 1545–1549. [Google Scholar] [CrossRef]
- Shi, C.; Deng, X.; Yang, Y.; Huang, X.; Wu, B. Precipitation Chemistry and Corresponding Transport Patterns of Influencing Air Masses at Huangshan Mountain in East China. Adv. Atmos. Sci. 2014, 31, 1157–1166. [Google Scholar] [CrossRef]
- Shaltout, M.; Omstedt, A. Recent sea surface temperature trends and future scenarios for the Mediterranean Sea. Oceanologia 2014, 56, 411–443. [Google Scholar] [CrossRef]
- Brown, B.E.; Dunne, R.P.; Chansang, H. Coral Bleaching Relative to Elevated Seawater Temperature in the Andaman Sea (Indian Ocean) over the Last 50 Years. Coral Reefs 1996, 15, 151–152. [Google Scholar] [CrossRef]
- Garreta, A.; Val-Moraes, S.P.; García-Fernández, Q.; Busquets, M.; Juan, C.; Oliver, A.; Ortiz, A.; Gaffney, B.J.; Fita, I.; Manresa, À.; et al. Structure and Interaction with Phospholipids of a Prokaryotic Lipoxygenase from Pseudomonas Aeruginosa. FASEB J. 2013, 27, 4811–4821. [Google Scholar] [CrossRef]

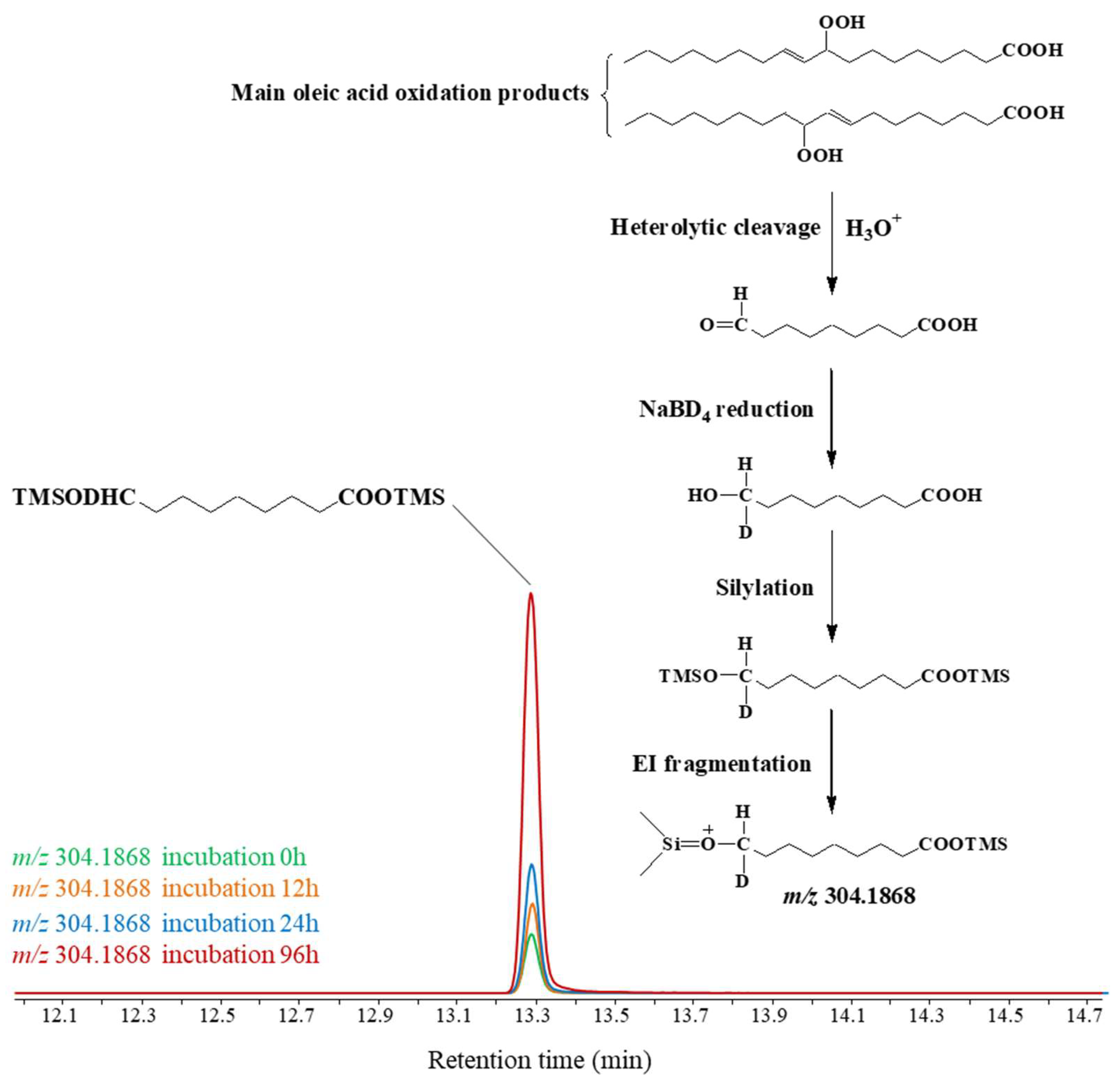

{kind=link}
{kind=link}
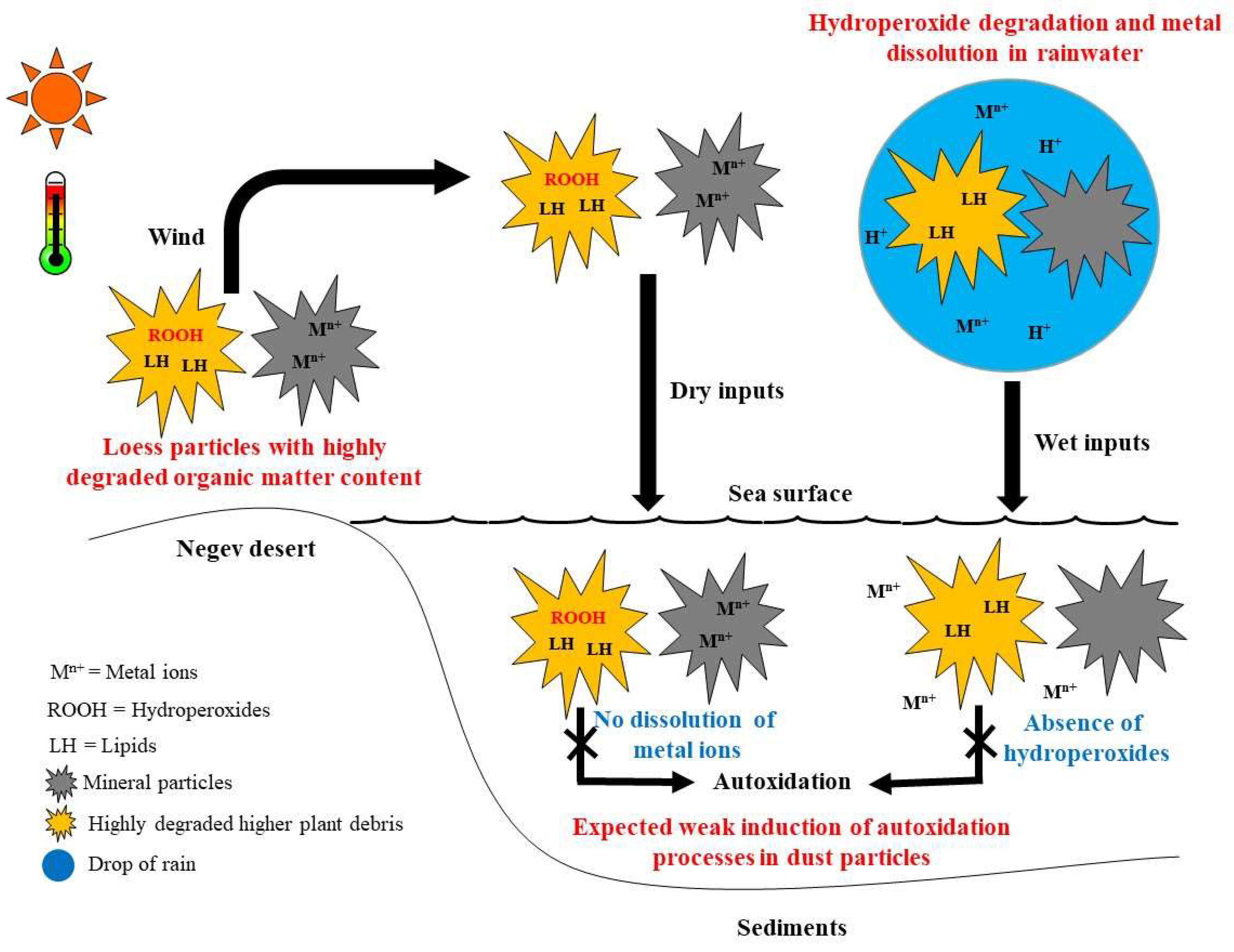
{kind=link}
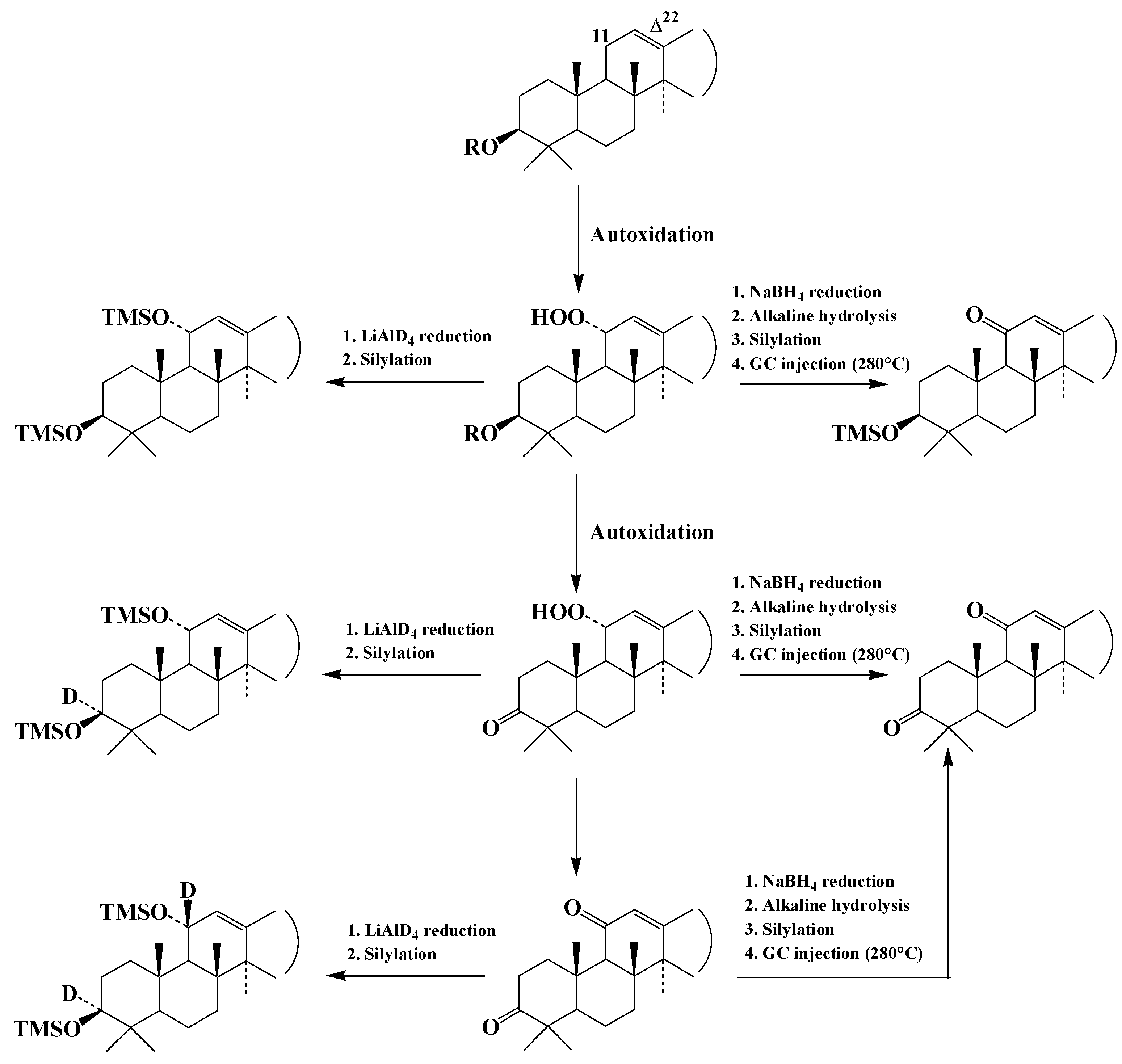
{kind=link}
{kind=link}
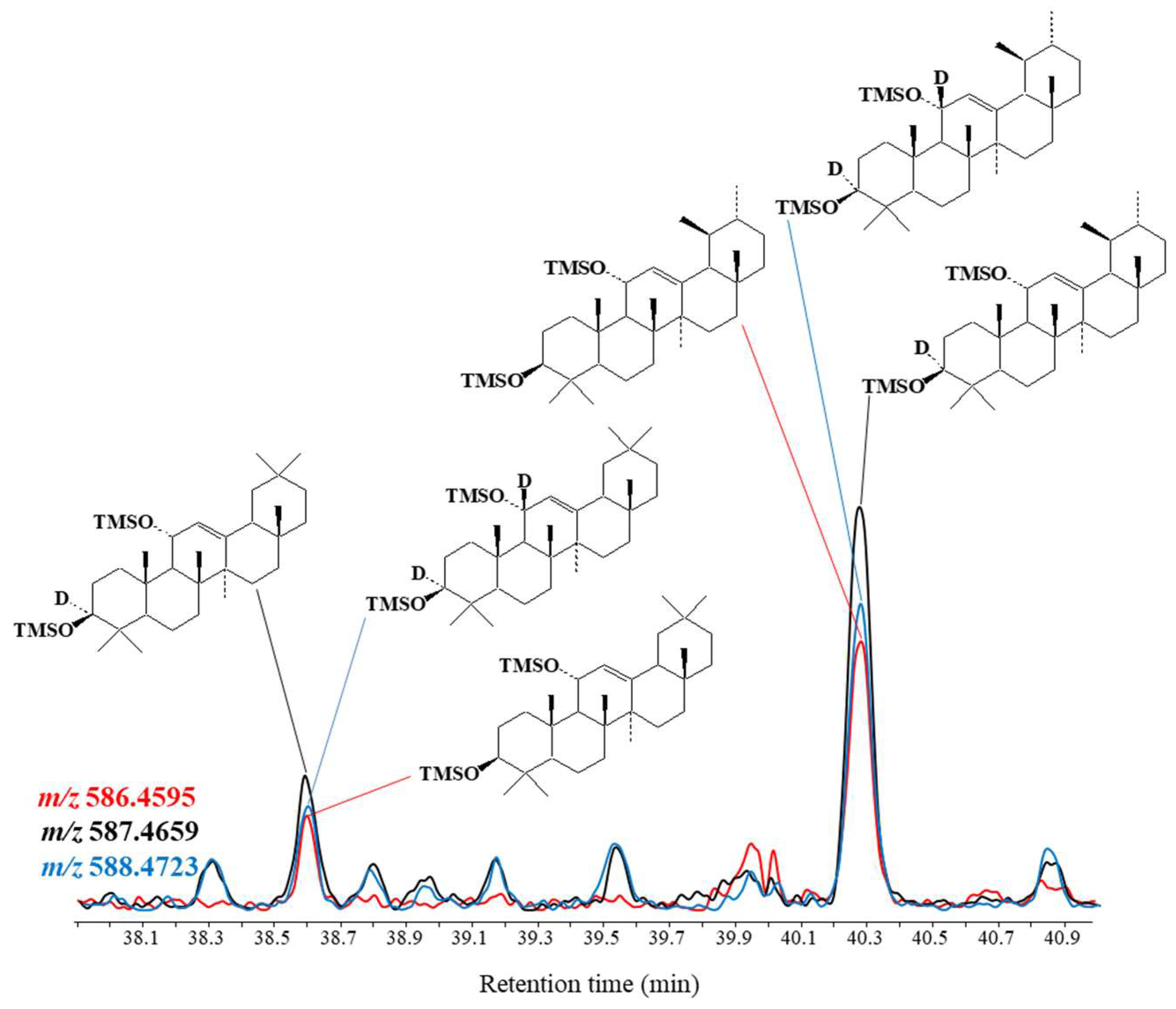
{kind=link}
{kind=link}
| Lipid | Concentration | Origin |
|---|---|---|
| C18:1Δ9 (oleic) acid | 43.0 ± 10.0 a | Plants, fungi, bacteria |
| C18:1Δ11 (vaccenic) acid | 5.1 ± 1.3 a | Bacteria |
| C18:2Δ9,12 (linoleic) acid | 8.2 ± 0.9 a | Plants, fungi |
| C22:1Δ13 (erucic) acid | 6.4 ± 1.3 a | Brassicaceae |
| ω,(9-10)-dihydroxy-C16:0 acids 9,10-epoxyoctadecanoic acid c Dehydroabietic acid | 22.3 ± 2.7 a 10.2 ± 1.9 a 4.1 ± 1.9 a | Higher plants (cuticular waxes) Fungi-infected plants Gymnosperms |
| p-coumaric acid | 0.6 ± 0.1 a | Higher plants (lignocellulose) |
| Ferulic acid | 2.4 ± 0.3 a | Higher plants (lignocellulose) |
| Cholesterol | 8.8 ± 3.1 a | Plants, fungi, animals |
| Campesterol | 0.5 ± 0.1 a | Higher plants |
| Sitosterol Ergosterol | 0.6 ± 0.1 a 0.3 ± 0.1 a | Higher plants Fungi, yeasts |
| α-amyrin | 24.2 ± 8.2 b | Angiosperms |
| β-amyrin | 26.6 ± 3.7 b | Angiosperms, fungi |
| Concentrations | Dust Sample | T0 | T12h | T24h | T96h |
|---|---|---|---|---|---|
| Ca | 137,988 ± 13,990 | 2.21 ± 0.13 | 4.02 ± 0.15 | 4.42 ± 0.10 | 4.11 ± 0.24 |
| Fe | 32,589 ± 3143 | 0.06 ± 0.01 | 0.68 ± 0.25 | 1.55 ± 0.51 | 0.02 ± 0.01 |
| Al | 41,357 ± 3580 | 0.10 ± 0.01 | 0.90 ± 0.21 | 1.84 ± 0.61 | 0.27 ± 0.01 |
| Zn | 77.1 ± 1.3 | 15.62 ± 3.77 | 10.86 ± 11.50 | 13.09 ± 11.85 | 2.73 ± 1.07 |
| Cu | 21.4 ± 1.9 | 3.43 ± 0.96 | 2.58 ± 0.69 | 3.44 ± 0.26 | 4.37 ± 2.87 |
| V | 112 ± 11 | 0.56 ± 0.02 | 2.12 ± 0.21 | 3.50 ± 0.32 | 2.73 ± 0.05 |
| Mn | 613 ± 63 | 0.21 ± 0.034 | 0.68 ± 0.29 | 1.29 ± 0.35 | 0.06 ± 0.02 |
| Co | 13.7 ± 1.5 | 0.17 ± 0.031 | 1.03 ± 0.35 | 1.91 ± 0.54 | 0.31 ± 0.06 |
| Cd | 0.63 ± 0.09 | 2.59 ± 1.11 | 2.01 ± 0.68 | 2.89 ± 1.08 | 1.13 ± 0.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rontani, J.-F.; Charriere, B.; Menniti, C.; Katra, I.; Aubert, D. Effects of Dry and Wet Negev Soil–Dust Deposition on the Induction of Autoxidation of Soil–Dust Lipid Components. Water 2022, 14, 4092. https://doi.org/10.3390/w14244092
Rontani J-F, Charriere B, Menniti C, Katra I, Aubert D. Effects of Dry and Wet Negev Soil–Dust Deposition on the Induction of Autoxidation of Soil–Dust Lipid Components. Water. 2022; 14(24):4092. https://doi.org/10.3390/w14244092
Chicago/Turabian StyleRontani, Jean-François, Bruno Charriere, Christophe Menniti, Itzhak Katra, and Dominique Aubert. 2022. "Effects of Dry and Wet Negev Soil–Dust Deposition on the Induction of Autoxidation of Soil–Dust Lipid Components" Water 14, no. 24: 4092. https://doi.org/10.3390/w14244092