

Challenges of Comparing Marine Microbiome Community Composition Data Provided by Different Commercial Laboratories and Classification Databases

 ,
, 
 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Isolation
2.3. 16S rRNA Amplicon Library Generation
2.4. Taxonomic Classification of the Bacterial 16S rRNA Gene
3. Results
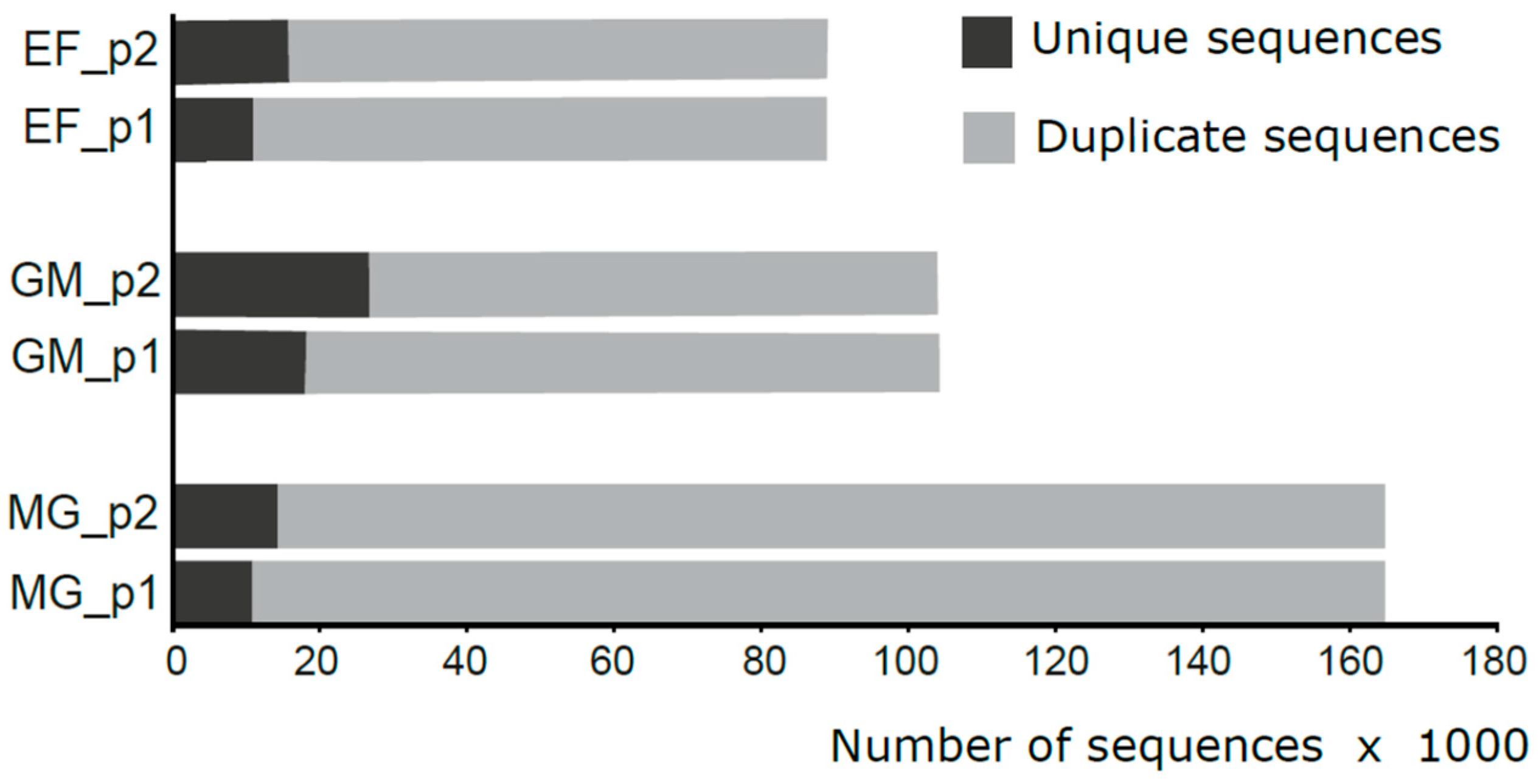
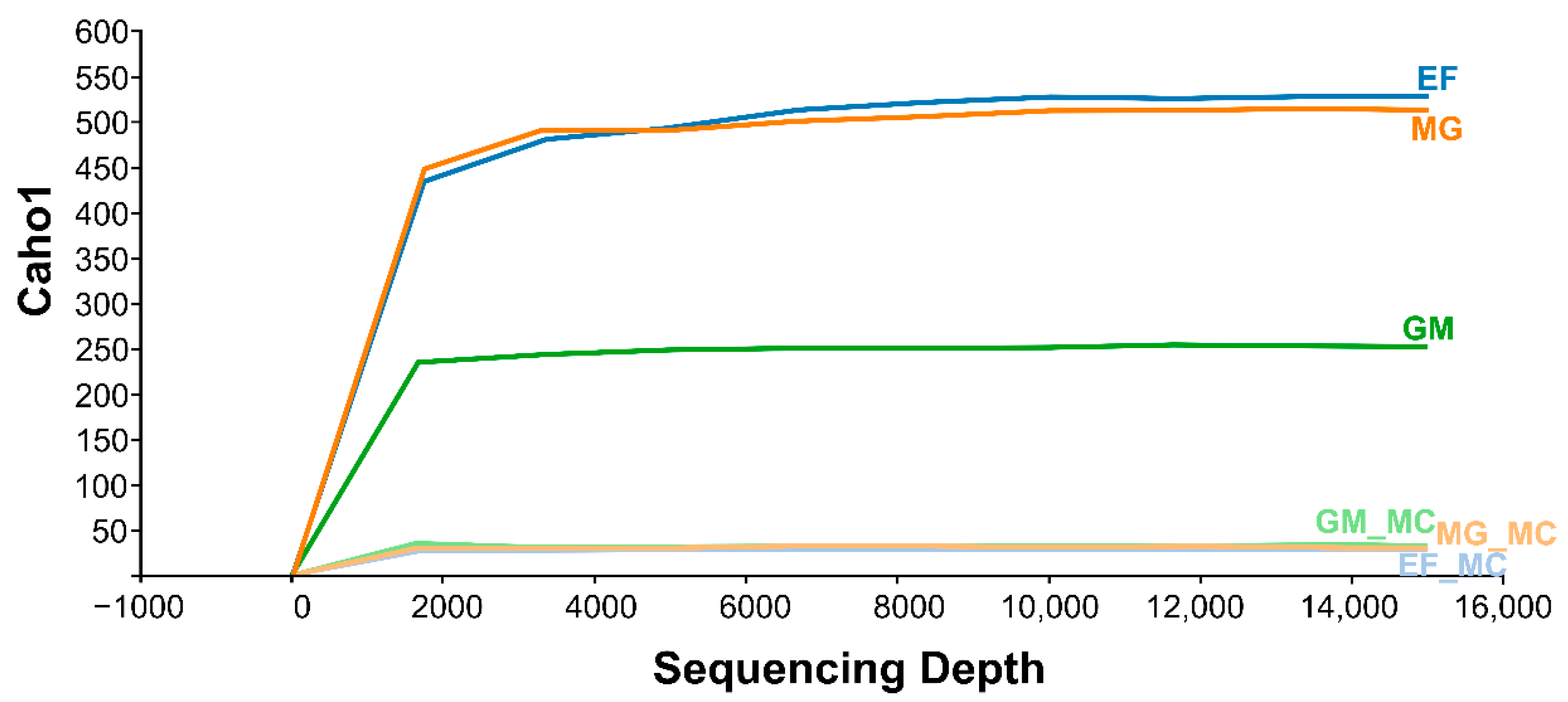
3.1. Overall Statistics, Trimming, and De-Multiplexing with DADA2
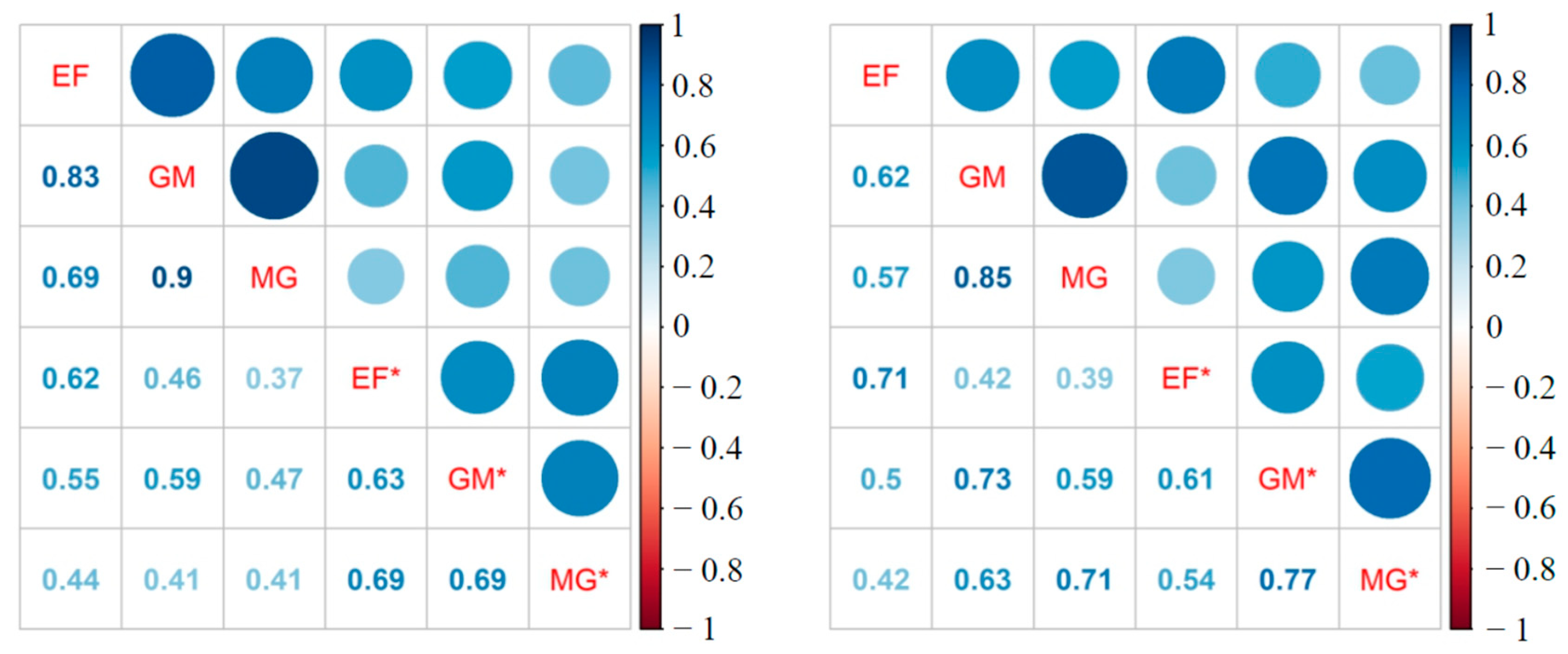
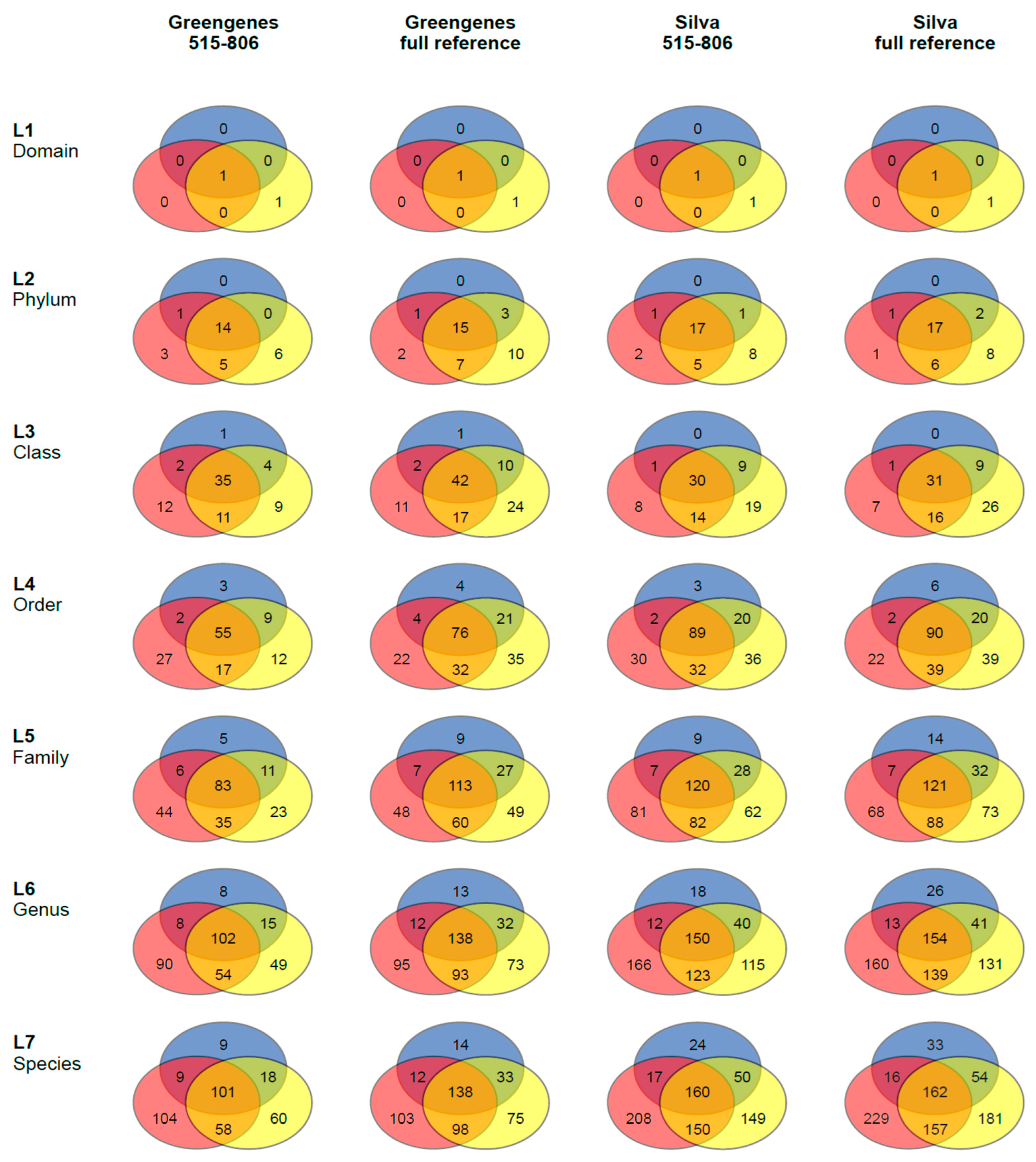
3.2. 16S rRNA Community Analysis
3.3. Putative Bacterial Endosymbionts
3.4. Mock Community
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arrigo, K.R. Marine microorganisms and global nutrient cycles. Nature 2005, 437, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, J. Metagenomics: Application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 2004, 4, 669–685. [Google Scholar] [CrossRef] [PubMed]
- DeLong, E.F. The microbial ocean from genomes to biomes. Nature 2009, 459, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Rinke, C.; Schwientek, P.; Sczyrba, A.; Ivanova, N.N.; Anderson, I.J.; Cheng, J.-F.; Darling, A.; Malfatti, S.; Swan, B.K.; Gies, E.A.; et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 2013, 499, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Aevarsson, A.; Kaczorowska, A.K.; Adalsteinsson, B.T.; Ahlqvist, J.; Al-Karadaghi, S.; Altenbuchner, J.; Arsin, H.; Átlasson, Ú.Á.; Brandt, D.; Cichowicz-Cieślak, M.; et al. Going to extremes—A metagenomic journey into the dark matter of life. FEMS Microbiol. Lett. 2021, 368, fnab067. [Google Scholar] [CrossRef] [PubMed]
- Stahl, D.A.; Lane, D.J.; Olsen, G.J.; Pace, N.R. Analysis of hydrothermal vent associated symbionts by ribosomal RNA sequences. Science 1984, 224, 409–411. [Google Scholar] [CrossRef]
- Stahl, D.A.; Lane, D.J.; Olsen, G.J.; Pace, N.R. Characterization of a Yellowstone hot spring microbial community by 5S rRNA sequences. Appl. Environ. Microbiol. 1985, 49, 1379–1384. [Google Scholar] [CrossRef]
- Bourlat, S.J.; Borja, A.; Gilbert, J.; Taylor, M.I.; Davies, N.; Weisberg, S.B.; Griffith, J.F.; Lettieri, T.; Field, D.; Benzie, J.; et al. Genomics in marine monitoring: New opportunities for assessing marine health status. Mar. Pollut. Bull. 2013, 74, 19–31. [Google Scholar] [CrossRef]
- Mioduchowska, M.; Zając, K.; Bartoszek, K.; Madanecki, P.; Kur, J.; Zając, T. 16S rRNA-based metagenomic analysis of the gut microbial community associated with the DUI species Unio crassus (Bivalvia: Unionidae). J. Zoolog. Syst. Evol. Res. 2020, 58, 615–623. [Google Scholar] [CrossRef]
- Wei, Z.; Gu, Y.; Friman, V.-P.; Kowalchuk, G.A.; Xu, Y.; Shen, Q.; Jousset, A. Initial soil microbiome composition and functioning predetermine future plant health. Sci. Adv. 2019, 5, eaaw0759. [Google Scholar] [CrossRef]
- Lee, C.S.; Kim, M.; Lee, C.; Yu, Z.; Lee, J. The microbiota of recreational freshwaters and the implications for environmental and public health. Front. Microbiol. 2016, 7, 1826. [Google Scholar] [CrossRef] [PubMed]
- Stal, J.L.; Cretoiu, M.S. The Marine Microbiome: An Untapped Source of Biodiversity and Biotechnological Potential; Springer: Cham, Switzerland, 2016; pp. 1–229. [Google Scholar]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, M.D.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar] [CrossRef] [PubMed]
- Fonselius, S.; Valderrama, J. One hundred years of hydrographic measurements in the Baltic Sea. J. Sea Res. 2003, 49, 229–241. [Google Scholar] [CrossRef]
- Hardeman, F.; Sjoling, S. Metagenomic approach for the isolation of a novel low-temperature-active lipase from uncultured bacteria of marine sediment. FEMS Microbiol. Ecol. 2007, 59, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Rheinheimer, G.; Gocke, K.; Hoppe, H.G. Vertical distribution of microbiological and hydrographic-chemical parameters in different areas of the Baltic Sea. Mar. Ecol. Prog. Ser. 1989, 52, 55–70. [Google Scholar] [CrossRef]
- Herlemann, D.P.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef]
- Hu, Y.O.; Karlson, B.; Charvet, S.; Andersson, A.F. Diversity of Pico-to mesoplankton along the 2000 km salinity gradient of the Baltic Sea. Front. Microbiol. 2016, 7, 679. [Google Scholar] [CrossRef]
- Hugerth, L.W.; Larsson, J.; Alneberg, J.; Lindh, M.V.; Legrand, C.; Pinhassi, J.; Andersson, A.F. Metagenome-assembled genomes uncover a global brackish microbiome. Genome Biol. 2015, 16, 279. [Google Scholar] [CrossRef]
- Alneberg, J.; Sundh, J.; Bennke, C.; Beier, S.; Lundin, D.; Hugerth, L.W.; Pinhassi, J.; Kisand, V.; Riemann, L.; Jürgens, K.; et al. BARM and BalticMicrobeDB, a reference metagenome and interface to meta-omic data for the Baltic Sea. Sci. Data 2018, 5, 180146. [Google Scholar] [CrossRef]
- Azam, F.; Fenchel, T.; Field, J.G.; Gray, J.S.; Meyer-Reil, L.A.; Thingstad, F. The ecological role of water-column microbes in the sea. Mar. Ecol. Prog. Ser. 1983, 10, 257–263. [Google Scholar] [CrossRef]
- Taylor, A.H.; Joint, I. A steady-state analysis of the ‘microbial loop’ in stratified systems. Mar. Ecol. Prog. Ser. 1990, 59, 1–17. [Google Scholar] [CrossRef]
- Guo, B.; Zhang, L.; Sun, H.; Gao, M.; Yu, N.; Zhang, O.; Mou, A.; Liu, Y. Microbial co-occurrence network topological properties link with reactor parameters and reveal the importance of low-abundance genera. Npj Biofilms Microbiomes 2022, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.A.; Webster, N.S.; Miller, D.J.; Bourne, D.G. Host-microbe coevolution: Applying evidence from model systems to complex marine invertebrate holobionts. mBio 2019, 10, e02241-18. [Google Scholar] [CrossRef]
- Pais, R.; Lohs, C.; Wu, Y.; Wang, J.; Aksoy, S. The obligate mutualist Wigglesworthia glossinidia influences reproduction, digestion, and immunity processes of its host, the tsetse fly. Appl. Environ. Microbiol. 2008, 74, 5965–5974. [Google Scholar] [CrossRef] [PubMed]
- Ruehland, C.; Blazejak, A.; Lott, C.; Loy, A.; Erséus, C.; Dubilier, N. Multiple bacterial symbionts in two species of co-occurring gutless oligochaete worms from Mediterranean Sea grass sediments. Environ. Microbiol. 2018, 10, 3404–3416. [Google Scholar] [CrossRef] [PubMed]
- Webster, N.S.; Thomas, T. The Sponge Hologenome. mBio 2016, 7, e00135-16. [Google Scholar] [CrossRef]
- Krediet, C.J.; Ritchie, K.B.; Paul, V.J.; Teplitski, M. Coral-associated micro-organisms and their roles in promoting coral health and thwarting diseases. Proc. R. Soc. B 2013, 280, 20122328. [Google Scholar] [CrossRef]
- Sergeant, M.J.; Constantinidou, C.; Cogan, T.; Penn, C.W.; Pallen, M.J. High-throughput sequencing of 16S rRNA gene amplicons: Effects of extraction procedure, primer length and annealing temperature. PLoS ONE 2012, 7, e38094. [Google Scholar] [CrossRef]
- Cruaud, P.; Vigneron, A.; Lucchetti-Miganeh, C.; Ciron, P.E.; Godfroy, A.; Cambon-Bonavita, M.A. Influence of DNA extraction method, 16S rRNA targeted hypervariable regions, and sample origin on microbial diversity detected by 454 pyrosequencing in marine chemosynthetic ecosystems. Appl. Environ. Microbiol. 2014, 80, 4626–4639. [Google Scholar] [CrossRef]
- Plummer, E.; Twin, J.; Bulach, D.M.; Garland, S.M.; Tabrizi, S.N. A comparison of three bioinformatics pipelines for the analysis of preterm gut microbiota using 16S rRNA gene sequencing data. J. Proteom. Bioinform. 2015, 8, 283–291. [Google Scholar] [CrossRef]
- Kennedy, J.; Flemer, B.; Jackson, S.A.; Morrissey, J.P.; O’Gara, F.; Dobson, A.D. Evidence of a putative deep sea specific microbiome in marine sponges. PLoS ONE 2014, 9, e91092. [Google Scholar] [CrossRef]
- Balvociute, M.; Huson, D.H. SILVA, RDP, Greengenes, NCBI and OTT—How do these taxonomies compare? BMC Genom. 2019, 18, 114. [Google Scholar]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, 643–648. [Google Scholar] [CrossRef]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Federhen, S. The NCBI taxonomy database. Nucleic Acids Res. 2012, 40, 136–143. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Gohl, D.M.; Vangay, P.; Garbe, J.; MacLean, A.; Hauge, A.; Becker, A.; Gould, T.J.; Clayton, J.B.; Johnson, T.J.; Hunter, R.; et al. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat. Biotechnol. 2016, 34, 942–949. [Google Scholar] [CrossRef]
- McGovern, E.; Waters, S.M.; Blackshields, G.; McCabe, M.S. Evaluating established methods for rumen 16S rRNA amplicon sequencing with mock microbial populations. Front. Microbiol. 2018, 9, 1365. [Google Scholar] [CrossRef]
- Yeh, Y.C.; Needham, D.M.; Sieradzki, E.T.; Fuhrman, J.A. Taxon disappearance from microbiome analysis reinforces the value of mock communities as a standard in every sequencing run. MSystems 2018, 3, e00023-18. [Google Scholar] [CrossRef]
- Gołębiewski, M.; Tretyn, A. Generating amplicon reads for microbial community assessment with next-generation sequencing. J. Appl. Microbiol. 2000, 128, 330–354. [Google Scholar] [CrossRef]
- Ibarbalz, F.M.; Pérez, M.V.; Figuerola, E.L.; Erijman, L. The bias associated with amplicon sequencing does not affect the quantitative assessment of bacterial community dynamics. PLoS ONE 2014, 9, e99722. [Google Scholar] [CrossRef]
- Piwosz, K.; Shabarova, T.; Pernthaler, J.; Posch, T.; Šimek, K.; Porcal, P.; Salcher, M.M. Bacterial and eukaryotic small-subunit amplicon data do not provide a quantitative picture of microbial communities, but they are reliable in the context of ecological interpretations. mSphere 2020, 5, e00052-20. [Google Scholar] [CrossRef] [PubMed]
- Moskot, M.; Kotlarska, E.; Jakóbkiewicz-Banecka, J.; Gabig-Cimińska, M.; Fari, K.; Węgrzyn, G.; Wróbel, B. Metal and antibiotic resistance of bacteria isolated from the Baltic Sea. Int Microbiol 2012, 15, 131–139. [Google Scholar]
- Toruńska-Sitarz, A.; Kotlarska, E.; Mazur-Marzec, H. Biodegradation of nodularin and other nonribosomal peptides by the Baltic bacteria. Int. Biodeterior. Biodegrad. 2018, 134, 48–57. [Google Scholar] [CrossRef]
- Turner, S.; Pryer, K.M.; Miao, V.P.W.; Palmer, J.D. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J. Eukaryot. Microbiol. 1999, 46, 327–338. [Google Scholar] [CrossRef]
- Kisand, V.; Cuadros, R.; Wikner, J. Phylogeny of culturable estuarine bacteria catabolizing riverine organic matter in the N Baltic. Appl. Environ. Microbiol. 2002, 68, 379–388. [Google Scholar] [CrossRef]
- Eiler, A.; Heinrich, F.; Bertilsson, S. Coherent dynamics and association networks among lake bacterioplankton taxa. ISME J. 2012, 6, 330–342. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplles, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 2013, 41, e1. [Google Scholar] [CrossRef]
- Andrews, S. FastQC A Quality Control tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fast-qc/ (accessed on 25 November 2014).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ 2018, 6, e27295v2. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria; Available online: https://www.R-project.org/ (accessed on 12 October 2022).
- Oren, A.; Garrity, G.M. Valid publication of the names of forty-two phyla of prokaryotes. Int. J. Syst. Evol. Microbiol. 2021, 71, 10. [Google Scholar] [CrossRef]
- Skerman, V.B.D.; McGowan, V.; Sneath, P.H.A. Approved lists of bacterial names. Int. J. Syst. Bacteriol. 1980, 30, 225–420. [Google Scholar] [CrossRef]
- Parte, A.C.; Sardà Carbasse, J.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbio. 2020, 70, 5607–5612. [Google Scholar] [CrossRef]
- The List of Prokaryotic names with Standing in Nomenclature (LPSN). Available online: https://lpsn.dsmz.de/ (accessed on 12 October 2022).
- Index Fungorum. Available online: http://www.indexfungorum.org/ (accessed on 12 October 2022).
- McIntyre, A.B.; Ounit, R.; Afshinnekoo, E.; Prill, R.I.; Hénaff, E.; Alexander, N.; Minot, S.S.; Danko, D.; Foox, J.; Ahsanuddin, S.; et al. Comprehensive benchmarking and ensemble approaches for metagenomic classifiers. Genome Biol. 2017, 18, 182. [Google Scholar] [CrossRef] [PubMed]
- Beaudry, M.S.; Wang, J.; Kieran, T.J.; Thomas, J.; Bayona-Vásquez, N.J.; Gao, B.; Devault, A.; Brunelle, B.; Lu, K.; Wang, J.-S.; et al. Improved microbial community characterization of 16S rRNA via metagenome hybridization capture enrichment. Front. Microbiol. 2021, 12, 644662. [Google Scholar] [CrossRef]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef]
- Bahram, M.; Anslan, S.; Hildebrand, F.; Bork, P.; Tedersoo, L. Newly designed 16S rRNA metabarcoding primers amplify diverse and novel archaeal taxa from the environment. Env. Microbiol. Rep. 2019, 11, 487–494. [Google Scholar] [CrossRef]
- Hamady, M.; Knight, R. Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome Res. 2009, 19, 1141–1152. [Google Scholar] [CrossRef]
- Baker, G.C.; Smith, J.J.; Cowan, D.A. Review and re-analysis of domain-specific 16S primers. J. Microbiol. Methods 2003, 55, 541–555. [Google Scholar] [CrossRef]
- Tringe, S.G.; Hugenholtz, P. A renaissance for the pioneering 16S rRNA gene. Curr. Opin. Microbiol. 2008, 11, 442–446. [Google Scholar] [CrossRef]
- Youssef, N.; Sheik, C.S.; Krumholz, L.R.; Najar, F.Z.; Roe, B.A.; Elshahed, M.S. Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys. Appl. Environ. Microbiol. 2009, 75, 5227–5236. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Jansson, J.K.; Knight, R. The Earth Microbiome project: Successes and aspirations. BMC Biol. 2014, 12, 69. [Google Scholar] [CrossRef]
- Wu, J.; Gao, W.; Johnson, R.H.; Zhang, W.; Meldrum, D.R. Integrated metagenomic and metatranscriptomic analyses of microbial communities in the meso- and bathypelagic realm of North Pacific Ocean. Mar. Drugs 2013, 11, 3777–3801. [Google Scholar] [CrossRef]
- Morris, R.M.; Rappe, M.S.; Connon, S.A.; Vergin, K.L.; Siebold, W.A.; Carlson, C.A.; Giovannoni, S.J. SAR11 clade dominates ocean surface bacterioplankton communities. Nature 2002, 420, 806–810. [Google Scholar] [CrossRef]
- Dupont, C.L.; Larsson, J.; Yooseph, S.; Ininbergs, K.; Goll, J.; Asplund-Samuelsson, J.; McCrowm, J.P.; Celepli, N.; Zeigler Allen, L.; Ekman, M.; et al. Functional tradeoffs underpin salinity-driven divergence in microbial community composition. PLoS ONE 2014, 9, e89549. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Field, D.; Swift, P.; Newbold, L.; Oliver, A.; Smyth, T.; Somerfield, P.J.; Huse, S.; Joint, I. The seasonal structure of microbial communities in the Western English Channel. Environ. Microbiol. 2009, 11, 3132–3139. [Google Scholar] [CrossRef]
- Shade, A.; Jones, S.E.; Caporaso, J.G.; Handelsman, J.; Knight, R.; Fierer, N.; Gilbert, J.A. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio 2014, 5, e01371-14. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.F.; Riemann, L.; Bertilsson, S. Pyrosequencing reveals contrasting seasonal dynamics of taxa within Baltic Sea bacterioplankton communities. ISME J. 2010, 4, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Lindh, M.V.; Figueroa, D.; Sjöstedt, J.; Baltar, F.; Lundin, D.; Andersson, A.; Legrand, C.; Pinhassi, J. Transplant experiments uncover Baltic Sea basin-specific responses in bacterioplankton community composition and metabolic activities. Front. Microbiol. 2015, 6, 223. [Google Scholar] [CrossRef] [PubMed]
- Kublanov, I.V.; Perevalova, A.A.; Slobodkina, G.B.; Lebedinsky, A.V.; Bidzhieva, S.K.; Kolganova, T.V.; Kaliberda, E.N.; Rumsh, L.D.; Haertlé, T.; Bonch-Osmolovskaya, E.A. Biodiversity of thermophilic prokaryotes with hydrolytic activities in hot springs of Uzon Caldera, Kamchatka (Russia). Appl. Environ. Microbiol. 2009, 75, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.A.; Caporaso, J.G.; Lauber, C.L.; Berg-Lyons, D.; Fierer, N.; Knight, R. PrimerProspector: De novo design and taxonomic analysis of barcoded polymerase chain reaction primers. Bioinformatics 2011, 27, 1159–1161. [Google Scholar] [CrossRef]
- Iasakova, T.R.; Kanapatskiy, T.A.; Toshchakov, S.V.; Korzhenkov, A.A.; Ulyanova, M.O.; Pimenov, N.V. The Baltic Sea methane pockmark microbiome: The new insights into the patterns of relative abundance and ANME niche separation. Mar. Environ. 2022, 173, 105533. [Google Scholar] [CrossRef] [PubMed]
- Dinasquet, J.; Kragh, T.; Schroter, M.L.; Søndergaard, M.; Riemann, L. Functional and compositional succession of bacterioplankton in response to a gradient in bioavailable dissolved organic carbon. EMI 2013, 15, 2616–2628. [Google Scholar] [CrossRef] [PubMed]
- Merhej, V.; Raoult, D. Rickettsial evolution in the light of comparative genomics. Biol. Rev. 2011, 86, 379–405. [Google Scholar] [CrossRef]
- Werren, J.H.; Baldo, L.; Clark, M.E. Wolbachia: Master manipulators of invertebrate biology. Nat. Rev. Microbiol. 2008, 6, 741–751. [Google Scholar] [CrossRef]
- Merhej, V.; Royer-Carenzi, M.; Pontarotti, P.; Raoult, D. Massive comparative genomic analysis reveals convergent evolution of specialized bacteria. Biol. Direct 2009, 4, 13. [Google Scholar] [CrossRef]
- Fournier, P.-E.; El Karkouri, K.; Leroy, Q.; Robert, C.; Giumelli, B.; Renesto, P.; Socolovschi, C.; Parola, P.; Audic, S.; Raoult, D. Analysis of the Rickettsia africae genome reveals that virulence acquisition in Rickettsia species may be explained by genome reduction. BMC Genomics 2009, 10, 166. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Audic, S.; Renesto-Audiffren, P.; Fournier, P.-E.; Barbe, V.; Samson, D.; Roux, V.; Cossart, P.; Weissenbach, J.; Claverie, J.-M.; et al. Mechanisms of evolution in Rickettsia conorii and R. prowazekii. Science 2001, 293, 2093–2098. [Google Scholar] [CrossRef] [PubMed]
- Renvoisé, A.; Merhej, V.; Georgiades, K.; Raoult, D. Intracellular Rickettsiales: Insights into manipulators of eukaryotic cells. Trends Mol. Med. 2011, 17, 573–583. [Google Scholar]
- Vandekerckhove, T.T.; Coomans, A.; Cornelis, K.; Baert, P.; Gillis, M. Use of the Verrucomicrobia-specific probe EUB338-III and fluorescent in situ hybridization for detection of “Candidatus Xiphinematobacter” cells in nematode hosts. Appl. Environ. Microbiol. 2002, 68, 3121–3125. [Google Scholar] [CrossRef]
- Vandekerckhove, T.T.; Navarro, J.B.; Coomans, A.; Hedlund, B.P. Genus II. Candidatus Xiphinematobacter. In Bergey’s Manual of Systematic Bacteriology; Krieg, N.R., Staley, J.T., Hedlund, B.P., Paster, B.J., Ward, N., Ludwig, W., Whitman, W.B., Eds.; Springer: New York, NY, USA, 2011; pp. 838–841. [Google Scholar]
- Dulski, T.; Kozłowski, K.; Ciesielski, S. Habitat and seasonality shape the structure of tench (Tinca tinca L.) gut microbiome. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Vandekerckhove, T.T.; Willems, A.; Gillis, M.; Coomans, A. Occurrence of novel verrucomicrobial species, endosymbiotic in Xiphinema americanum-group species (Nematoda, Longidoridae) and associated with parthenogenesis. Int. J. Syst. Evol. Microbiol. 2000, 50, 2197–2205. [Google Scholar] [CrossRef]
- Bing, X.L.; Yang, J.; Zchori-Fein, E.; Wang, X.W.; Liu, S.S. Characterization of a newly discovered symbiont of the whitefly Bemisia tabaci (Hemiptera: Aleyrodidae). Appl. Environ. Microbiol. 2013, 79, 569–575. [Google Scholar] [CrossRef]
- Giovannoni, S.J.; Rappe, M. Chapter 3: Evolution, diversity, and molecular ecology of marine prokaryotes. In Microbial Ecology of the Oceans; Kirchman, D.L., Ed.; Wiley-Liss, Inc.: Hoboken, NJ, USA, 2000; pp. 47–84. [Google Scholar]
- Lamendella, R.; Strutt, S.; Borglin, S.; Chakraborty, R.; Tas, N.; Mason, O.U.; Hultman, J.; Prestat, E.; Hazen, T.C.; Jansson, J.K. Assessment of the deepwater horizon oil spill impact on Gulf Coast microbial communities. Front. Microbiol. 2014, 5, 130. [Google Scholar] [CrossRef]
- Moisander, P.H.; Sexton, A.D.; Daley, M.C. Stable associations masked by temporal variability in the marine copepod microbiome. PLoS ONE 2015, 10, e0138967. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Species Cultured in the Laboratory | ||
|---|---|---|
| Expected Taxon | Accession Number (Literature Data if Available) | Source of Isolation |
| Acinetobacter johnsonii (**) | KPD 1303 | Rainwater |
| Aeromonas sp. (*) | IOMB 800 | Seawater, the Baltic Sea |
| Aliivibrio fischeri (previously Vibrio fischeri) (**) | KPD 141 | Seawater |
| Alteromonas haloplanktis (*) | IOMB 474 [45] | Seawater, the Baltic Sea |
| Chryseobacterium indoltheticum (**) | KPD 1306 | Rainwater |
| Enterobacter asburiae (**) | KPD 1375 | Pond water |
| Erwinia billingiae (**) | KPD 1325 | Rainwater |
| Klebsiella pneumoniae (*) | IOMB 754 | Seawater, the Baltic Sea |
| Marinomonas sp. (*) | IOMB 406 [45] | Seawater, the Baltic Sea |
| Micrococcus luteus (**) | KPD 778 | Palm surface |
| Novosphingobium resinovorum (**) | KPD 1310 | Rainwater |
| Ochrobactrum sp. (*) | CCNP0038 [46] | Sediment, the Baltic Sea |
| Pantoea vagans (**) | KPD 1311 | Rainwater |
| Paracoccus sp. (*) | IOMB 231 [45] | Seawater, the Baltic Sea |
| Photobacterium sp. (*) | IOMB 384 [45] | Seawater, the Baltic Sea |
| Pseudomonas chlororaphis (**) | KPD 1374 | Pond water |
| Psychrobacter immobilis (**) | KPD 1363 | Fish from Lake Żarnowieckie, Poland |
| Raoultella terrigena (**) | KPD 1302 | Rainwater |
| Rathayibacter caricis (*) | IOMB 359 [45] | Seawater, the Baltic Sea |
| Rheinheimera aquamaris (*) | CCNP0045 [46] | Sediment, the Baltic Sea |
| Serratia liquefaciens (*) | IOMB 517 [45] | Seawater, the Baltic Sea |
| Shewanella baltica (*) | IOMB 300 [45] | Seawater, the Baltic Sea |
| Vibrio harveyi (**) | KPD 143 | Seawater |
| Yersinia massiliensis (**) | KPD 1318 | Rainwater |
| ZymoBIOMICS™ Microbial Community DNA Standard | ||
| Expected taxon | Accession number | Source of extracted DNA |
| Bacillus subtilis | B-354 | Bacteria |
| Cryptococcus neoformans | Y-2534 | Yeast |
| Enterococcus faecalis | B-537 | Bacteria |
| Escherichia coli | B-1109 | Bacteria |
| Lactobacillus fermentum | B-1840 | Bacteria |
| Listeria monocytogenes | B-33116 | Bacteria |
| Pseudomonas aeruginosa | B-3509 | Bacteria |
| Saccharomyces cerevisiae | Y-567 | Yeast |
| Salmonella enterica | B-4212 | Bacteria |
| Staphylococcus aureus | B-41012 | Bacteria |
| Commercial Laboratory | EF | GM | MG |
|---|---|---|---|
| Region of bacterial 16S rRNA | V3–V4 | V3–V4 | V3–V4 |
| Primers | fwd and rev [47,48] * | 341F and 785R [49] ** | V3-F and V4-R [50] *** |
| Type of read | Paired-end | Paired-end | Paired-end |
| Library protocol | In-house sequencing library preparation protocol | 16S metagenomic library preparation guide | 16S metagenomic sequencing library preparation part # 15,044,223 Rev. B |
| Automatic de-multiplexing of raw reads and primary sequence analysis | MiSeq | MiSeq | MiSeq |
| Samples | Sequence Length | Total Sequences | Not Trimmed | Optimal Trimmed | Equal Trimmed |
|---|---|---|---|---|---|
| EF | 284–285 | 88,135 | 59,933 | 64,691 | 66,140 |
| GM | 251 | 103,726 | 19,513 | NA | 17,668 |
| MG | 301 | 164,764 | 6167 | 56,617 | 27,059 |
| Commercial Laboratory | EF | GM | MG |
|---|---|---|---|
| Number of sequences | 88,135 | 103,726 | 164,764 |
| Length of sequences | forward: 285; reverse: 284 | forward and reverse: 251 | forward and reverse: 301 |
| %GC | forward: 53.8; reverse: 54.7 | forward: 56.1; reverse: 53.6 | forward: 54.9; reverse: 54.0 |
| Number of ASVs * | 272/351/535/564 | 137/197/251/265 | 237/344/509/554 |
| Classification rate summary (at least 97% sequence similarity)—domain level | Bacteria: 66,140; archaea: 0 | Bacteria: 17,668; archaea: 0 | Bacteria: 27,032; archaea: 27 |
| Phylum Names From Databases/Updated According to the work of Oren and Garrity [58] | Commercial Laboratories | ||
|---|---|---|---|
| EF | GM | MG | |
| Acidobacteria/ Acidobacteriota corrig. phyl. nov. | 112/1231/1227/1231 | 114/737/748/748 | 152/925/972/931 |
| Actinobacteria/N | 14069/14144/14134/14144 | 2511/2505/2511/2511 | 2532/2583/2566/2583 |
| Armatimonadetes/ Armatimonadota corrig. phyl. nov. | 8/8/8/8 | 0/0/0/0 | 12/12/12/12 |
| Bacteroidetes/ Bacteroidota corrig. phyl. nov. | 10764/10764/11164/11146 | 1624/1624/1744/1742 | 3833/3827/4160/4126 |
| Caldithrix/ Calditrichota corrig. phyl. nov. | 59/93/0/0 | 0/29/0/0 | 0/53/0/0 |
| Calditrichaeota/N | 0/0/75/93 | 0/0/29/29 | 0/0/5362 |
| Chlorobi/ Chlorobiota corrig. phyl. nov. | 220/382/0/0 | 22/116/0/0 | 24/291/0/0 |
| Chloroflexi/ Chloroflexota corrig. phyl. nov. | 396/396/396/396 | 477/675/595/675 | 1415/1862/1693/1865 |
| Cyanobacteria/N | 398/420/420/420 | 610/649/649/649 | 1303/1341/1372/1370 |
| Dadabacteria/N | 0/0/0/0 | 0/0/0/0 | 0/0/0/5 |
| Deinococcus-Thermus/ Deinococcota corrig. phyl. nov. | 0/0/0/0 | 0/0/0/0 | 0/0/0/4 |
| Dependentiae/N | 0/0/0/82 | 0/0/0/0 | 0/0/0/19 |
| Epsilonbacteraeota/N | 0/0/328/328 | 0/0/75/75 | 0/0/170/170 |
| Fibrobacteres/ Fibrobacterota corrig. phyl. nov. | 22/22/22/22 | 0/0/0/0 | 15/15/15/15 |
| Firmicutes/N | 113/41/41/41 | 43/30/72/30 | 172/101/147/101 |
| Fusobacteria/ Fusobacteriota corrig. phyl. nov. | 20/20/20/20 | 12/12/12/12 | 0/0/0/0 |
| GN02/N | 9/9/0/0 | 0/0/0/0 | 0/0/0/0 |
| GN04/N | 5/5/0/0 | 0/0/0/0 | 10/10/0/0 |
| Gemmatimonadetes/ Gemmatimonadota corrig. phyl. nov. | 113/302/288/302 | 16/94/94/94 | 22/181/181/181 |
| Kiritimatiellaeota/ Kirimitatiellota corrig. phyl. nov. | 0/0/15/15 | 0/0/26/26 | 0/0/49/51 |
| Latescibacteria/N | 0/0/28/41 | 0/0/4/4 | 0/0/68/72 |
| Modulibacteria/N | 0/0/0/0 | 0/0/0/0 | 0/0/6/6 |
| Nitrospinae/ Nitrospinota corrig. phyl. nov. | 0/0/0/0 | 0/0/46/46 | 0/0/64/64 |
| Nitrospirae/ Nitrospirota corrig. phyl. nov. | 172/213/207/213 | 97/112/112/112 | 60/92/92/92 |
| Omnitrophicaeota/N | 0/0/7/7 | 0/0/0/0 | 0/0/0/2 |
| Patescibacteria/N | 0/0/9/9 | 0/0/20/20 | 0/0/103/108 |
| Planctomycetes/ Planctomycetota corrig. phyl. nov. | 61/111/123/111 | 1606/1651/1627/1651 | 2604/2997/2854/3000 |
| Poribacteria/N | 0/0/0/0 | 0/0/0/0 | 0/0/2/2 |
| Proteobacteria/N | 37162/37548/37000/37220 | 8365/8443/8318/8322 | 10546/10773/10527/10554 |
| Spirochaetes/ Spirochaetota corrig. phyl. nov. | 14/14/14/14 | 0/0/0/0 | 0/0/0/0 |
| TM6/N | 82/82/0/0 | 0/0/0/0 | 6/19/0/0 |
| TM7/N | 0/0/0/0 | 0/0/0/0 | 9/65/0/0 |
| Tenericutes/N | 0/0/35/35 | 0/0/0/0 | 0/0/38/38 |
| Verrucomicrobia/ Verrucomicrobiota corrig. phyl. nov. | 223/242/223/223 | 821/939/915/913 | 1281/1565/1514/1514 |
| WS3/N | 14/41/0/0 | 0/4/0/7 | 17/74/0/14 |
| Zixibacteria/N | 0/0/380/5 | 0/0/71/0 | 0/0/354/10 |
| Putative Bacterial Endosymbiont | Commercial Laboratory | ||
|---|---|---|---|
| EF* | GM* | MG* | |
| “Candidatus Xiphinematobacter” | P(28)/N/N/N | N/N/N/N | N/N/N/N |
| Rickettsiales | P(127)/P(74)/P(16)/P(177) | N/P(14)/N/N | P (13)/P(28)/P(13)/P(13) |
| “Candidatus Portiera” | N/P(509)/N/N | N/P(107)/N/N | N/P(95)/N/N |
| Bacterial Species Cultured in the Laboratory | ||||||
|---|---|---|---|---|---|---|
| Expected Taxon | The Concentration of DNA (ng/µL) | Identified Taxon Using Greengenes/SILVA Databases | The Theoretical Microbial Composition in “Mock Communities” (%) | Results from Three Laboratories (%) | ||
| EF | GM | MG | ||||
| Acinetobacter johnsonii, Bouvet and Grimont 1986 | 9.7 | Acinetobacter johnsonii | 2.57 | 7.90 | 7.31 | 5.29 |
| Aeromonas sp., Stanier 1943 (Approved Lists 1980) | 9.00 | Gammaproteobacteria sp.1/Aeromonas sp. | 2.39 | 2.03 | 1.59 | 1.47 |
| Aliivibrio fischeri (previously Vibrio fischeri) (Beijerinck 1889), Urbanczyk et al., 2007 (previously Vibrio fischeri (Beijerinck 1889), Lehmann and Neumann 1896 (Approved Lists 1980)) | 7.2 | Vibrio sp. | 1.91 | 0.00 | 0.02 | 0.03 |
| Alteromonas haloplanktis (ZoBell and Upham 1944), Reichelt and Baumann 1973 | 8.50 | Alteromonadaceae sp./Alteromonas sp. | 2.25 | 0.02 | 0.01 | 0.01 |
| Chryseobacterium indoltheticum (Campbell and Williams 1951), Vandamme et al., 1994 | 38.4 | Chryseobacterium sp./Chryseobacterium indoltheticum | 10.19 | 33.05 | 25.62 | 28.90 |
| Enterobacter asburiae, Brenner et al., 1988 | 17.1 | Enterobacteriaceae sp.1/Enterobacter asburiae | 4.54 | 8.36 | 8.28 | 10.16 |
| Erwinia billingiae, Mergaert et al., 1999 | 11.4 | Erwinia sp./Erwinia billingiae | 3.02 | 0.05 | 0.14 | 0.33 |
| Klebsiella pneumoniae (Schroeter 1886), Trevisan 1887 (Approved Lists 1980) | 23.10 | Klebsiella sp./Klebsiella pneumoniae | 6.13 | 0.07 | 0.30 | 0.41 |
| Marinomonas sp., Landschoot and De Ley 1984 | 32.30 | Marinomonas sp. | 8.57 | 2.61 | 5.61 | 8.10 |
| Micrococcus luteus (Schroeter 1872), Cohn 1872 (Approved Lists 1980) | 9.6 | Micrococcus luteus | 2.55 | 0.28 | 2.04 | 1.13 |
| Novosphingobium resinovorum (Delaporte and Daste 1956), Lim et al., 2007 | 13.3 | Novosphingobium resinovorum | 3.53 | 1.00 | 2.47 | 0.47 |
| Ochrobactrum sp., Holmes et al., 1988 | 4.00 | Ochrobactrum sp. | 1.06 | 0.09 | 0.23 | 0.05 |
| Pantoea vagans, Brady et al., 2009 | 12.9 | Gammaproteobacteria sp.2/Pantoea vagans | 3.42 | 4.24 | 3.32 | 3.61 |
| Paracoccus sp., Davis 1969 (Approved Lists 1980) | 6.10 | Paracoccus sp. | 1.62 | 0.12 | 0.47 | 0.13 |
| Photobacterium sp., Beijerinck 1889 (Approved Lists 1980) | 11.40 | Photobacterium sp. | 3.02 | 1.03 | 1.84 | 1.62 |
| Pseudomonas chlororaphis (Guignard and Sauvageau 1894), Bergey et al., 1930 (Approved Lists 1980) | 10.6 | Pseudomonas sp./Pseudomonas chlororaphis | 2.81 | 0.07 | 0.61 | 0.83 |
| Psychrobacter immobilis, Juni and Heym 1986 | 14.5 | Psychrobacter sp./Psychrobacter immobilis | 3.85 | 13.84 | 12.52 | 10.20 |
| Raoultella terrigena (Izard et al., 1981), Drancourt et al., 2001 | 19.7 | Enterobacteriaceae sp.2/Raoultella terrigena | 5.23 | 4.64 | 3.39 | 4.34 |
| Rathayibacter caricis, Dorofeeva et al., 2002 | 7.50 | Rathayibacter caricis | 1.99 | 0.00 | 0.08 | 0.01 |
| Rheinheimera aquamaris, Yoon et al., 2007 | 44.70 | Rheinheimera sp. | 11.86 | 2.07 | 2.22 | 2.76 |
| Serratia liquefaciens (Grimes and Hennerty 1931), Bascomb et al., 1971 (Approved Lists 1980) | 13.10 | Serratia sp./Serratia liquefaciens | 3.47 | 0.00 | 0.05 | 0.06 |
| Shewanella baltica, Ziemke et al., 1998 | 16.70 | Shewanella sp./Shewanella baltica | 4.43 | 3.54 | 3.73 | 3.37 |
| Vibrio harveyi (Johnson and Shunk 1936), Baumann et al., 1981 | 9 | Vibrio sp.2/Vibrio harveyi | 2.39 | 2.12 | 2.00 | 1.30 |
| Yersinia massiliensis, Merhej et al., 2008 | 17.2 | Yersinia sp./Yersinia massiliensis | 4.56 | 5.54 | 5.67 | 6.01 |
| ZymoBIOMICS™ Microbial Community DNA Standard | ||||||
| Expected Taxon * | The Theoretical Concentration of DNA (ng/µL) | Identified Taxon Using the SILVA/Greengenes Databases | The Theoretical Microbial Composition in “Mock Communities” (%) | Results from Three Laboratories (%) | ||
| EF | GM | MG | ||||
| Bacillus subtilis (Ehrenberg 1835), Cohn 1872 (Approved Lists 1980) | 1.2 | Bacillus sp./Bacillus subtilis | 0.46 | 0.93 | 1.48 | 1.32 |
| Cryptococcus neoformans (San Felice), Vuill. 1901 | 0.2 | n/a | n/a | 0.00 | 0.00 | 0.00 |
| Enterococcus faecalis (Andrewes and Horder 1906), Schleifer and Kilpper-Bälz 1984 | 1.2 | Enterococcus sp./Enterococcus faecalis | 0.26 | 0.68 | 1.32 | 0.62 |
| Escherichia coli (Migula 1895), Castellani and Chalmers 1919 (Approved Lists 1980) | 1.2 | Escherichia coli | 0.27 | 0.12 | 0.35 | 0.36 |
| Lactobacillus fermentum, Beijerinck 1901 (Approved Lists 1980) | 1.2 | Lactobacillus sp./Lactobacillus fermentum | 0.49 | 0.65 | 1.27 | 0.84 |
| Listeria monocytogenes (Murray et al., 1926), Pirie 1940 (Approved Lists 1980) | 1.2 | Listeria sp./Listeria monocytogenes | 0.38 | 0.51 | 1.48 | 1.00 |
| Pseudomonas aeruginosa (Schroeter 1872), Migula 1900 (Approved Lists 1980) | 1.2 | Pseudomonas sp./Pseudomonas aeruginosa | 0.11 | 0.07 | 0.11 | 0.15 |
| Saccharomyces cerevisiae (Desm.), Meyen 1838 | 0.2 | n/a | n/a | 0.00 | 0.00 | 0.00 |
| Salmonella enterica (ex Kauffmann and Edwards 1952), Le Minor and Popoff 1987 | 1.2 | Salmonella enterica | 0.28 | 3.75 | 3.33 | 4.30 |
| Staphylococcus aureus, Rosenbach 1884 (Approved Lists 1980) | 1.2 | Staphylococcus aureus | 0.41 | 0.63 | 1.17 | 0.86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mioduchowska, M.; Iglikowska, A.; Jastrzębski, J.P.; Kaczorowska, A.-K.; Kotlarska, E.; Trzebny, A.; Weydmann-Zwolicka, A. Challenges of Comparing Marine Microbiome Community Composition Data Provided by Different Commercial Laboratories and Classification Databases. Water 2022, 14, 3855. https://doi.org/10.3390/w14233855
Mioduchowska M, Iglikowska A, Jastrzębski JP, Kaczorowska A-K, Kotlarska E, Trzebny A, Weydmann-Zwolicka A. Challenges of Comparing Marine Microbiome Community Composition Data Provided by Different Commercial Laboratories and Classification Databases. Water. 2022; 14(23):3855. https://doi.org/10.3390/w14233855
Chicago/Turabian StyleMioduchowska, Monika, Anna Iglikowska, Jan P. Jastrzębski, Anna-Karina Kaczorowska, Ewa Kotlarska, Artur Trzebny, and Agata Weydmann-Zwolicka. 2022. "Challenges of Comparing Marine Microbiome Community Composition Data Provided by Different Commercial Laboratories and Classification Databases" Water 14, no. 23: 3855. https://doi.org/10.3390/w14233855
APA StyleMioduchowska, M., Iglikowska, A., Jastrzębski, J. P., Kaczorowska, A.-K., Kotlarska, E., Trzebny, A., & Weydmann-Zwolicka, A. (2022). Challenges of Comparing Marine Microbiome Community Composition Data Provided by Different Commercial Laboratories and Classification Databases. Water, 14(23), 3855. https://doi.org/10.3390/w14233855