
Comparison of DNA Extraction Methods for the Direct Quantification of Bacteria from Water Using Quantitative Real-Time PCR

Abstract
:1. Introduction
2. Materials and Methodology
2.1. Growth and Maintenance of Bacterial Strains
2.2. Comparison of Optimised DNA Extraction Method with Commercial Water-Testing Kits
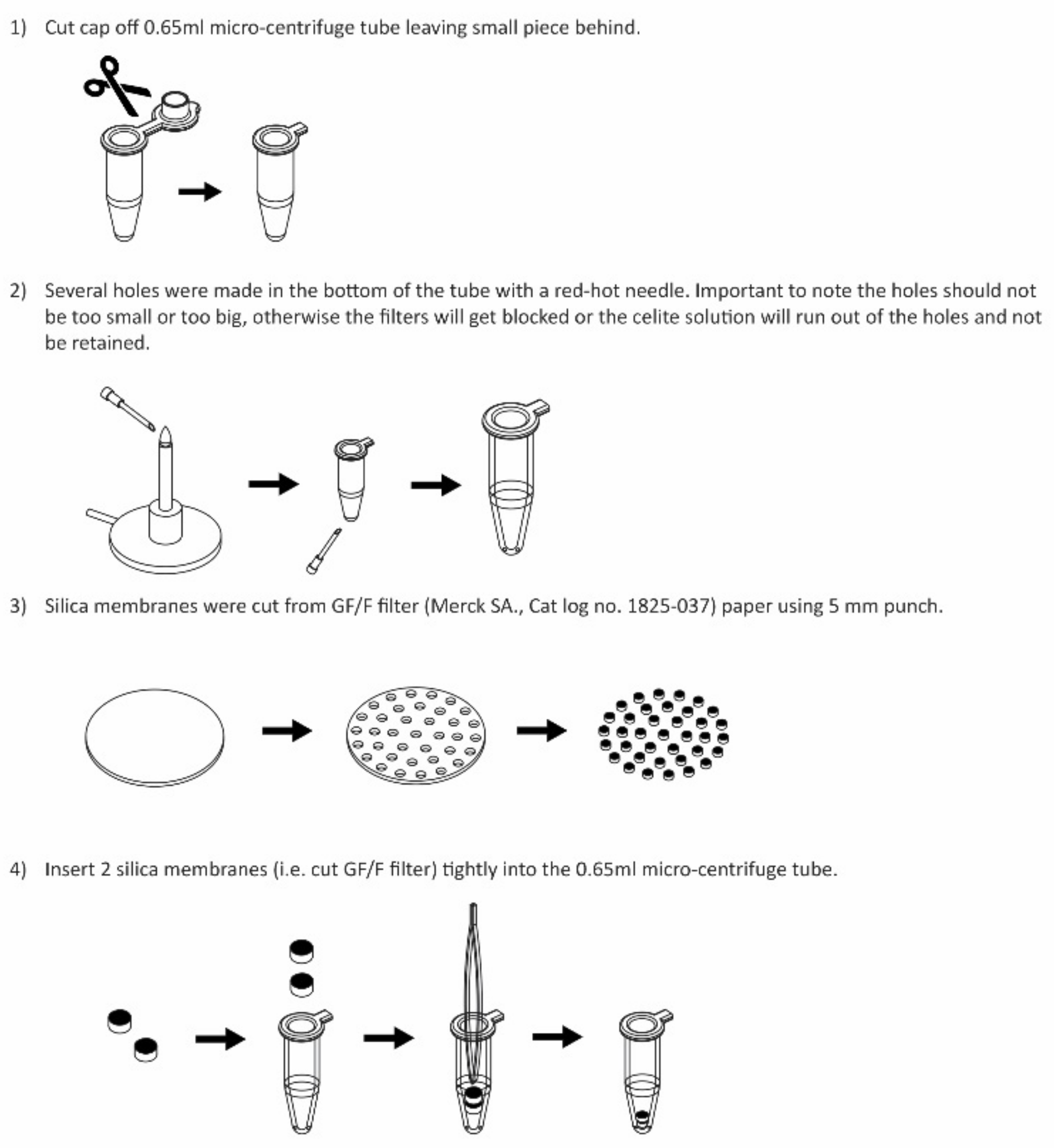
2.2.1. DNA Extraction
Buffer Preparations
2.2.2. Quantitative Real-Time Polymerase Chain Reaction (q-PCR)
2.3. Statistical Analysis
3. Results and Discussion
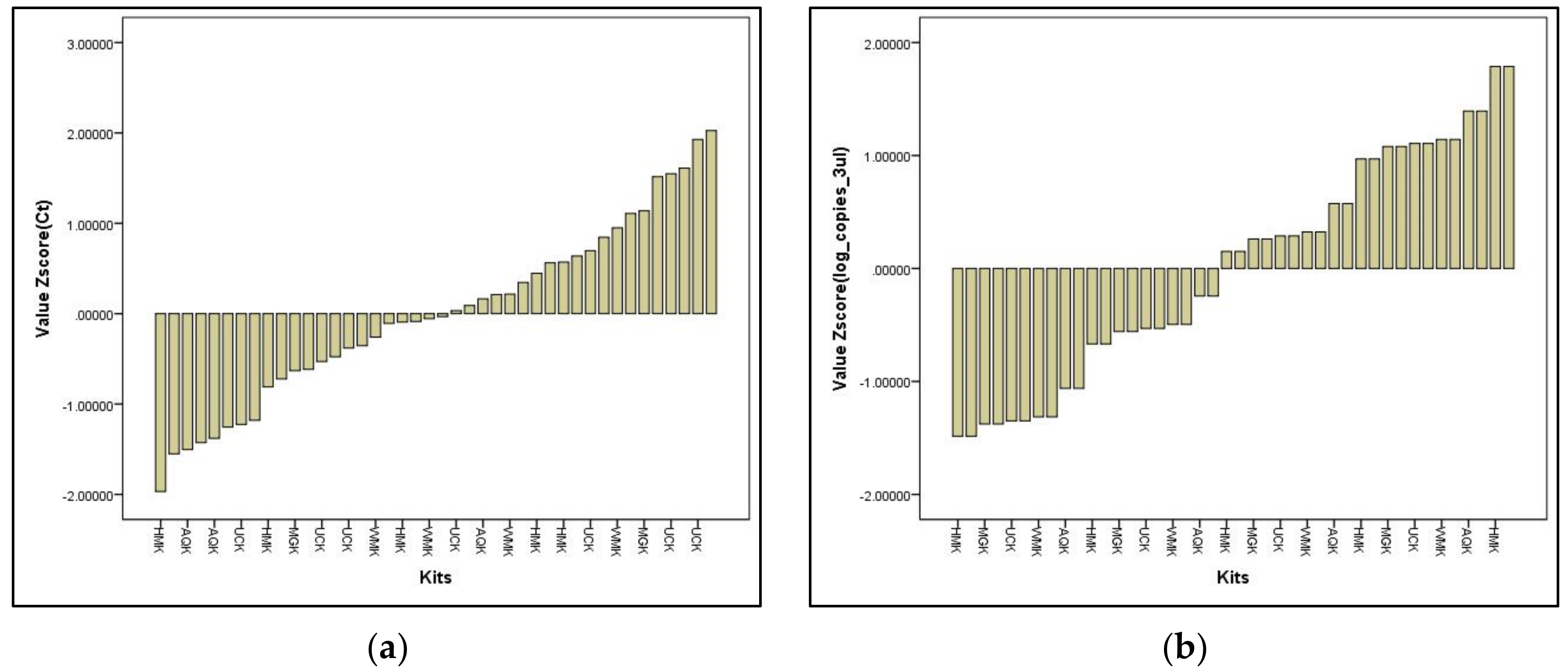
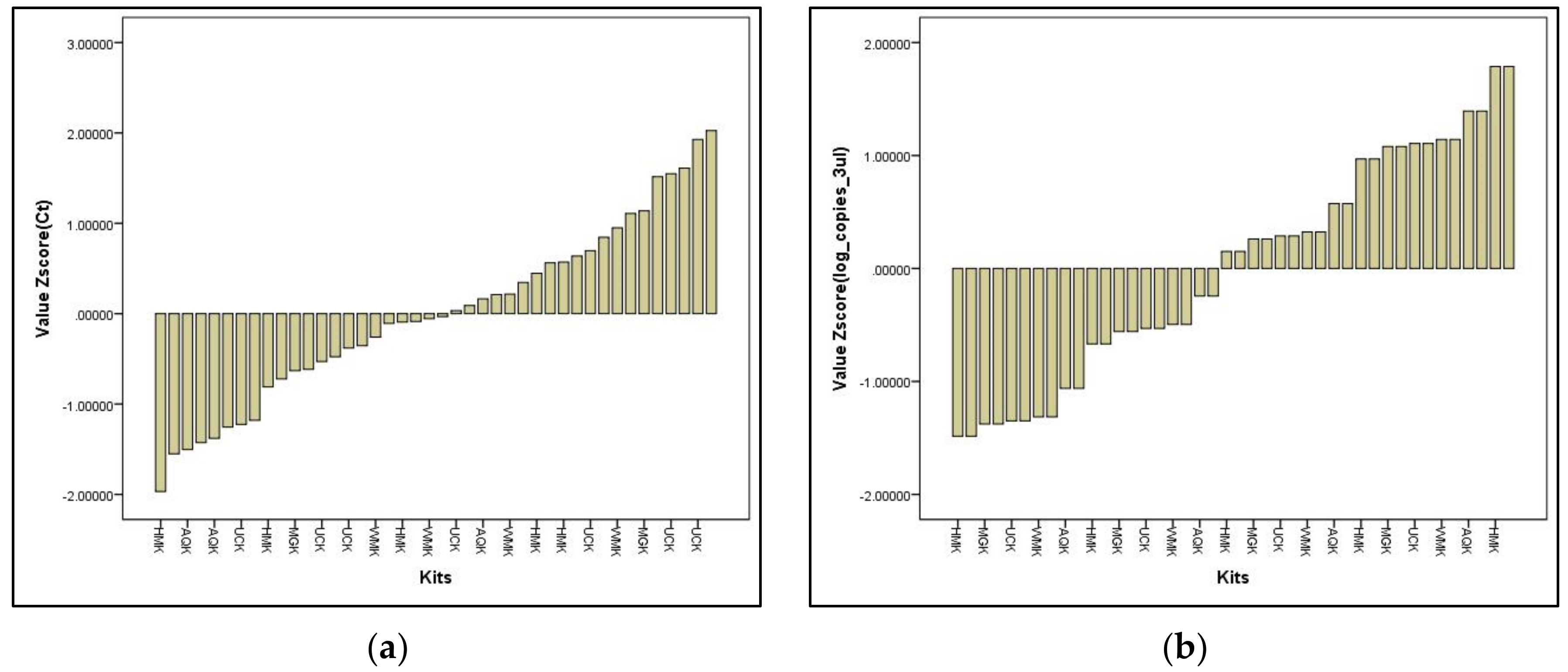
Validation of the In-House DNA Extraction Method against Commercial Water-Testing Kits
- Satisfactory performance
- Questionable performance
- Unsatisfactory performance
- (1)
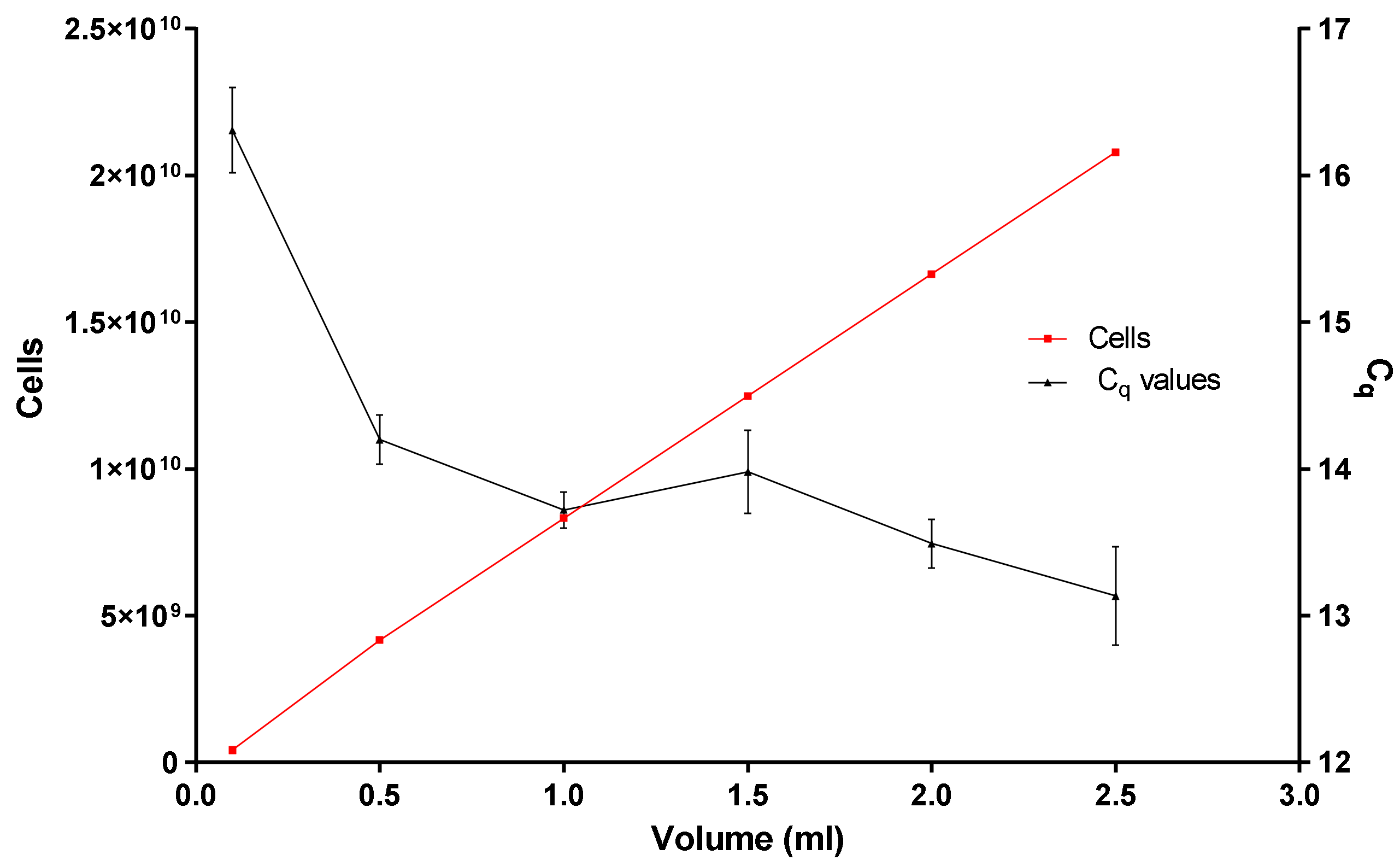
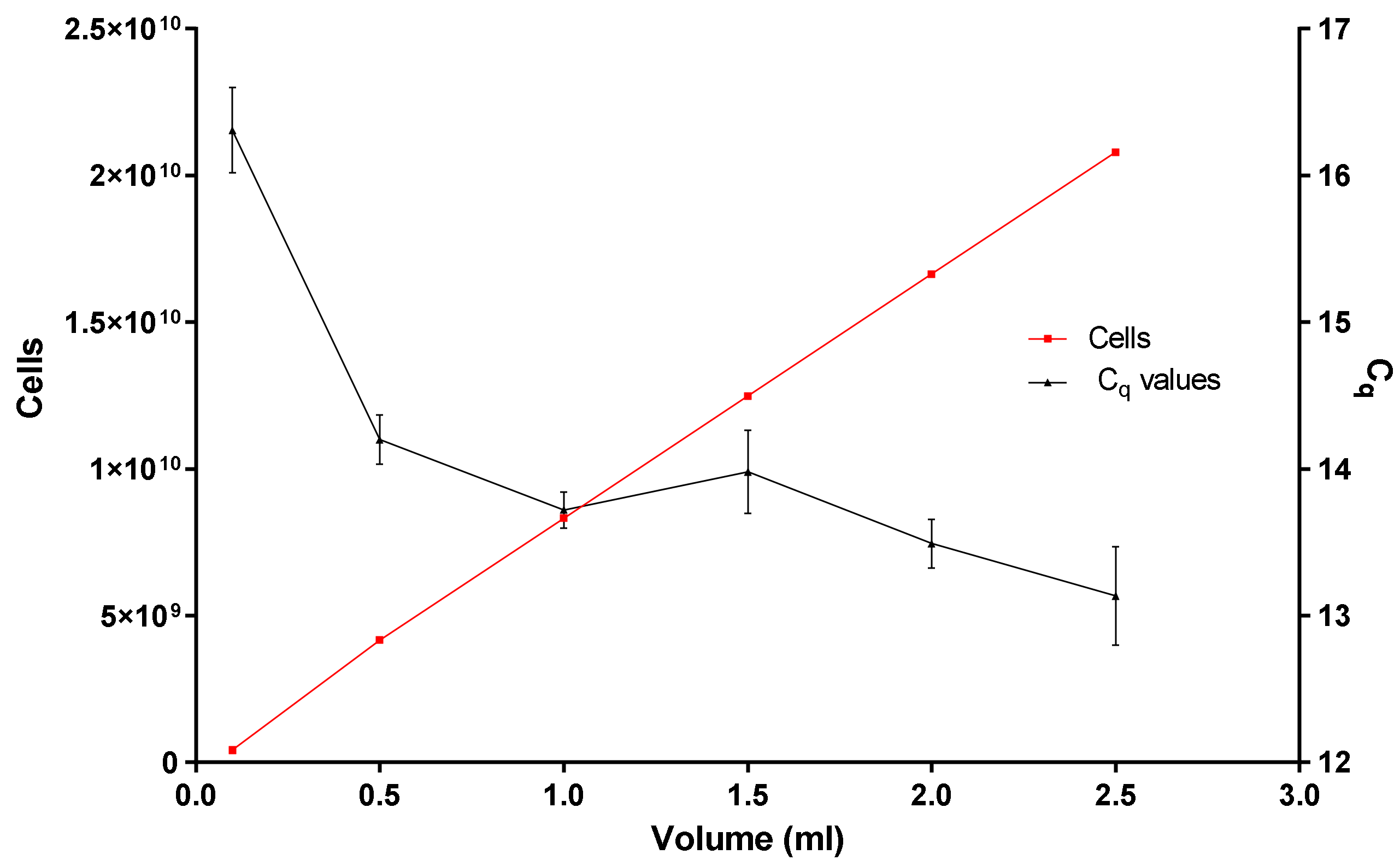
- Is the binding and loading capacity of the in-house DNA extraction method (Figure 5) responsible for a low copy number? The binding and loading capacity of the optimized DNA extraction method indicates that if you pool together concentrated E. coli cells for 0.1 or even 2.5 mL, the q-PCR Cq values remain in the region of 12.5 and 16.5, even though there is an exponential increase in the concentration of E. coli cells (Figure 5).
- (2)
- Why is there no substantial difference in the q-PCR values? It could be due to variables or limiting factors in the PCR reaction. Typically, a PCR reaction begins exponentially, then enters a quasi-linear phase, then plateaus. Several factors have been presumed to contribute to this plateau: (1) utilization of substrates (dNTPs and primer concentration), (2) thermal inactivation and limiting concentration of DNA polymerase, (3) the effect of DNA concentration as well as the effect of background DNA [22,23].
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mendes Silva, D.; Domingues, L. On the track for an efficient detection of Escherichia coli in water: A review on PCR-based methods. Ecotoxicol. Environ. Saf. 2015, 113, 400–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, R.G.; Wildeboer, D. E. coli as an Indicator of Contamination and Health Risk in Environmental Waters. In Escherichia coli—Recent Advances on Physiology, Pathogenesis and Biotechnological Applications; InTech: London, UK, 2017. [Google Scholar]
- Jafari, A.; Aslani, M.; Bouzari, S. Escherichia coli: A brief review of diarrheagenic pathotypes and their role in diarrheal diseases in Iran. Iran. J. Microbiol. 2012, 4, 102–117. [Google Scholar] [PubMed]
- Lemarchand, K.; Berthiaume, F.; Maynard, C.; Harel, J.; Payment, P.; Bayardelle, P.; Masson, L.; Brousseau, R. Optimization of microbial DNA extraction and purification from raw wastewater samples for downstream pathogen detection by microarrays. J. Microbiol. Methods 2005, 63, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, X.; Wu, J.; Coin, L.; O’Brien, J.; Hai, F.; Jiang, G. Molecular Methods for Pathogenic Bacteria Detection and Recent Advances in Wastewater Analysis. Water 2021, 13, 3551. [Google Scholar] [CrossRef]
- Banu, O.K.; George, B.T.; Paul, J. Development of a competitive PCR assay for the quantification of total Escherichia coli DNA in water. Afr. J. Biotechnol. 2010, 9, 564–572. Available online: http://www.academicjournals.org/AJB (accessed on 14 November 2022).
- Omar, K.B.; Barnard, T.G. Detection of diarrhoeagenic Escherichia coli in clinical and environmental water sources in South Africa using single-step 11-gene m-PCR. World J. Microbiol. Biotechnol. 2014, 30, 2663–2671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkurt, M. Comparison between modified DNA extraction protocols and commercial isolation kits in grapevine (Vitis vinifera L.). Genet. Mol. Res. GMR 2012, 11, 2343–2351. [Google Scholar] [CrossRef] [PubMed]
- McOrist, A.L.; Jackson, M.; Bird, A.R. A comparison of five methods for extraction of bacterial DNA from human faecal samples. J. Microbiol. Methods 2002, 50, 131–139. [Google Scholar] [CrossRef]
- Hu, Q.; Liu, Y.; Yi, S.; Huang, D. A comparison of four methods for PCR inhibitor removal. Forensic Sci. Int. Genet. 2015, 16, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Boom, R.; Sol, C.; Beld, M.; Weel, J.; Goudsmit, J.; Wertheim-Van Dillen, P. Improved Silica-Guanidiniumthiocyanate DNA Isolation Procedure Based on Selective Binding of Bovine Alpha-Casein to Silica Particles. J. Clin. Microbiol. 1999, 37, 615–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoorzook, K.B.; Barnard, T.G. Culture independent DNA extraction method for bacterial cells concentrated from water. MethodsX 2022, 9, 101653. [Google Scholar] [CrossRef] [PubMed]
- Omar, K.B.; Potgieter, N.; Barnard, T.G. Development of a rapid screening method for the detection of pathogenic Escherichia coli using a combination of Colilert® Quanti-Trays/2000 and PCR. Water Sci. Technol. Water Supply 2010, 10, 7–13. [Google Scholar] [CrossRef]
- Borodina, T.A.; Lehrach, H.; Soldatov, A. DNA purification on homemade silica spin-columns. Anal. Biochem. 2003, 321, 135–137. [Google Scholar] [CrossRef]
- Lee, C.L.; Ow, D.S.W.; Oh, S.K.W. Quantitative real-time polymerase chain reaction for determination of plasmid copy number in bacteria. J. Microbiol. Methods 2006, 65, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.A.; Weagant, S.D.; Feng, P. Glutamate Decarboxylase genes as a pre-screening marker for detection of pathogenic Escherichia coli groups. Appl. Environ. Microbiol. 2001, 67, 3110–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, A.; Robertson, M.L.; Lowden, M.; Ibarra, J.A.; Puente, J.L.; Finlay, B.B. Transcriptional inhibitor of virulence factors in enteropathogenic Escherichia coli. Antimicrob. Agents Chemother. 2005, 49, 4101–4109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlen, Y.; McNair, A.; Perseguers, S.; Mazza, C.; Mermod, N. Statistical significance of quantitative PCR. BMC Bioinform. 2007, 20, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.S.; Shipley, G.L.; Pfaffl, M.W. Real-Time PCR; Dorak, M., Ed.; Taylor & Francis: Abingdon, UK, 2007. [Google Scholar]
- Chandelier, A.; Ivors, K.; Garbelotto, M.; Zini, J.; Laurent, F.; Cavelier, M. Validation of a Real-Time PCR Method for the Detection of Phytophthora Ramorum 1. 2006. Available online: http://www.eppo.org/QUARANTINE/ (accessed on 14 November 2022).
- Ellison, S.L.R.; Barwick, V.J.; Farrant, T.J.D. Practical Statistics for the Analytical Scientist; Royal Society of Chemistry: Cambridge, UK, 2009. [Google Scholar]
- Kainz, P. The PCR plateau phase—Towards an understanding of its limitations. Biochim. Biophys. Acta-Gene Struct. Expr. 2000, 1494, 23–27. [Google Scholar] [CrossRef]
- Lee, D.Y.; Lauder, H.; Cruwys, H.; Falletta, P.; Beaudette, L.A. Development and application of an oligonucleotide microarray and real-time quantitative PCR for detection of wastewater bacterial pathogens. Sci. Total Environ. 2008, 398, 203–211. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Sequence (5′-3′) | Amplicon Size | Patho-Type | Reference |
|---|---|---|---|---|
| gadAB-F gadAB-R P | GCGGAAGTCCCAGACGATATCC GCTACACGTACAGCTACAGCTA r-CGGTGRCMGGAMGCRA-q | 670 bp | All E. coli strains | Designed by Sophi Breniere (Sigma France) [16] |
| Commercial Kits | Initial Concentration (ng/µL) | Copies/3 µL |
|---|---|---|
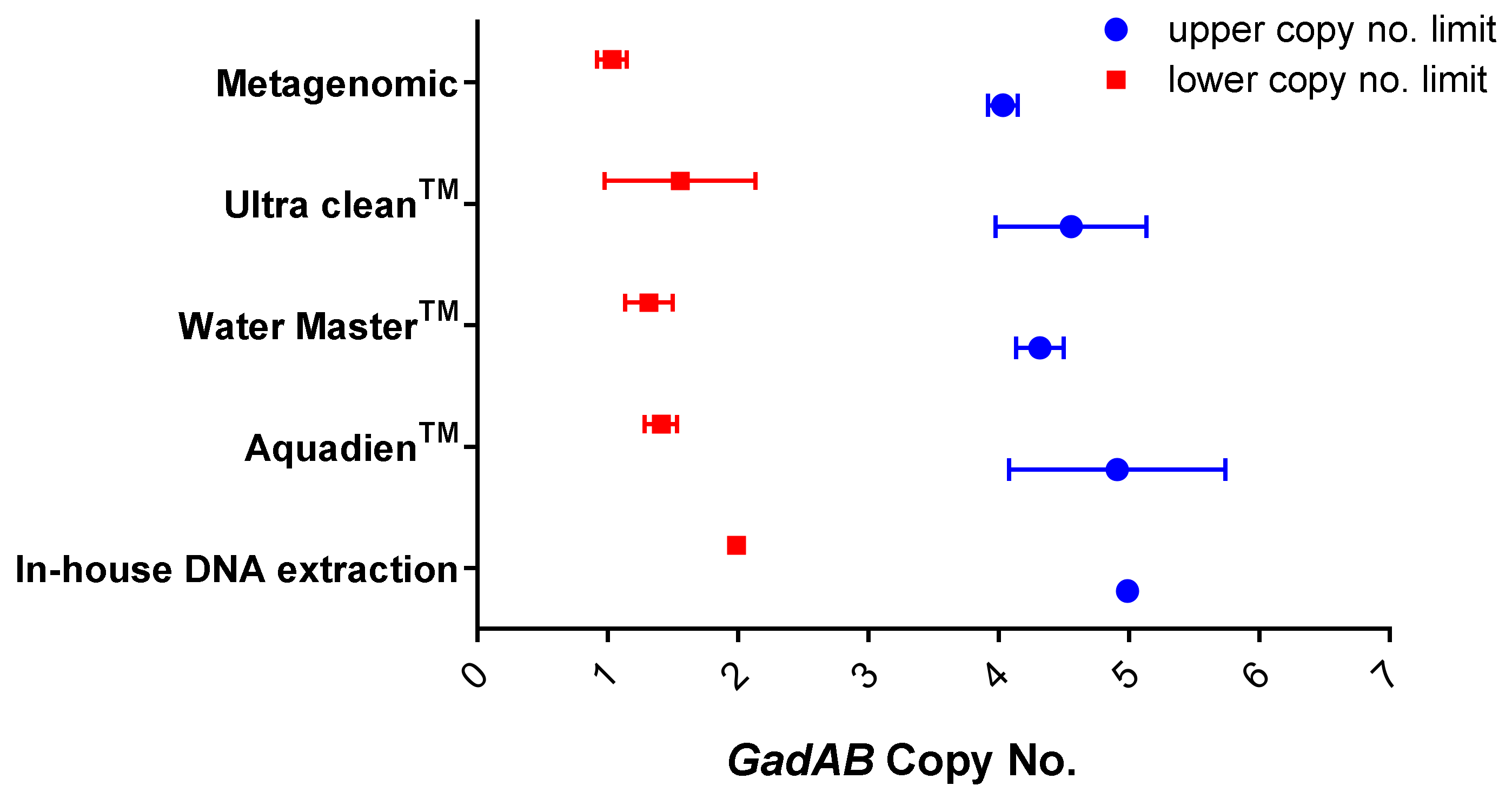
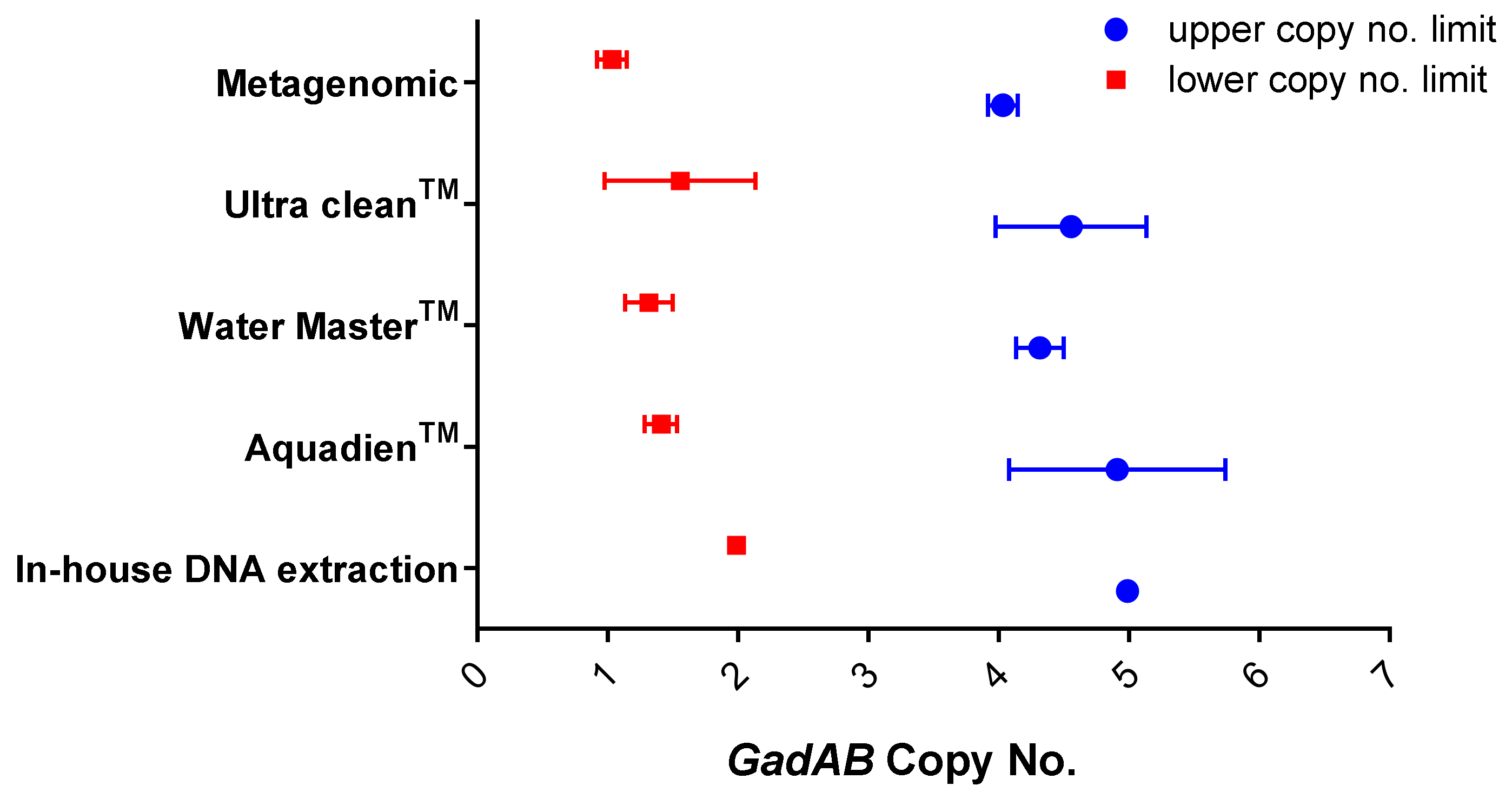
| In-house DNA extraction | 4.93 | 1.9 × 106 |
| AquadienTM kit | 5.4 | 2.7 × 106 |
| Ultra CleanTM water DNA isolation kit | 1.6 | 7.3 × 105 |
| Water MasterTM DNA purification kit | 4.79 | 1.9 × 105 |
| Metagenomic DNA isolation kit | 1.5 | 5.9 × 104 |
| In-House DNA Extraction | Ultra CleanTM Water | AquadienTM | Metagen-Omic | Water MasterTM | ||
|---|---|---|---|---|---|---|
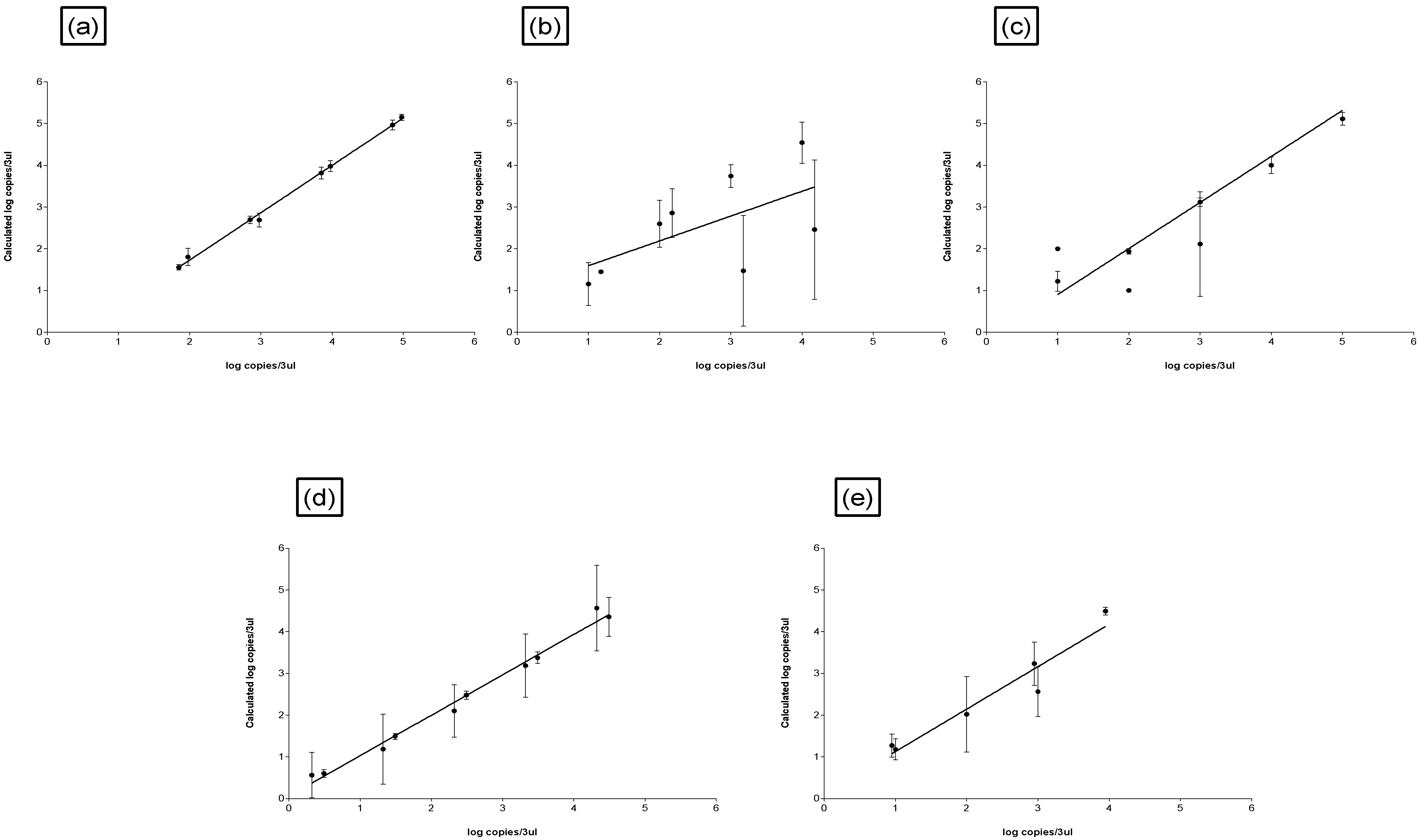
| Input DNA | R2 | 0.99 | 0.98 | 0.92 | 0.65 | 0.34 |
| Slope | −3.48 to −3.65 | −3.9 | −3.6 to −2.1 | −2.5 | −5.7 | |
| Y-intercept | 31.5 to 34 | 32 to 39 | 32 to 33 | 28 to 33 | 36 | |
| Input DNA vs. Calc. DNA | R2 | 0.99 | 0.43 | 0.92 | 0.81 | 0.24 |
| Slope | 1.1 | 0.49 to 1.72 | 0.99 | 0.8 | 0.5 | |
| Y-intercept | −0.41 | −2.2 to 1.8 | −0.36 to 0.35 | −0.6 to 0.8 | −0.59 to 2.1 |
| Treatment Name | Mean | Std. Dev | Std. Error | p-Value | CV |
|---|---|---|---|---|---|
| In-house DNA extraction | 0.991 | 0.00354 | 0% | ||
| Ultra CleanTM kit | 0.598 | 0.534 | 89.57% | ||
| In-house DNA extraction vs. Ultra CleanTM kit | 0.393 | 0.538 | 2.228 | 0.159 ** | |
| AquadienTM kit | 0.904 | 0.105 | 11.72% | ||
| In-house DNA extraction vs. AquadienTM kit | 0.0868 | 0.108 | −2.321 | 0.142 ** | |
| Water MasterTM kit | 0.64 | 0.147 | 22.10% | ||
| In-house DNA extraction vs. Water MasterTM kit | 0.351 | 0.143 | 3.663 | 0.021 * | |
| Metagenomic kit | 0.707 | 0.0823 | 11.95% | ||
| In-house DNA extraction vs. Metagenomic kit | 0.284 | 0.0788 | −1.036 | 0.0512 ** |
| Name | Cost per Kit | Cost/Reaction | Volume Eluted (uL) | Time Taken for 24 Samples | Additional Equipment/Reagents (Not Supplied) | Cautionary for Method |
|---|---|---|---|---|---|---|
| Ultra CleanTM water DNA isolation kit (0.45 μm) | ~R18895 plus ~R13216 water filters for 25 reactions, excl. VAT | R520 | 3000 | ~5 h |
|
|
| AquadienTM kit (Discontinued) | ~R12834 for 100 reactions excl. VAT | R150 | 100 | ~4 h |
|
|
| Metagenomic DNA isolation kits for water | ~R3900 for 20 reactions excl. VAT | R170 | 50 | ~4 h |
|
|
| Water MasterTM DNA purification kit (Discontinued) | ~R3410 for 20 reactions, excl. VAT | R205 | 60 | ~4 h |
|
|
| In-house DNA extraction method | ~R900 for 25 reactions incl. VAT | R40 | 100 | ~3 h |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoorzook, K.B.; Barnard, T.G. Comparison of DNA Extraction Methods for the Direct Quantification of Bacteria from Water Using Quantitative Real-Time PCR. Water 2022, 14, 3736. https://doi.org/10.3390/w14223736
Hoorzook KB, Barnard TG. Comparison of DNA Extraction Methods for the Direct Quantification of Bacteria from Water Using Quantitative Real-Time PCR. Water. 2022; 14(22):3736. https://doi.org/10.3390/w14223736
Chicago/Turabian StyleHoorzook, Kousar Banu, and Tobias George Barnard. 2022. "Comparison of DNA Extraction Methods for the Direct Quantification of Bacteria from Water Using Quantitative Real-Time PCR" Water 14, no. 22: 3736. https://doi.org/10.3390/w14223736