
Application of Coagulation and Foam Concentration Method to Quantify Waterborne Pathogens in River Water Samples

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
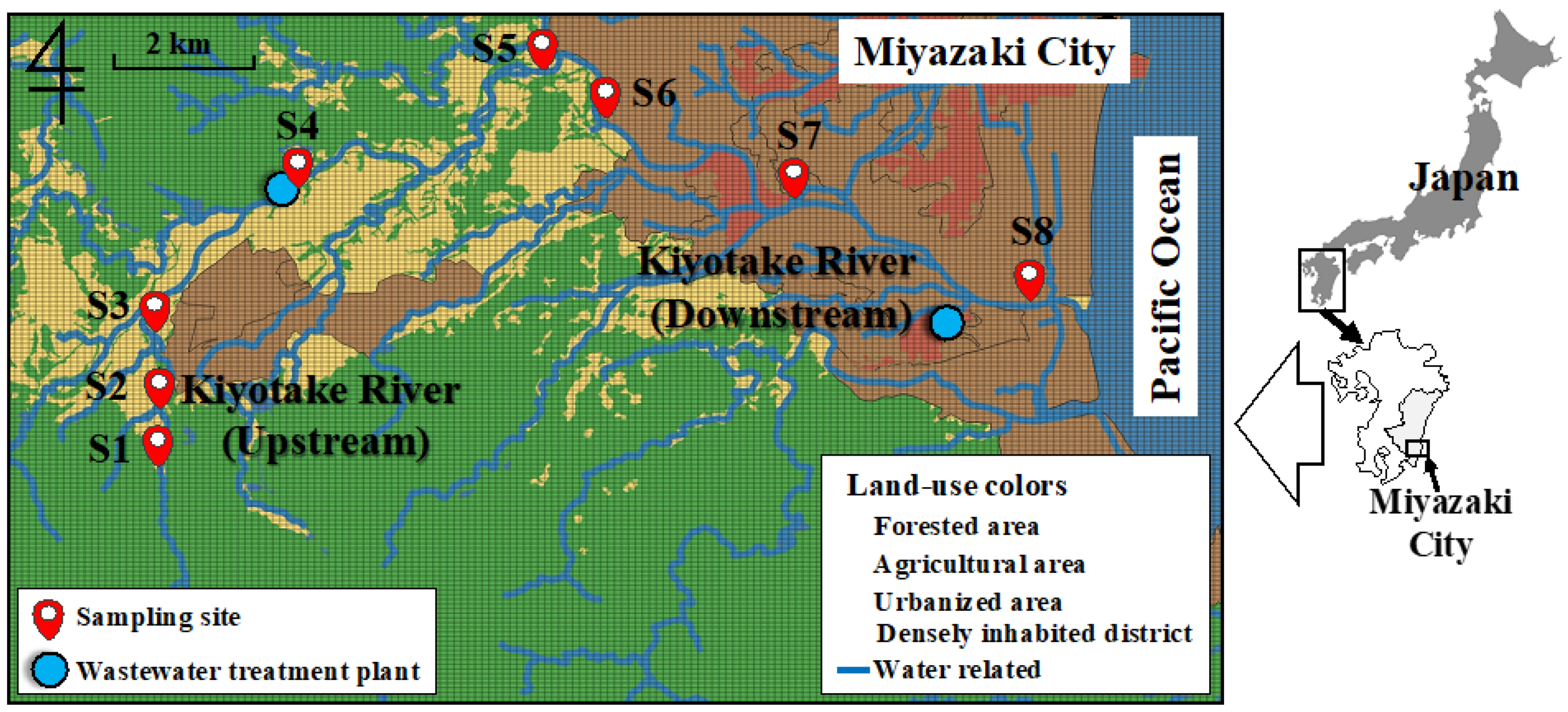
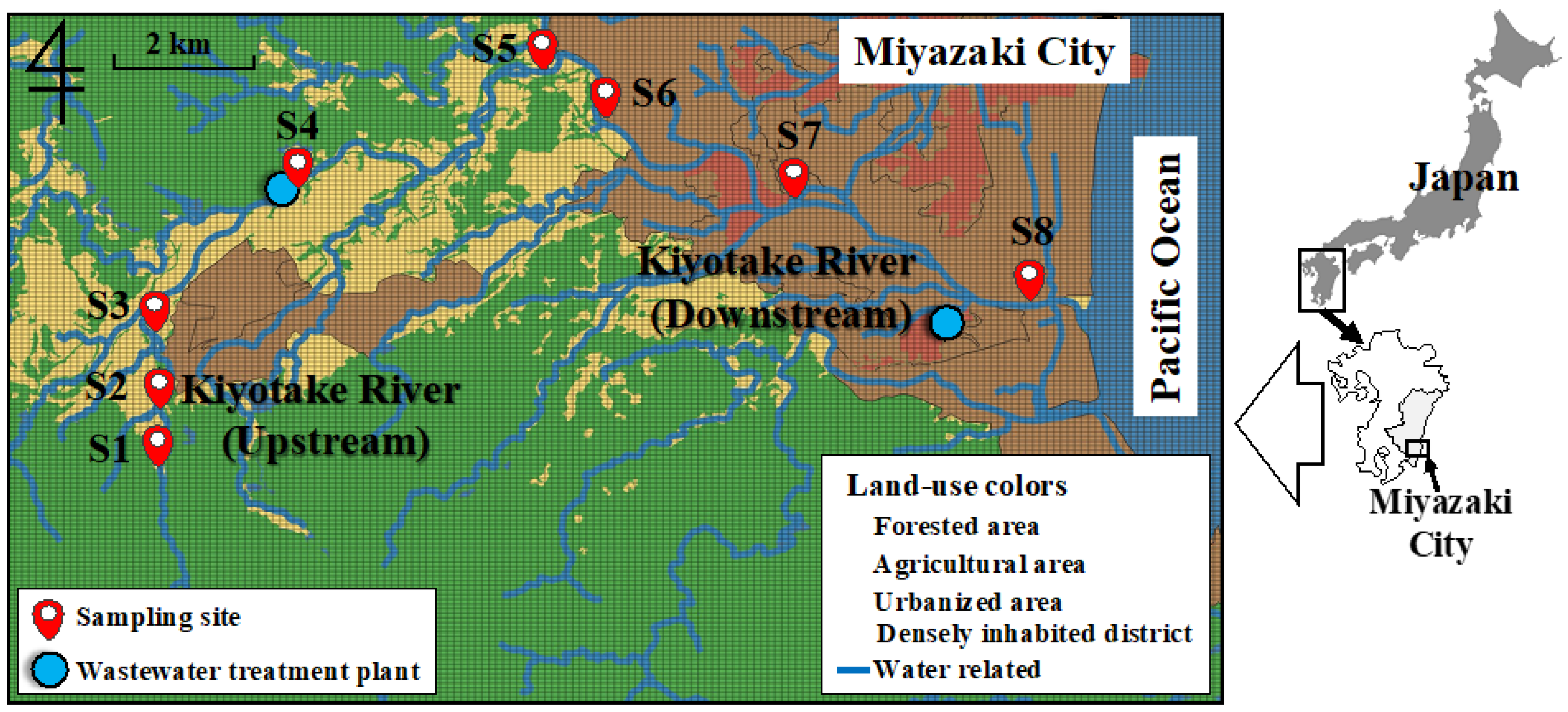
2.1. Water Sample Collection
2.2. Analysis of General Water Quality
2.3. Bacterial Cell Concentration and DNA Extraction
2.4. 16S rRNA-Gene Sequencing
2.5. Digital PCR
2.6. Statistical Analysis
3. Results and Discussion
3.1. General Water Quality
3.2. DNA Concentrations along the River
3.3. Detection of Pathogen Genes
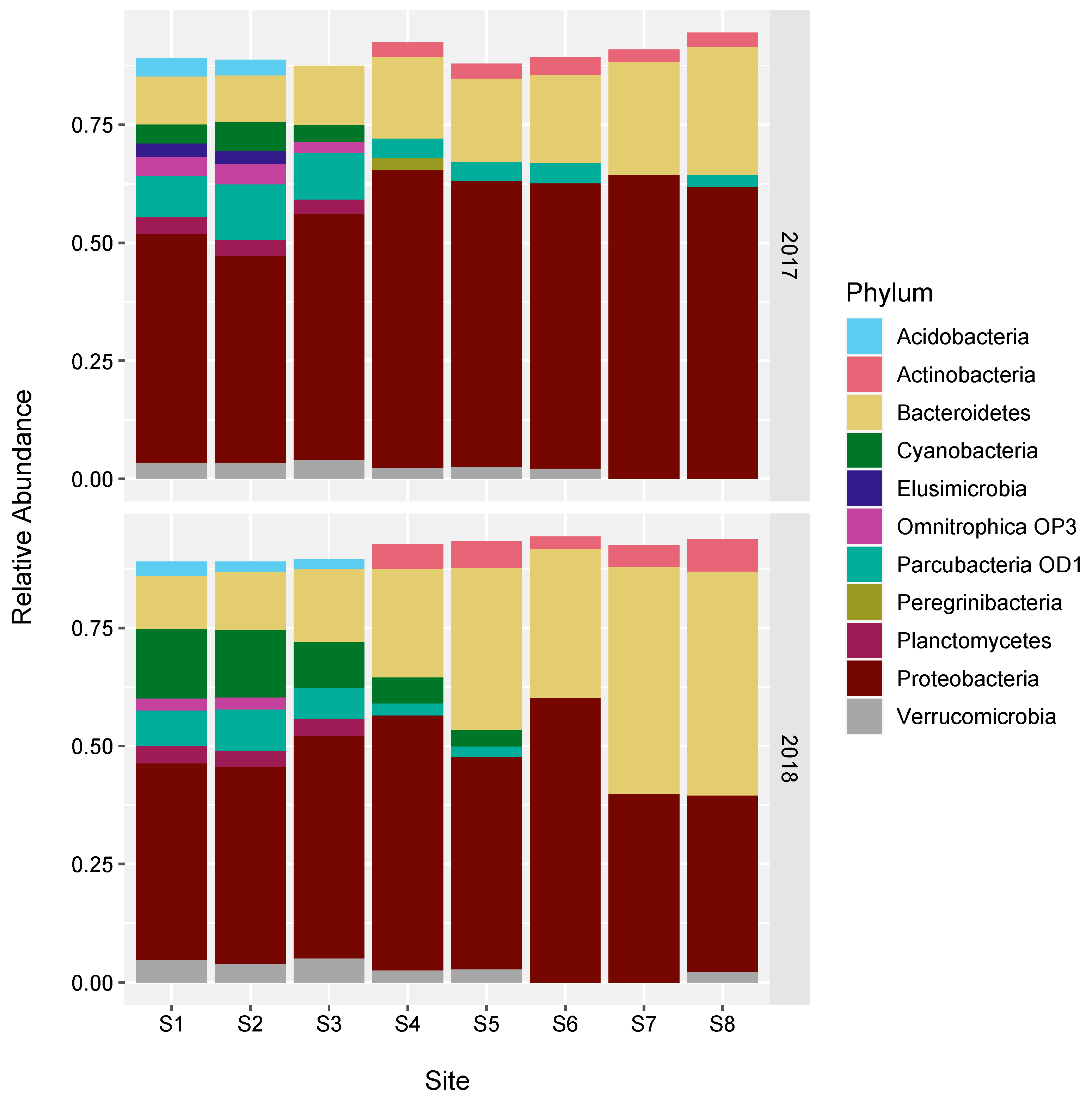
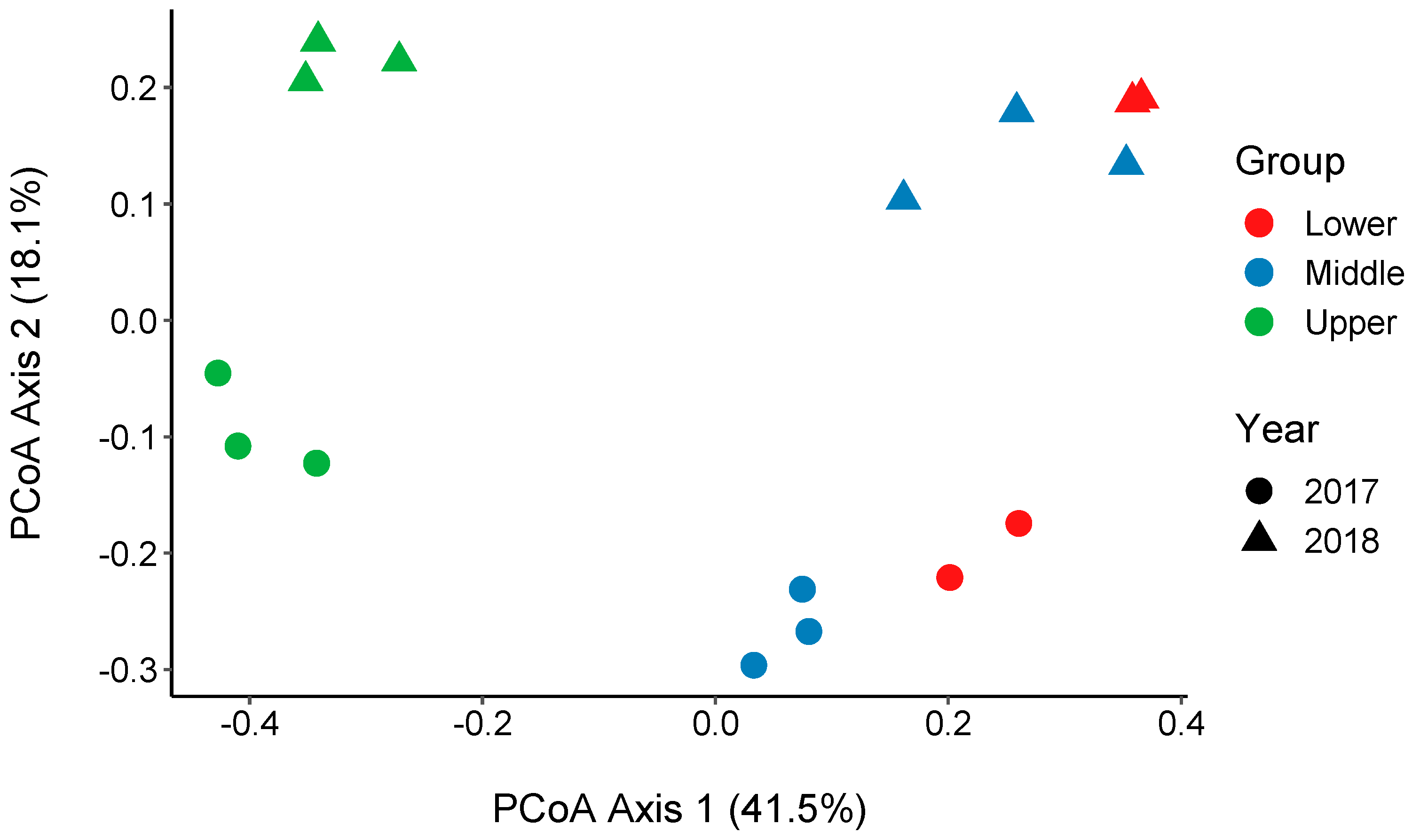
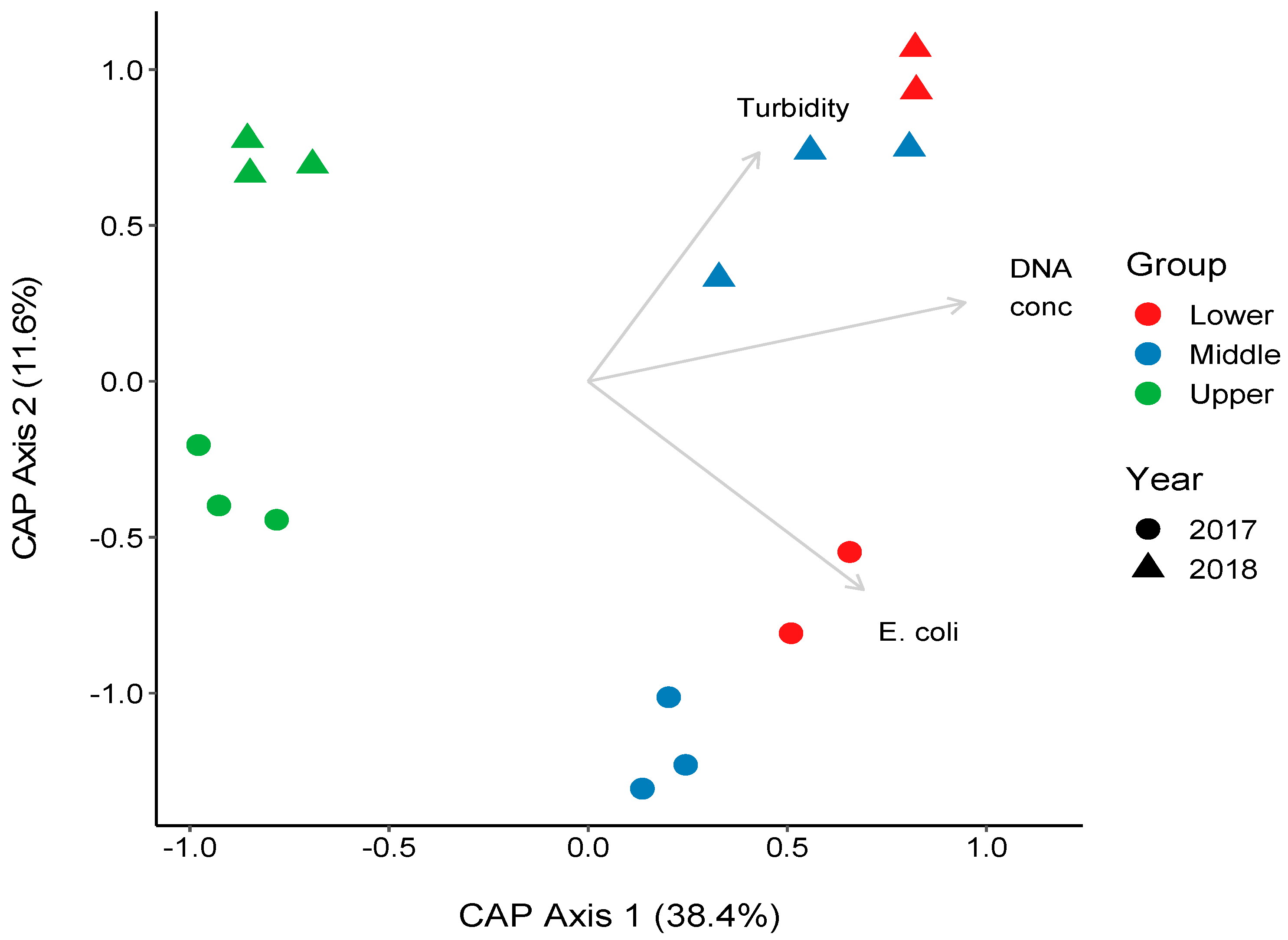
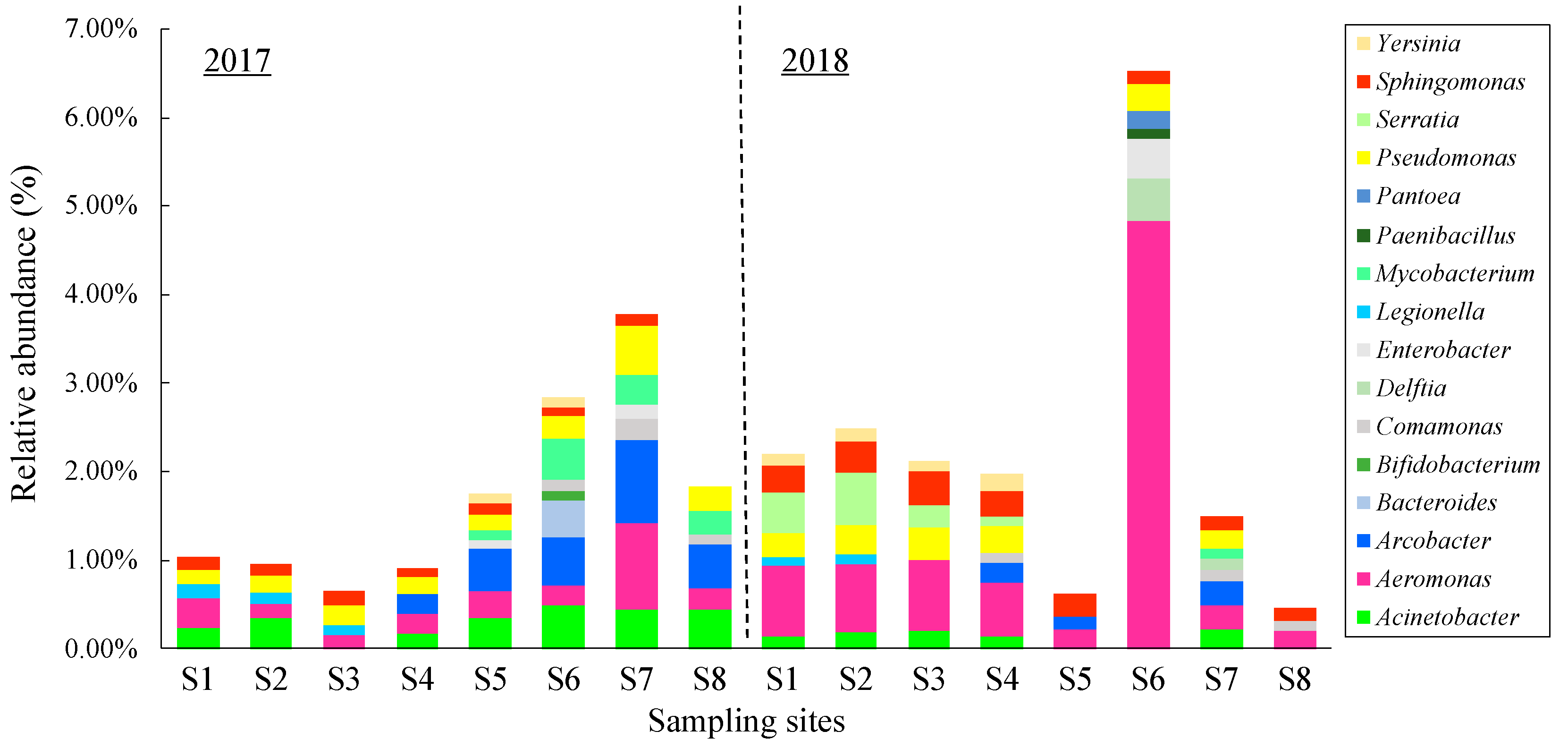
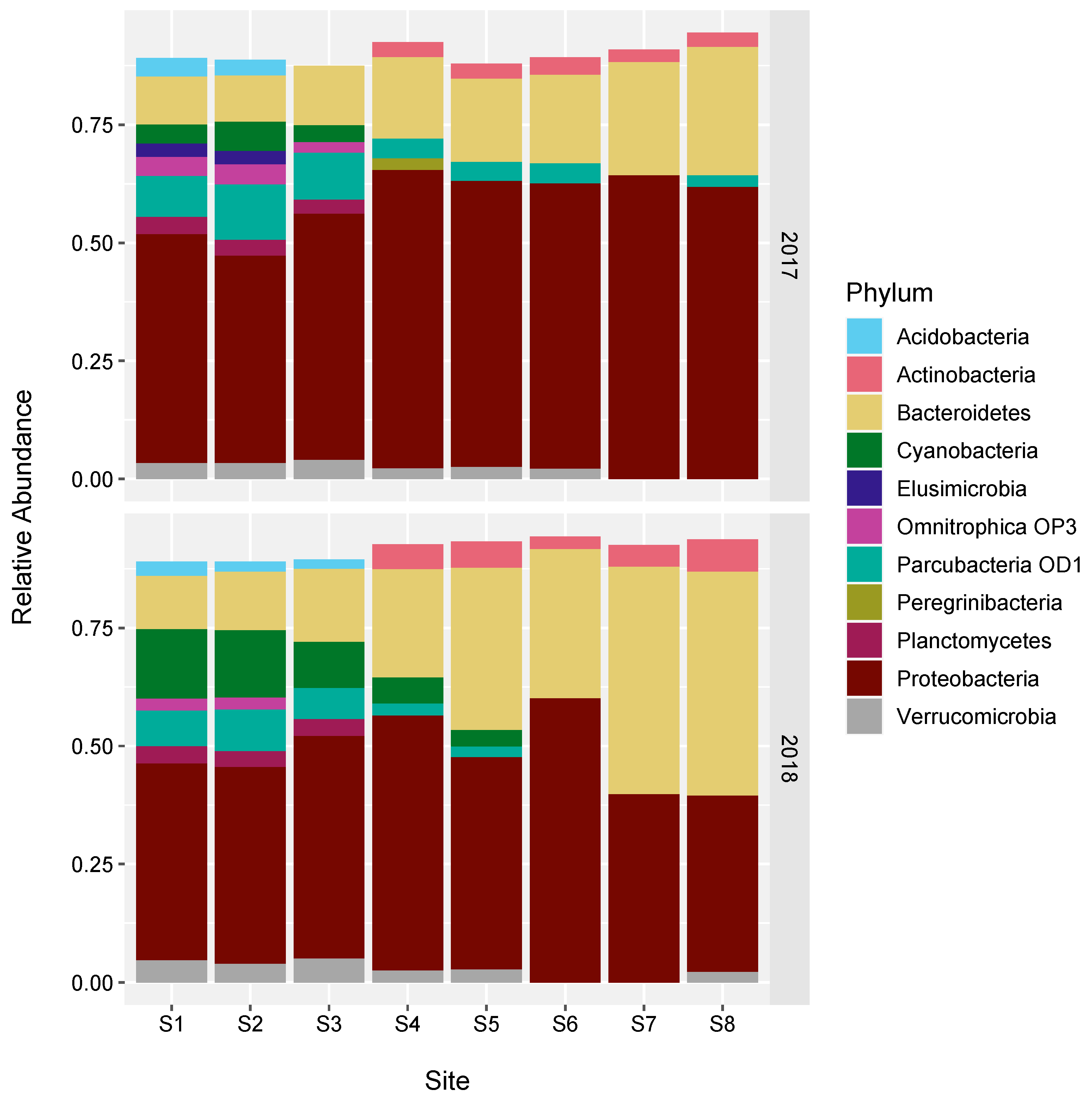
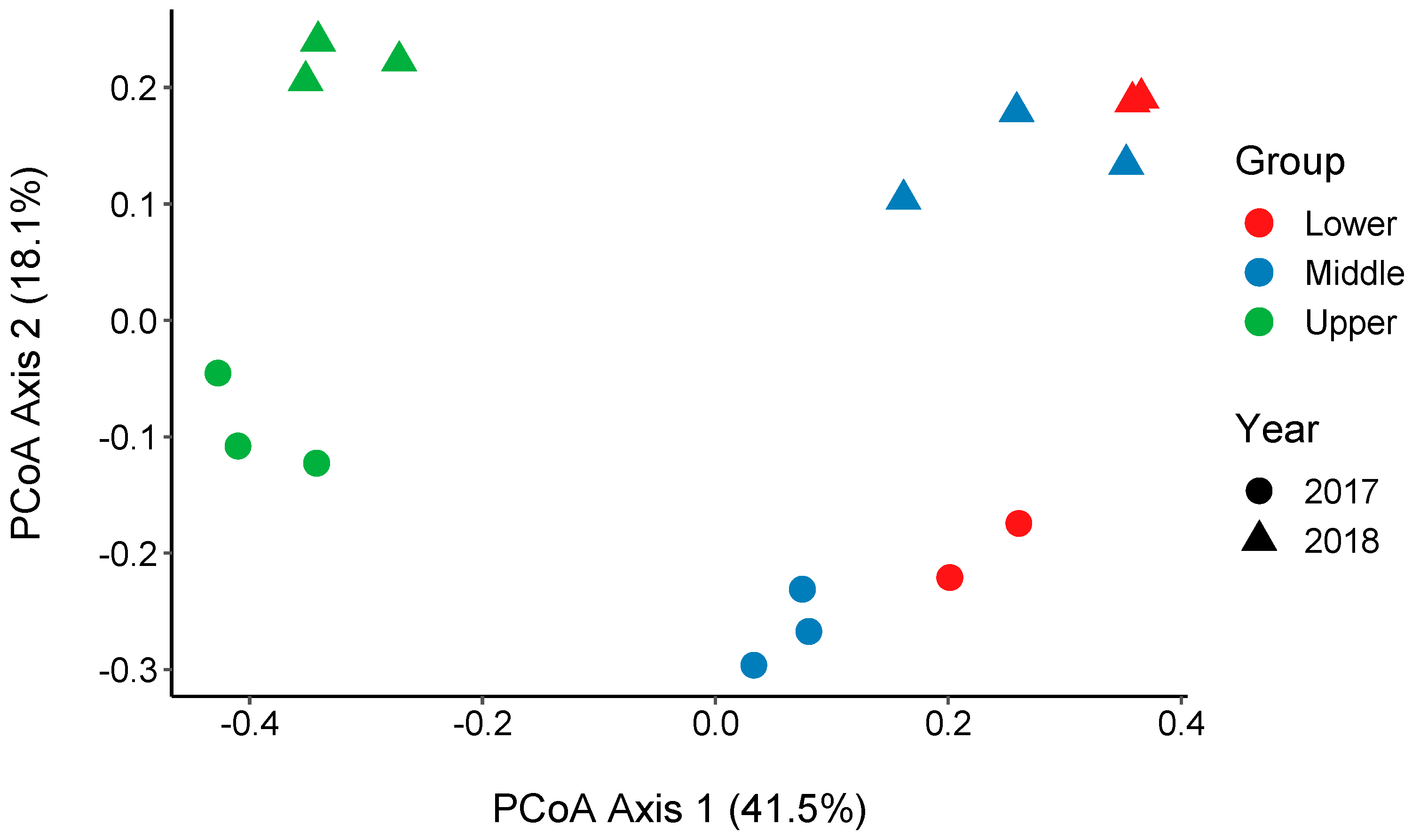
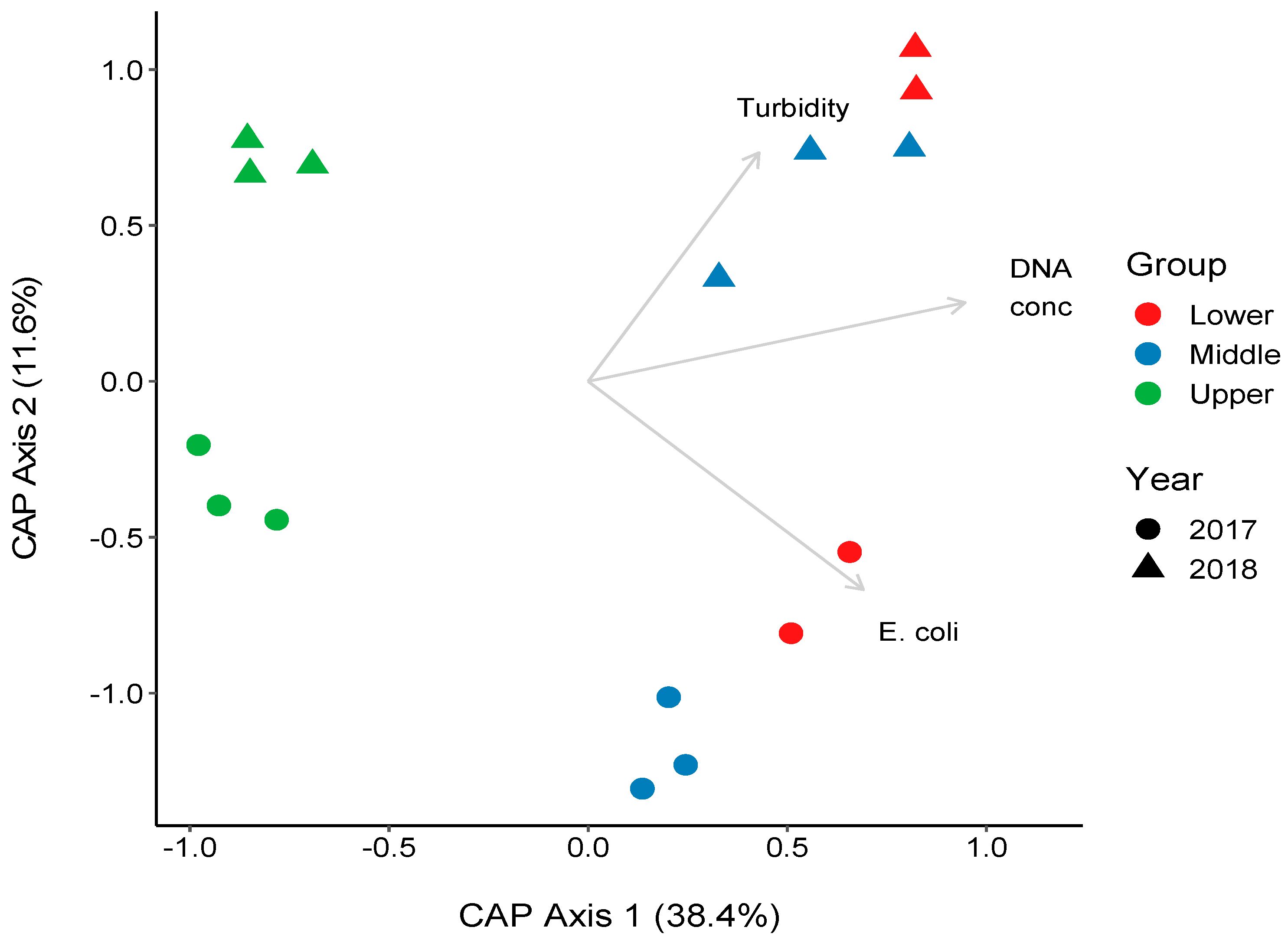
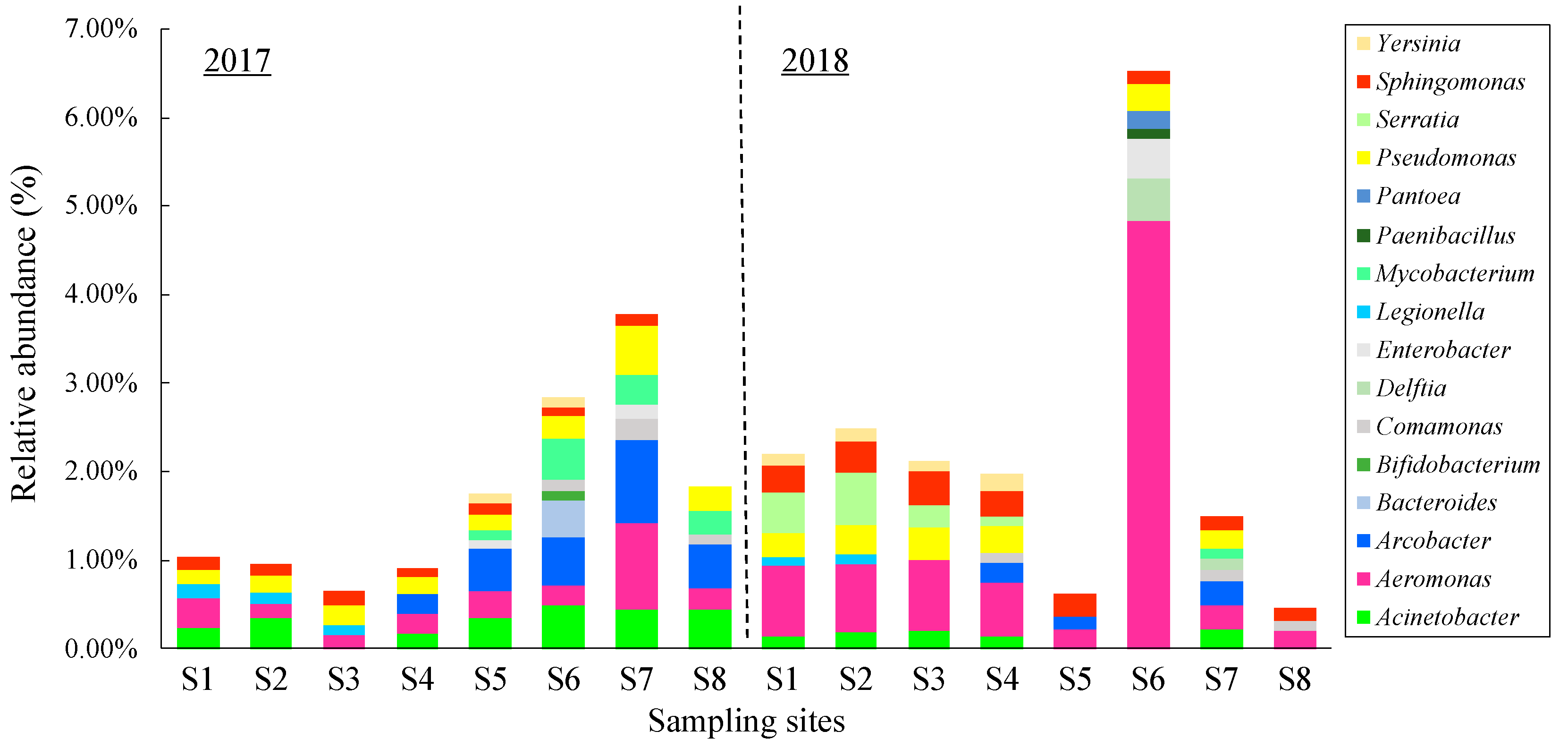
3.4. Changes in the Bacterial Community along the River
3.5. Occurrence of Potential Pathogens
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. WHO Global Water, Sanitation and Hygiene Annual Report 2018. Available online: https://apps.who.int/iris/bitstream/handle/10665/327118/WHO-CED-PHE-WSH-19.147-eng.pdf?sequence=1&isAllowed=y (accessed on 20 January 2019).
- Ministry of Health, Labour and Welfare. Japan. Available online: https://www.mhlw.go.jp/stf/seisakunitsuite/bunya/topics/bukyoku/kenkou/suido/kikikanri/03.html (accessed on 11 November 2019).
- Brookes, J.D.; Antenucci, J.; Hipsey, M.; Burch, M.D.; Ashbolt, N.J.; Ferguson, C. Fate and transport of pathogens in lakes and reservoirs. Environ. Int. 2004, 30, 741–759. [Google Scholar] [CrossRef] [PubMed]
- Alegbeleye, O.O.; Sant’Ana, A.S. Manure-borne pathogens as an important source of water contamination: An update on the dynamics of pathogen survival/transport as well as practical risk mitigation strategies. Int. J. Hyg. Environ. Health. 2020, 227, 113524. [Google Scholar] [CrossRef] [PubMed]
- Amarasiri, M.; Furukawa, T.; Nakajima, F.; Sei, K. Pathogens and disease vectors/hosts monitoring in aquatic environments: Potential of using eDNA/eRNA based approach. Sci. Total Environ. 2021, 796, 148810. [Google Scholar] [CrossRef] [PubMed]
- González-Fernández, A.; Symonds, E.M.; Gallard-Gongora, J.F.; Mull, B.; Lukasik, J.O.; Navarro, P.R.; Aguilar, A.B.; Peraud, J.; Brown, M.L.; Alvarado, D.M.; et al. Relationships among microbial indicators of fecal pollution, microbial source tracking markers, and pathogens in Costa Rican coastal waters. Water Res. 2021, 188, 116507. [Google Scholar] [CrossRef] [PubMed]
- Harwood, V.J.; Levine, A.D.; Scott, T.M.; Chivukula, V.; Lukasik, J.; Farrah, S.R.; Rose, J.B. Validity of the indicator organism paradigm for pathogen reduction in reclaimed water and public health protection. Appl. Environ. Microbiol. 2005, 71, 3163–3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aw, T.G.; Rose, J.B. Detection of pathogens in water: From phylochips to qPCR to pyrosequencing. Curr. Opin. Biotechnol. 2012, 23, 422–430. [Google Scholar] [CrossRef]
- Salipante, S.J.; Jerome, K.R. Digital PCR-An Emerging Technology with Broad Applications in Microbiology. Clin. Chem. 2020, 66, 117–123. [Google Scholar] [CrossRef]
- Deshmukh, R.A.; Joshi, K.; Bhand, S.; Roy, U. Recent developments in detection and enumeration of waterborne bacteria: A retrospective minireview. Microbiol. Open. 2016, 5, 901–922. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.; Cunha, M.Â. Assessment of the microbiological quality of recreational waters: Indicators and methods. Euro-Mediterr. J. Environ. Integr. 2017, 2, 25. [Google Scholar] [CrossRef] [Green Version]
- Mathai, P.P.; Dunn, H.M.; Magnone, P.; Zhang, Q.; Ishii, S.; Chun, C.L.; Sadowsky, M.J. Association between submerged aquatic vegetation and elevated levels of Escherichia coli and potential bacterial pathogens in freshwater lakes. Sci. Total Environ. 2019, 657, 319–324. [Google Scholar] [CrossRef]
- Zhang, Y.; Riley, L.K.; Lin, M.; Hu, Z. Determination of low-density Escherichia coli and Helicobacter pylori suspensions in water. Water Res. 2012, 46, 2140–2148. [Google Scholar] [CrossRef]
- Gonzales-Gustavson, E.; Cárdenas-Youngs, Y.; Calvo, M.; da Silva, M.F.; Hundesa, A.; Amorós, I.; Moreno, Y.; Moreno-Mesonero, L.; Rosell, R.; Ganges, L.; et al. Characterization of the efficiency and uncertainty of skimmed milk flocculation for the simultaneous concentration and quantification of water-borne viruses, bacteria and protozoa. J. Microbiol. Methods 2017, 134, 46–53. [Google Scholar] [CrossRef]
- Jansson, L.; Eriksson, R.; Hedman, J.; Lavander, M. Evaluation and modification of lanthanum-based flocculation for isolation of bacteria from water samples. Biotechnol. Rep. 2018, 19, e00267. [Google Scholar] [CrossRef]
- Suzuki, Y.; Imafuku, Y.; Nishiyama, M.; Teranishi, K.; Jikumaru, A.; Nukazawa, K.; Ogura, Y. A highly efficient method for concentrating DNA from river water by combined coagulation and foam separation. Sep. Sci. Technol. 2019, 54, 3128–3134. [Google Scholar] [CrossRef]
- Jikumaru, A.; Ishii, S.; Fukudome, T.; Kawahara, Y.; Iguchi, A.; Masago, Y.; Nukazawa, K.; Suzuki, Y. Fast, sensitive, and reliable detection of waterborne pathogens by digital PCR after coagulation and foam concentration. J. Biosci. Bioeng. 2020, 130, 76–81. [Google Scholar] [CrossRef]
- United States Environmental Protection Agency. Method 1600: Enterococci in Water by Membrane Filtration Using membrane-Enterococcus Indoxyl-β-D-Glucoside Agar (mEI); U.S. EPA: Washington, DC, USA, 2002.
- Suzuki, Y.; Shimizu, H.; Kuroda, T.; Takada, Y.; Nukazawa, K. Plant debris are hotbeds for pathogenic bacteria on recreational sandy beaches. Sci. Rep. 2021, 11, 11496. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package, R Package Version 2.5-6. 2019. Available online: https://CRAN.Rproject.org/package=vegan (accessed on 20 January 2021).
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.J.; Willis, T.J. Canonical analysis of principal coordinates: A useful method of constrained ordination for ecology. Ecology 2003, 84, 511–525. [Google Scholar] [CrossRef]
- Noto, Y.; Yasuda, M. The synthetic evaluation of water quality of rivers. J. JSCE 1983, 338, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Hata, A.; Katayama, H.; Kasuga, I. Consecutive ultrafiltration and silica adsorption for recovery of extracellular antibiotic resistance genes from an urban river. Environ. Pollut. 2020, 260, 114062. [Google Scholar] [CrossRef] [PubMed]
- Jokinen, C.C.; Cook, S.R.; Reuter, T.; Tymensen, L. Assessing enterococci as an alternative fecal indicator for irrigation water quality. Agric. Water Manag. 2020, 233, 106098. [Google Scholar] [CrossRef]
- Bussi, G.; Whitehead, P.G.; Thomas, A.R.C.; Masante, D.; Jones, L.; Cosby, B.J.; Emmett, B.A.; Malham, S.K.; Prudhomme, C.; Prosser, H. Climate and land-use change impact on faecal indicator bacteria in a temperate maritime catchment (the River Conwy, Wales). J. Hydrol. 2017, 553, 248–261. [Google Scholar] [CrossRef] [Green Version]
- Fang, T.; Cui, Q.; Huang, Y.; Dong, P.; Wang, H.; Liu, W.T.; Ye, Q. Distribution comparison and risk assessment of free-floating and particle-attached bacterial pathogens in urban recreational water: Implications for water quality management. Sci. Total Environ. 2018, 613–614, 428–438. [Google Scholar] [CrossRef]
- McLellan, S.L.; Huse, S.M.; Mueller-Spitz, S.R.; Andreishcheva, E.N.; Sogin, M.L. Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent. Environ Microbiol. 2010, 12, 378–392. [Google Scholar] [CrossRef] [Green Version]
- Baumann, P. Isolation of Acinetobacter from soil and water. J. Bacteriol. 1968, 96, 39–42. [Google Scholar] [CrossRef] [Green Version]
- Brandi, G.; Sisti, M.; Schiavano, G.F.; Salvaggio, L.; Albano, A. Survival of Aeromonas hydrophila, Aeromonas caviae and Aeromonas sobria in soil. J. Appl. Microbiol. 1996, 81, 439–444. [Google Scholar] [CrossRef]
- Peix, A.; Ramírez-Bahena, M.; Velázquez, E. The current status on the taxonomy of Pseudomonas revisited: An update. Infect. Genet. Evol. 2018, 57, 106–116. [Google Scholar] [CrossRef]
- Suzuki, Y.; Kajii, S.; Nishiyama, M.; Iguchi, A. Susceptibility of Pseudomonas aeruginosa isolates collected from river water in Japan to antipseudomonal agents. Sci. Total Environ. 2013, 450–451, 148–154. [Google Scholar] [CrossRef]
- Tabata, K.; Kasuya, K.; Abe, H.; Masuda, K.; Doi, Y. Poly(Aspartic Acid) Degradation by a Sphingomonas sp. Isolated from Freshwater. Appl. Environ. 1999, 65, 4268–4270. [Google Scholar] [CrossRef] [Green Version]
- Vachée, A.; Mossel, D.A.; Leclerc, H. Antimicrobial activity among Pseudomonas and related strains of mineral water origin. J. Appl. Microbiol. 1997, 83, 652–658. [Google Scholar] [CrossRef]
- Steele, T.W.; Moore, C.V.; Sangster, N. Distribution of Legionella longbeachae serogroup 1 and other legionellae in potting soils in Australia. Appl. Environ. Microbiol. 1990, 56, 2984–2988. [Google Scholar] [CrossRef] [Green Version]
- Fisher, J.C.; Levican, A.; Figueras, M.J.; McLellan, S.L. Population dynamics and ecology of Arcobacter in sewage. Front. Microbiol. 2014, 5, 525. [Google Scholar] [CrossRef] [Green Version]
- Li, W.C.; Tse, H.F.; Fok, L. Plastic waste in the marine environment: A review of sources, occurrence and effects. Sci. Total Environ. 2016, 566–567, 333–349. [Google Scholar] [CrossRef]
- Wagner, J.; Coupland, P.; Browne, H.P.; Lawley, T.D.; Francis, S.C.; Parkhill, J. Evaluation of PacBio sequencing for full-length bacterial 16S rRNA gene classification. BMC Microbiol. 2016, 16, 274. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Ahdoh, Y.; Maruyama, T.; Mitsuyama, M.; Shimotsu, Y.; Gomi, K.; Mori, H.; Kunikane, S. Removal of di-2-ethylhexyl phthalate (DEHP) form river water using dispersed bubbles. J. JSWE 2006, 29, 29–35. [Google Scholar] [CrossRef] [Green Version]
- Chui, L.; Couturier, M.R.; Chiu, T.; Wang, G.; Olson, A.B.; McDonald, R.R.; Antonishyn, N.A.; Horsman, G.; Gilmour, M.W. Comparison of Shiga toxin-producing Escherichia coli detection methods using clinical stool samples. J. Mol. Diagn. 2010, 12, 469–475. [Google Scholar] [CrossRef]
- Vu, D.T.; Sethabutr, O.; Von Seidlein, L.; Tran, V.T.; Do, G.C.; Bui, T.C.; Le, H.T.; Lee, H.; Houng, H.S.; Hale, T.L.; et al. Detection of Shigella by a PCR assay targeting the ipaH gene suggests increased prevalence of shigellosis in Nha Trang, Vietnam. J. Clin. Microbiol. 2004, 42, 2031–2035. [Google Scholar] [CrossRef] [Green Version]
- Vondrakova, L.; Pazlarova, J.; Demnerova, K. Detection, identification and quantification of Campylobacter jejuni, coli and lari in food matrices all at once using multiplex qPCR. Gut. Pathog. 2014, 6, 12. [Google Scholar] [CrossRef]
- Silkie, S.S.; Tolcher, M.P.; Nelson, K.L. Reagent decontamination to eliminate false-positives in Escherichia coli qPCR. J. Microbiol. Methods 2008, 72, 275–282. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Water Temperature | pH | Electric Conductivity | Turbidity | TOC | Fecal Bacteria (Mean CFU/100 mL) ** | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (°C) | (-) | (μS/cm) | (Degree *) | (mg/L) | Total Coliforms | Escherichia coli | Enterococcus | ||||||||
| 2017 | 2018 | 2017 | 2018 | 2017 | 2018 | 2017 | 2018 | 2017 | 2017 | 2018 | 2017 | 2018 | 2017 | 2018 | |
| S1 | 17.5 | 22.4 | 7.10 | 7.49 | 75.8 | 93.2 | 0.49 | 1.55 | 0.33 | 1.6 × 102 | 7.4 × 102 | 5.0 × 100 | 3 × 10−1 | 3.7 × 100 | 2.0 × 100 |
| S2 | 16.8 | 23.0 | 7.01 | 7.64 | 77.4 | 86.7 | 0.57 | 0.74 | 0.21 | 1.8 × 102 | 6.3 × 102 | 1.0 × 100 | 4.0 × 100 | 1.3 × 100 | 3.3 × 100 |
| S3 | 16.1 | 24.0 | 6.98 | 7.80 | 87.6 | 87.0 | 0.71 | 1.14 | 0.21 | 3.5 × 102 | 1.4 × 103 | 4.0 × 100 | 4.0 × 100 | 2.9 × 101 | 5.8 × 101 |
| S4 | 17.2 | 25.2 | 6.94 | 7.79 | 100.8 | 105.6 | 0.41 | 1.21 | 0.33 | 1.6 × 103 | 5.0 × 103 | 5.8 × 101 | 3.1 × 101 | 5.7 × 101 | 7.4 × 101 |
| S5 | 17.0 | 25.2 | 7.10 | 7.80 | 109.1 | 119.1 | 0.71 | 1.19 | 0.37 | 1.1 × 103 | 4.9 × 103 | 4.6 × 101 | 4.1 × 101 | 4.2 × 101 | 8.0 × 101 |
| S6 | 18.3 | 26.4 | 7.03 | 7.78 | 123.9 | 132.7 | 0.89 | 1.01 | 0.40 | 1.3 × 103 | 6.8 × 103 | 9.0 × 101 | 1.9 × 101 | 2.0 × 101 | 8.2 × 101 |
| S7 | 19.4 | 26.8 | 7.22 | 7.68 | 123.4 | 128.1 | 1.22 | 1.04 | 0.36 | 1.4 × 103 | 4.2 × 103 | 5.3 × 101 | 4.7 × 101 | 1.2 × 102 | 1.1 × 102 |
| S8 | 19.1 | 27.4 | 6.87 | 7.56 | 720.0 | 166.0 | 1.07 | 1.97 | 0.44 | 1.0 × 103 | 2.0 × 103 | 5.7 × 101 | 2.0 × 101 | 1.9 × 101 | 1.6 × 101 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki, Y.; Jikumaru, A.; Tamai, S.; Nukazawa, K.; Masago, Y.; Ishii, S. Application of Coagulation and Foam Concentration Method to Quantify Waterborne Pathogens in River Water Samples. Water 2022, 14, 3642. https://doi.org/10.3390/w14223642
Suzuki Y, Jikumaru A, Tamai S, Nukazawa K, Masago Y, Ishii S. Application of Coagulation and Foam Concentration Method to Quantify Waterborne Pathogens in River Water Samples. Water. 2022; 14(22):3642. https://doi.org/10.3390/w14223642
Chicago/Turabian StyleSuzuki, Yoshihiro, Atsushi Jikumaru, Soichiro Tamai, Kei Nukazawa, Yoshifumi Masago, and Satoshi Ishii. 2022. "Application of Coagulation and Foam Concentration Method to Quantify Waterborne Pathogens in River Water Samples" Water 14, no. 22: 3642. https://doi.org/10.3390/w14223642