
A Green Approach Based on Micro-X-ray Fluorescence for Arsenic, Micro- and Macronutrients Detection in Pteris vittata

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
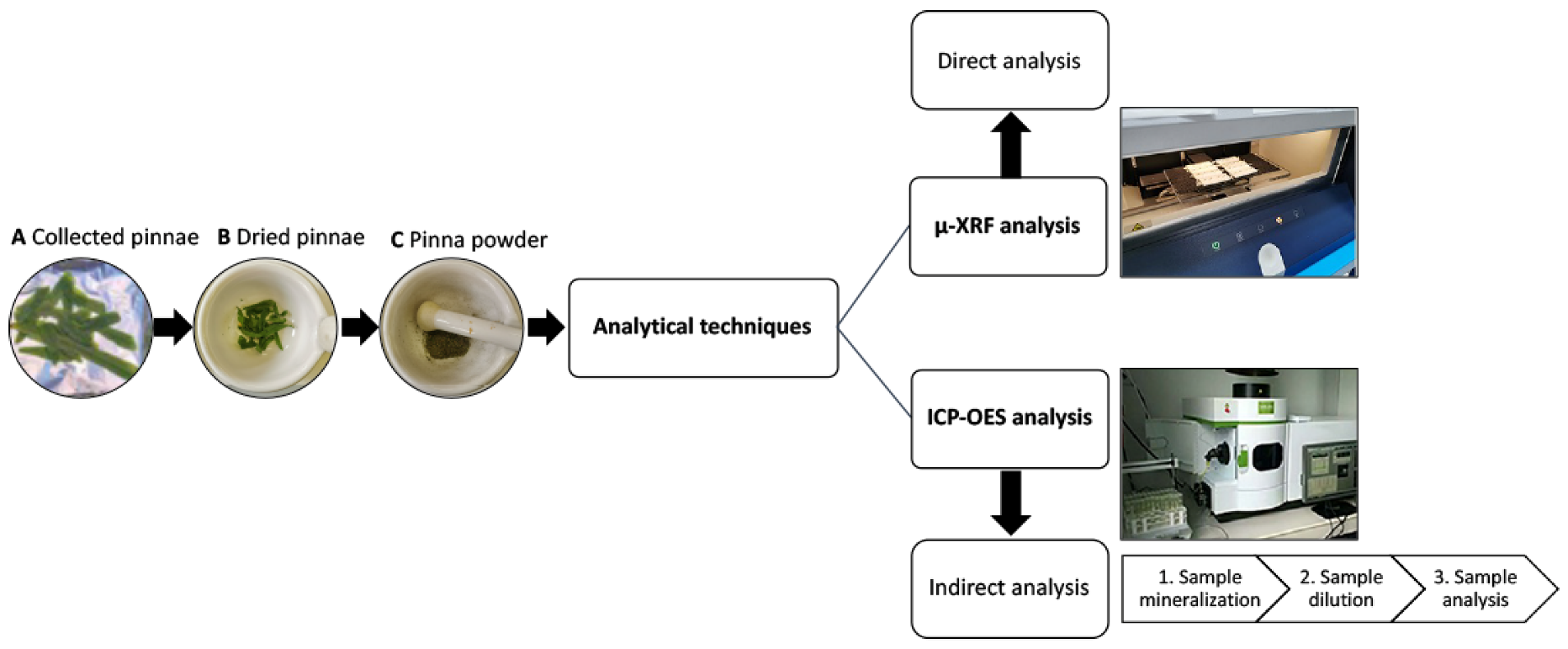
2.1. Plant Growth and Pinna Powder Preparation
2.2. The μXRF Device
2.3. μXRF Analysis on Dried Pinna Powder
2.4. Elements Quantification by ICP-OES
2.5. μXRF Measurement Strategies
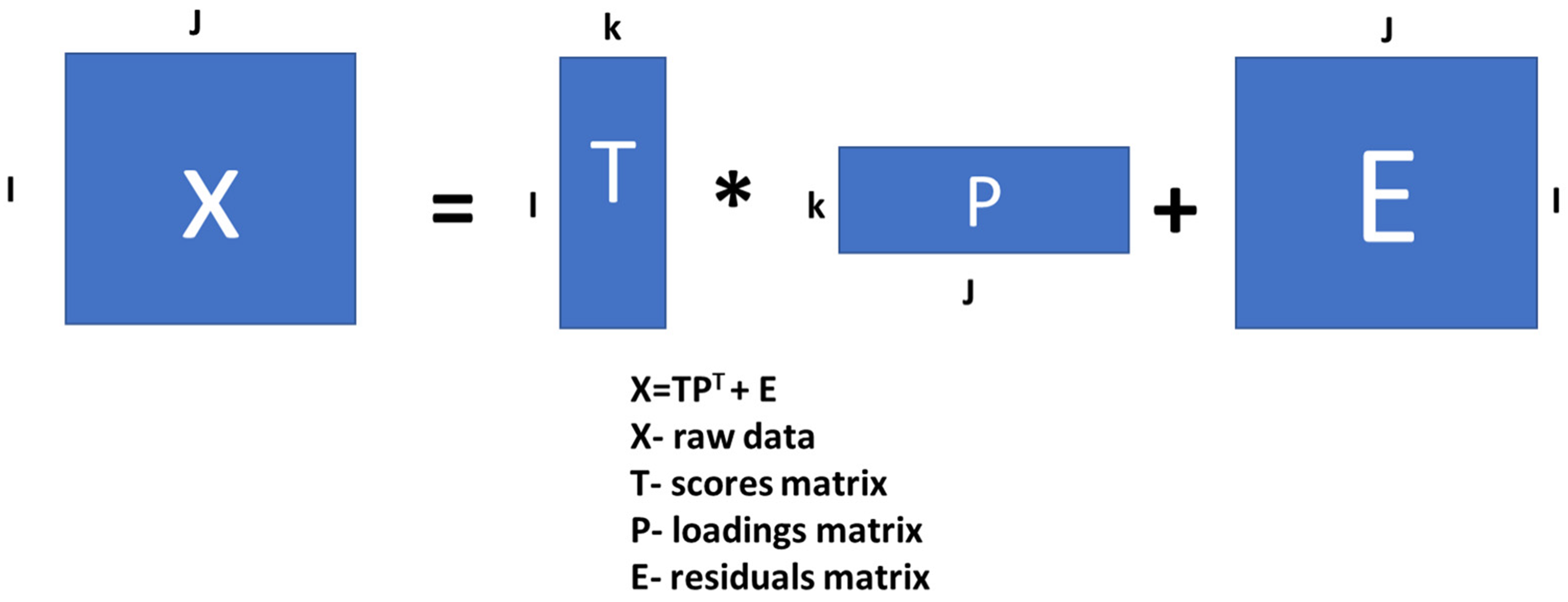
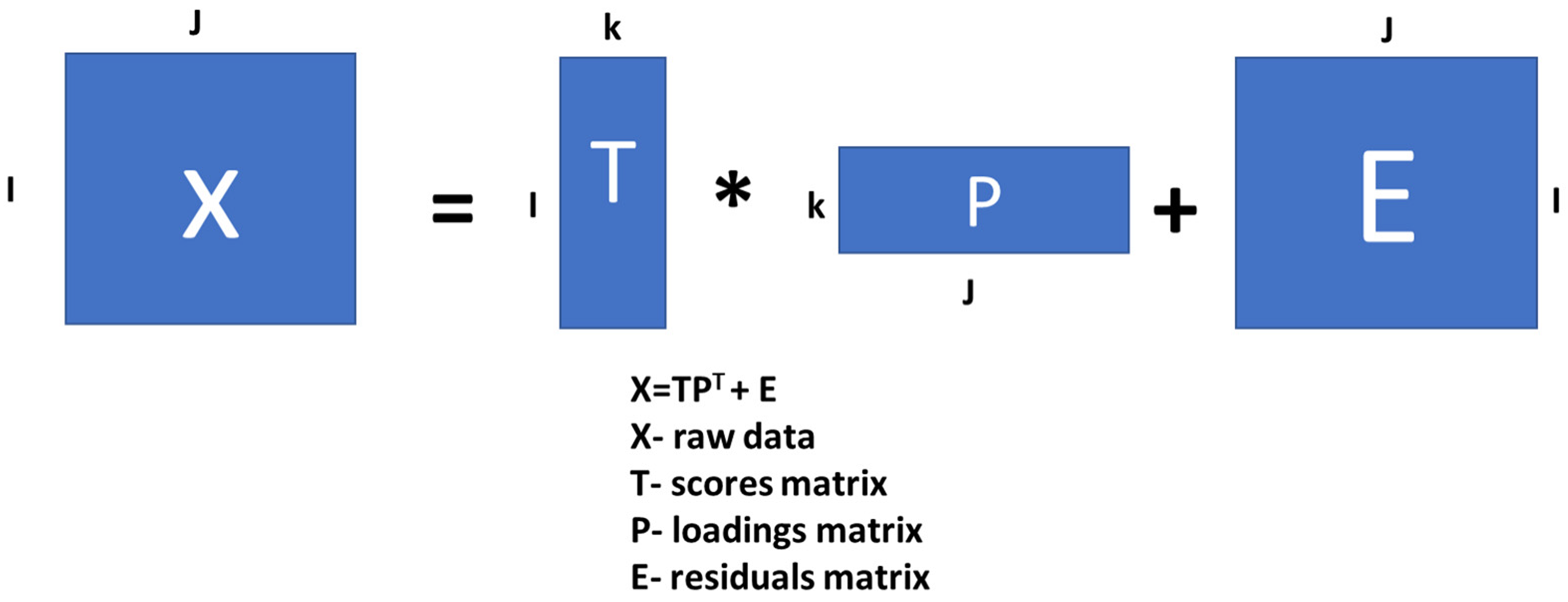
2.6. Principal Component Analysis (PCA)
3. Results
3.1. Arsenic and Micro- and Macronutrient Concentrations Detected by µXRF and ICP–OES Analyses
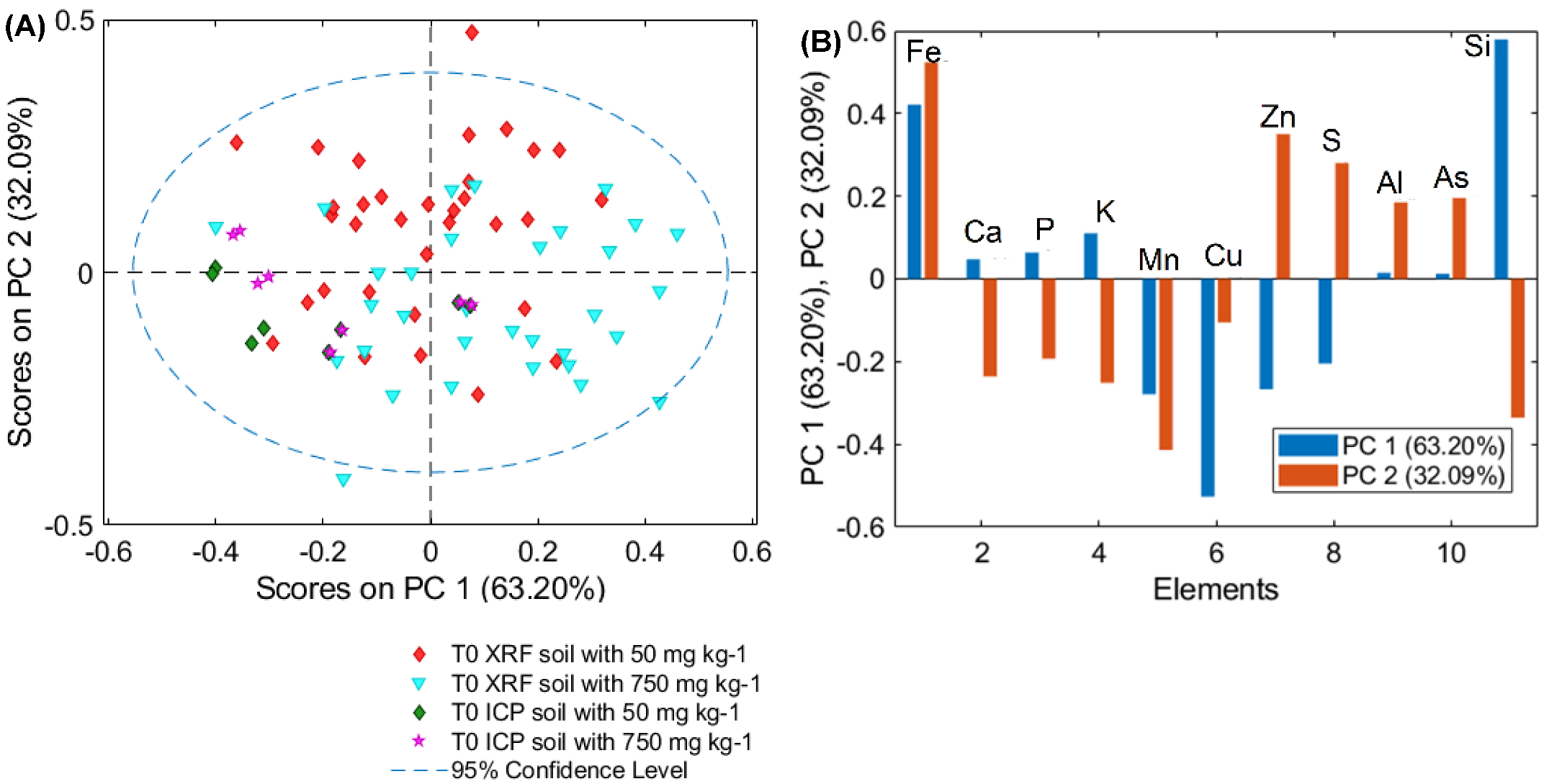
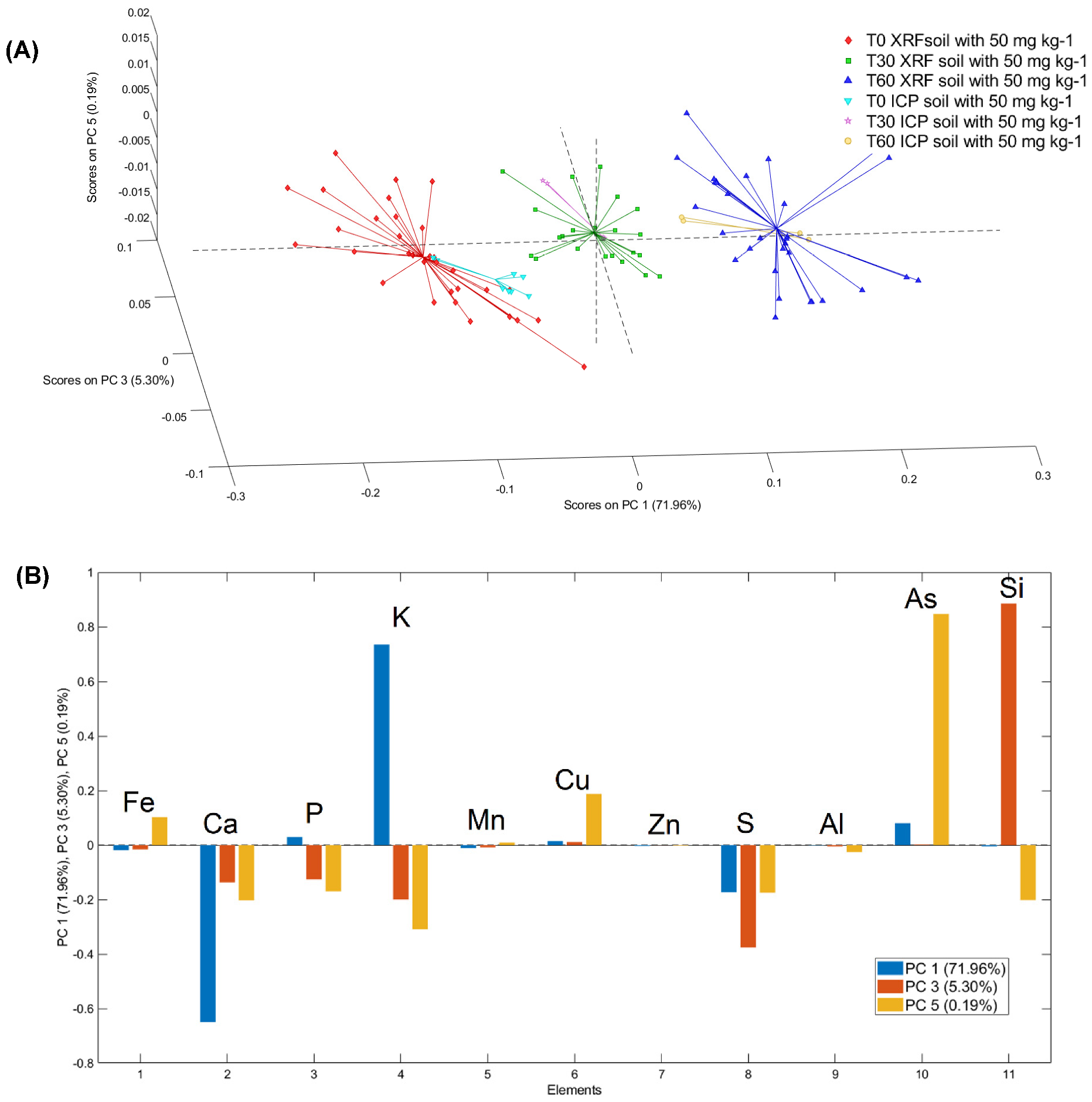
3.2. PCA Models of T0 Samples for μXRF and ICP–OES Analysis
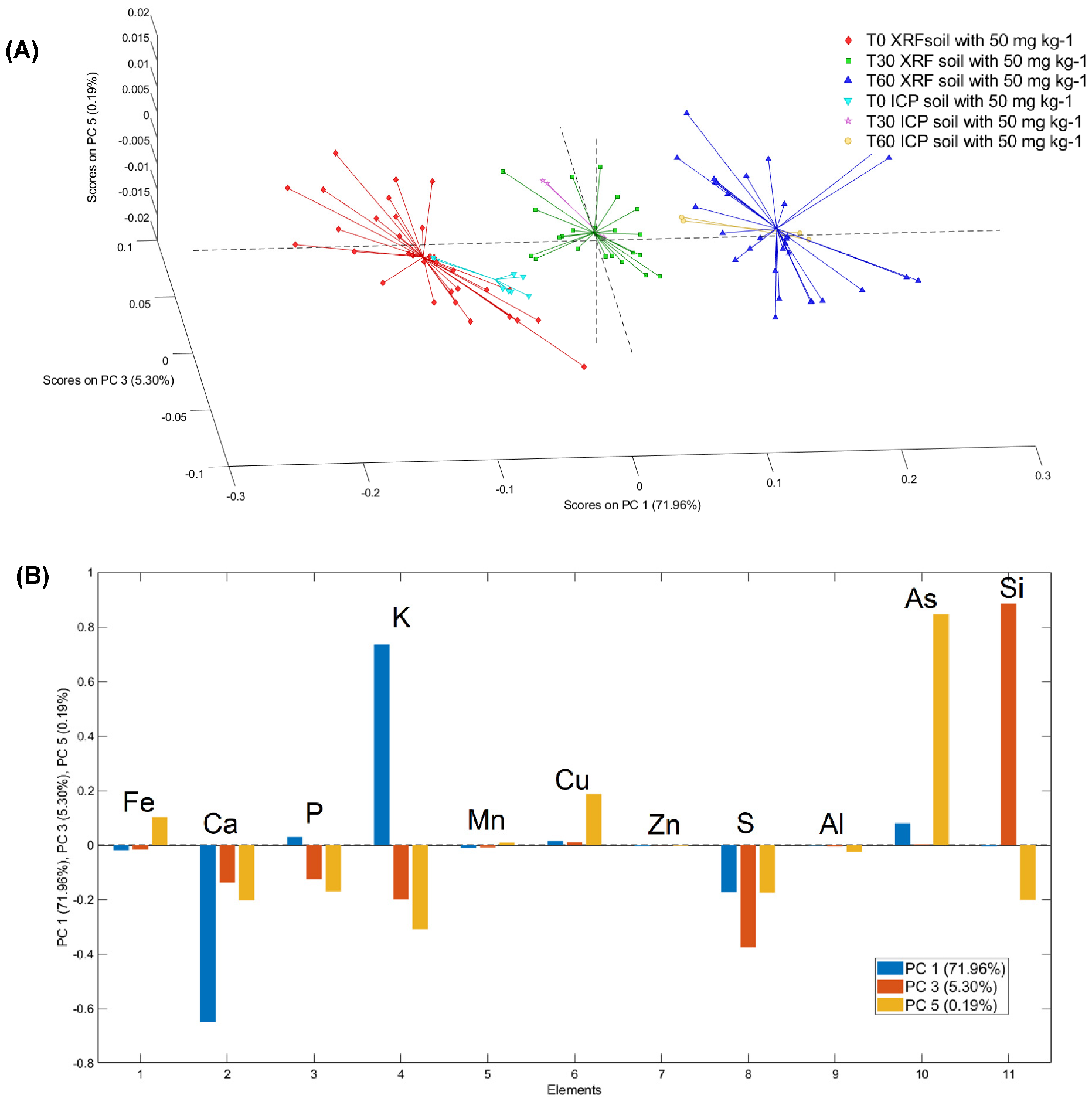
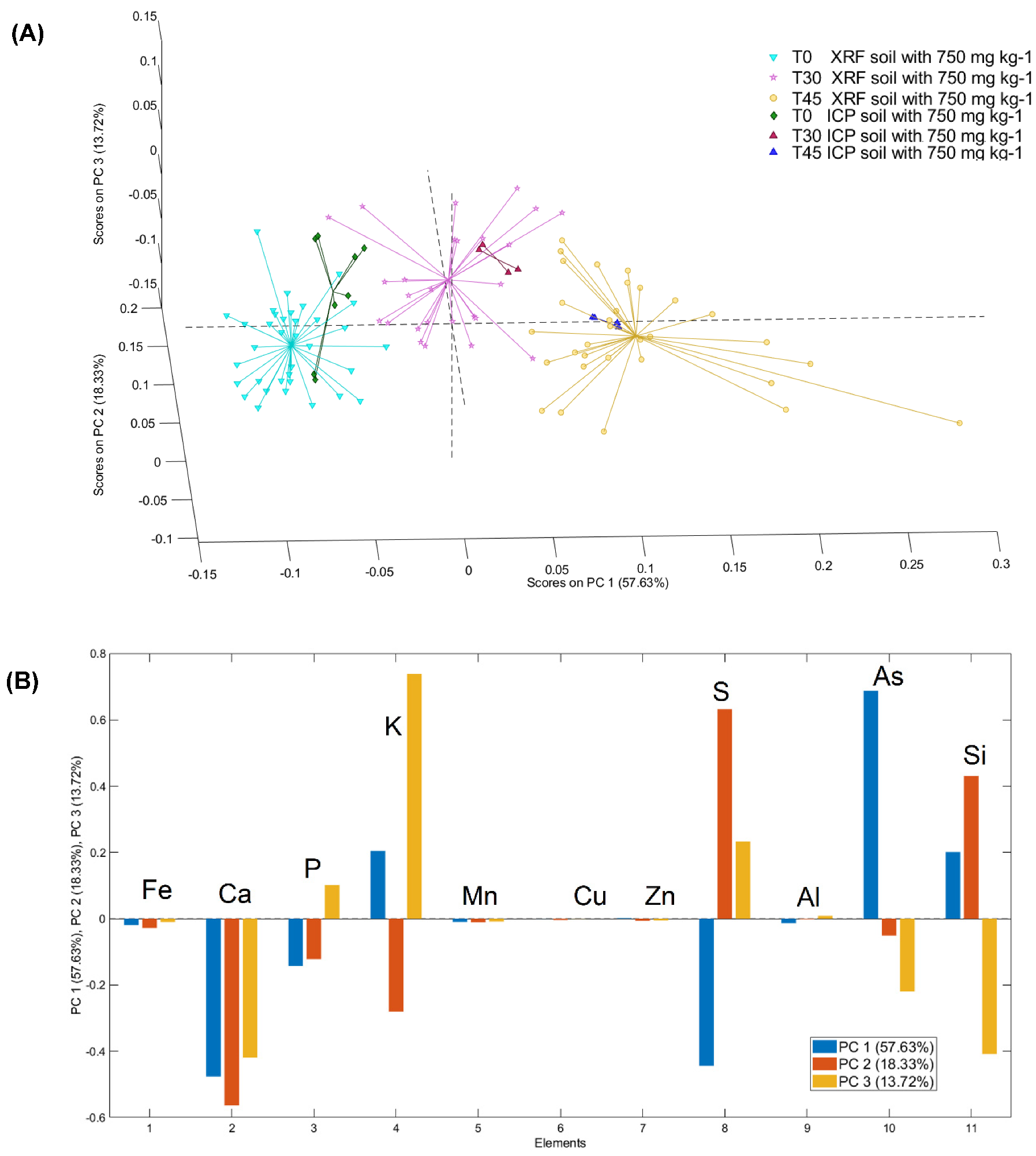
3.3. PCA Models of μXRF and ICP–OES Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ma, L.Q.; Komar, K.M.; Tu, C.; Zhang, W.; Cai, Y.; Kennelley, E.D. A fern that hyperaccumulates arsenic. Nature 2001, 409, 579. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.H.; Liu, X.; Rathinasabapathi, B.; Li, H.B.; Chen, Y.; Ma, L.Q. Mechanisms of efficient As solubilization in soils and As accumulation by As-hyperaccumulator Pteris vittata. Environ. Pollut. 2017, 227, 569–577. [Google Scholar] [CrossRef]
- Eze, V.C.; Harvey, A.P. Extractive recovery and valorisation of arsenic from contaminated soil through phytoremediation using Pteris cretica. Chemosphere 2018, 208, 484–492. [Google Scholar] [CrossRef] [Green Version]
- Melamed, D. Monitoring arsenic in the environment: A review of science and technologies with the potential for field measurements. Anal. Chim. Acta 2005, 532, 1–13. [Google Scholar] [CrossRef]
- Gómez-Ariza, J.L.; Sánchez-Rodas, D.; Giráldez, I.; Morales, E. A comparison between ICP-MS and AFS detection for arsenic speciation in environmental samples. Talanta 2000, 51, 257–268. [Google Scholar] [CrossRef]
- Armenta, S.; Garrigues, S.; de la Guardia, M. Green analytical chemistry. TrAC Trends Anal. Chem. 2008, 27, 497–511. [Google Scholar] [CrossRef]
- Obeidat, S.; Al-Momani, I.; Haddad, A.; Yasein, M.B. Combination of ICP-OES, XRF and XRD techniques for analysis of several dental ceramics and their identification using chemometrics. Spectroscopy 2011, 26, 141–149. [Google Scholar] [CrossRef]
- Marguí, E.; Ricketts, P.; Fletcher, H.; Karydas, A.G.; Migliori, A.; Leani, J.J.; Hidalgoa, M.; Queralt, I.; Voutchkov, M. Total reflection X-ray fluorescence as a fast multielemental technique for human placenta sample analysis. Spectrochim. Acta Part B At. Spectrosc. 2017, 130, 53–59. [Google Scholar] [CrossRef]
- Fiamegos, Y.; de la Calle Guntiñas, M.B. Validation strategy for an ed-xrf method to determine trace elements in a wide range of organic and inorganic matrices based on fulfilment of performance criteria. Spectrochim. Acta Part B At. Spectrosc. 2018, 150, 59–66. [Google Scholar] [CrossRef]
- Heredia, J.; Wagner, M.; Jofré, F.C.; Savio, M.; Azcarate, S.M.; Camiña, J.M. An overview on multi-elemental profile integrated with chemometrics for food quality assessment: Toward new challenges. Crit. Rev. Food Sci. Nutr. 2022, 1–21. [Google Scholar] [CrossRef]
- Liang, J.H.; Liu, P.P.; Chen, Z.; Sun, G.X.; Li, H. Rapid evaluation of arsenic contamination in paddy soils using field portable X-ray fluorescence spectrometry. J. Environ. Sci. 2018, 64, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Fittschen, U.E.A.; Falkenberg, G. Trends in environmental science using microscopic X-ray fluorescence. Spectrochim. Acta Part B At. Spectrosc. 2011, 66, 567–580. [Google Scholar] [CrossRef]
- Chojnacka, K.; Samoraj, M.; Tuhy, Ł.; Michalak, I.; Mironiuk, M.; Mikulewicz, M. Using XRF and ICP-OES in Biosorption Studies. Molecules 2018, 23, 2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuparina, E.; Aisueva, T.S. Determination of heavy metal levels in medicinal plant Hemerocallis minor Miller by X-ray fluorescence spectrometry. Environ. Chem. Lett. 2009, 9, 19–23. [Google Scholar] [CrossRef]
- Gallardo, H.; Queralt, I.; Tapias, J.; Guerra, M.; Carvalho, M.L.; Marguí, E. Possibilities of low-power X-ray fluorescence spectrometry methods for rapid multielemental analysis and imaging of vegetal foodstuffs. J. Food Compos. Anal. 2016, 50, 1–9. [Google Scholar] [CrossRef]
- Byers, H.L.; McHenry, L.J.; Grundl, T.J. XRF techniques to quantify heavy metals in vegetables at low detection limits. Food Chem. X 2019, 1, 100001. [Google Scholar] [CrossRef]
- Coetzee, P.P.; Hoffmann, P.; Speer, R.; Lieser, K.H. Comparison of trace element determination in powdered soil and grass samples by energy-dispersive XRF and by ICP-AES. Anal. Bioanal. Chem. 1986, 323, 254–256. [Google Scholar] [CrossRef]
- Capobianco, G.; Brunetti, P.; Bonifazi, G.; Costantino, P.; Cardarelli, M.; Serranti, S. The use of micro-energy dispersive X-ray fluorescence spectrometry combined with a multivariate approach to determine element variation and distribution in tobacco seedlings exposed to arsenate. Spectrochim. Acta Part B At. Spectrosc. 2018, 147, 132–140. [Google Scholar] [CrossRef]
- Antenozio, M.L.; Giannelli, G.; Marabottini, R.; Brunetti, P.; Allevato, E.; Marzi, D.; Capobianco, G.; Bonifazi, G.; Serranti, S.; Visioli, G.; et al. Phytoextraction efficiency of Pteris vittata grown on a naturally As-rich soil and characterization of As-resistant rhizosphere bacteria. Sci. Rep. 2021, 11, 6794. [Google Scholar] [CrossRef]
- Demirdöğen, R.E. Green Analytical Chemistry: A Useful Tool to Provide the Necessary Green Energy. J. Eng. Sci. 2014, 2, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Chojnacka, K.; Mikulewicz, M. Green analytical methods of metals determination in biosorption studies. TrAC Trends Anal. Chem. 2019, 116, 254–265. [Google Scholar] [CrossRef]
- Espino, M.; Fernández, M.; Gomez, F.J.V.; Silva, M.F. Natural designer solvents for greening analytical chemistry. TrAC Trends Anal. Chem. 2016, 76, 126–136. [Google Scholar] [CrossRef]
- Parsons, C.; Grabulosa, E.M.; Pili, E.; Floor, G.H.; Roman-Ross, G.; Charlet, L. Quantification of trace arsenic in soils by field-portable X-ray fluorescence spectrometry: Considerations for sample preparation and measurement conditions. J. Hazard. Mater. 2013, 262, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Padilla, J.T.; Hormes, J.; Selim, H.M. Use of portable XRF: Effect of thickness and antecedent moisture of soils on measured concentration of trace elements. Geoderma 2019, 337, 143–149. [Google Scholar] [CrossRef]
- Marzi, D.; Antenozio, M.L.; Vernazzaro, S.; Sette, C.; Veschetti, E.; Lucentini, L.; Daniele, G.; Brunetti, P.; Cardarelli, M. Advanced Drinking Groundwater As Phytofiltration by the Hyperaccumulating Fern Pteris vittata. Water 2021, 13, 2187. [Google Scholar] [CrossRef]
- De Winter, N.J.; Sinnesael, M.; Makarona, C.; Vansteenberge, S.; Claeys, P. Trace element analyses of carbonates using portable and micro-X-ray fluorescence: Performance and optimization of measurement parameters and strategies. J. Anal. At. Spectrom. 2017, 32, 1211–1223. [Google Scholar] [CrossRef]
- Sherman, J. The theoretical derivation of fluorescent X-ray intensities from mixtures. Spectrochim. Acta 1955, 7, 283–306. [Google Scholar] [CrossRef]
- Terzano, R.; Alfeld, M.; Janssens, K.; Vekemans, B.; Schoonjans, T.; Vincze, L.; Tomasi, N.; Pinton, E.; Cesco, S. Spatially resolved (semi) quantitative determination of iron (Fe) in plants by means of synchrotron micro X-ray fluorescence. Anal. Bioanal. Chem. 2013, 405, 3341–3350. [Google Scholar] [CrossRef]
- Markowicz, A. An overview of quantification methods in energy-dispersive X-ray fluorescence analysis. Pramana 2011, 76, 321–329. [Google Scholar] [CrossRef]
- Margui, E.; Van Grieken, R. X-Ray Fluorescence Spectrometry and Related Techniques: An Introduction; Momentum Press: New York, NY, USA, 2013. [Google Scholar]
- Ensina, A.; Carvalho, P.M.; Machado, J.; Carvalho, M.L.; Casal, D.; Pais, D.; Santos, J.P.; Dias, A.A.; Pessanha, S. Analysis of human tissues using Energy Dispersive X Ray Fluorescence–Dark matrix determination for the application to cancer research. J. Trace Elem. Med. Biol. 2021, 68, 126837. [Google Scholar] [CrossRef]
- Shrivastava, A.; Gupta, V.B. Methods for the determination of limit of detection and limit of quantitation of the analytical methods. Chron. Young Sci. 2011, 2, 21–25. [Google Scholar] [CrossRef]
- Stazi, S.R.; Cassaniti, C.; Marabottini, R.; Giuffrida, F.; Leonardi, C. Arsenic uptake and partitioning in grafted tomato plants. Hortic. Environ. Biotechnol. 2016, 57, 241–247. [Google Scholar] [CrossRef]
- Croudace, I.W.; Rindby, A.; Rothwell, R.G. ITRAX: Description and evaluation of a new multi-function X-ray core scanner. Geol. Soc. Lond. Spec. Publ. 2006, 267, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Haschke, M. Laboratory Micro-X-ray Fluorescence Spectroscopy; Springer: Cham, Switzerland, 2014; Volume 55, ISBN 978-3-319-04864-2. [Google Scholar]
- Panchuk, V.; Yaroshenko, I.; Legin, A.; Semenov, V.; Kirsanov, D. Application of chemometric methods to XRF-data–A tutorial review. Anal. Chim. Acta 2018, 1040, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, G.; Pelosi, C.; Agresti, G.; Bonifazi, G.; Santamaria, U.; Serranti, S. X-ray fluorescence investigation on yellow pigments based on lead, tin and antimony through the comparison between laboratory and portable instruments. J. Cult. Herit. 2018, 29, 19–29. [Google Scholar] [CrossRef]
- Bro, R.; Smilde, A.K. Principal component analysis. Anal. Methods 2014, 6, 2812–2831. [Google Scholar] [CrossRef] [Green Version]
- Jolliffe, I.T. Principal Component Analysis for Special Types of Data; Springer: New York, NY, USA, 2002; pp. 338–372. [Google Scholar]
- Smilde, A.K.; Geladi, P.; Bro, R. Multi-Way Analysis: Applications in the Chemical Sciences; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- Wissmann, D. XRF in Agronomy Applications Analysis of Plant Tissues and Fertilizers; Spectroscopy, Special Issues 2019, 34. Available online: https://www.spectroscopyonline.com/view/xrf-agronomy-applications-analysis-plant-tissues-and-fertilizers (accessed on 5 June 2022).
- Queralt, I.; Ovejero, M.; Carvalho, M.L.; Marques, A.F.; Llabrés, J.M. Quantitative determination of essential and trace element content of medicinal plants and their infusions by XRF and ICP techniques. X-ray Spectrom. Int. J. 2005, 34, 213–217. [Google Scholar] [CrossRef]
- Fleming, D.E.B.; Foran, K.A.; Kim, J.S.; Guernsey, J.R. Portable x-ray fluorescence for assessing trace elements in rice and rice products: Comparison with inductively coupled plasma-mass spectrometry. Appl. Radiat. Isot. 2015, 104, 217–223. [Google Scholar] [CrossRef]
- Rodrigues, E.S.; Gomes, M.H.; Duran, N.M.; Cassanji, J.G.; da Cruz, T.N.M.; Sant’Anna Neto, A.; Savassa, S.M.; de Almeida, E.; Carvalho, H.W. Laboratory microprobe X-ray fluorescence in plant science: Emerging applications and case studies. Front Plant Sci. 2018, 9, 1588. [Google Scholar] [CrossRef]
- Campos, N.V.; Guerra, M.B.B.; Mello, J.W.V.; Schaefer, C.E.G.; Krug, F.J.; Alves, E.E.; Azevedo, A.A. Accumulation and spatial distribution of arsenic and phosphorus in the fern Pityrogramma calomelanos evaluated by micro X-ray fluorescence spectrometry. J. Anal. At. Spectrom. 2015, 30, 2375–2383. [Google Scholar] [CrossRef]
- Szalóki, I.; Gerényi, A.; Fodor, F.; Radócz, G.; Czech, V.; Vincze, L. Improved Micro-X-ray Fluorescence Confocal Imaging of Two-Dimensional Distribution of Arsenic Concentration in Cucumber Hypocotyls Using Synchrotron Radiation. Anal. Chem. 2021, 93, 11660–11668. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Ma, L.Q. Effects of arsenic on concentration and distribution of nutrients in the fronds of the arsenic hyperaccumulator Pteris vittata L. Environ. Pollut. 2005, 135, 333–340. [Google Scholar] [CrossRef]
- Carbonell-Barrachina, A.A.; Burló-Carbonell, F.; Mataix-Beneyto, J. Effect of sodium arsenite and sodium chloride on bean plant nutrition (macronutrients). J. Plant Nutr. 1997, 20, 1617–1633. [Google Scholar] [CrossRef]
- Carbonell-Barrachina, A.A.; Burló, F.; López, E.; Mataix, J. Tomato plant nutrition as affected by arsenic concentration. J. Plant Nutr. 1998, 21, 235–244. [Google Scholar] [CrossRef]
- Lombi, E.; Zhao, F.J.; Fuhrmann, M.; Ma, L.Q.; McGrath, S.P. Arsenic distribution and speciation in the fronds of the hyperaccumulator Pteris vittata. New Phytol. 2002, 156, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.R.; Zhao, F.J.; Meharg, A.A.; Raab, A.; Feldmann, J.; McGrath, S. Mechanisms of arsenic hyper-accumulator in Pteris vittata: Uptake kinetics, interactions with phosphate and arsenic speciation. Plant Physiol. 2002, 130, 1552–1561. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.Y.; McGrath, S.P.; Zhao, F.J. Rapid reduction of arsenate in the medium mediated by plant roots. New Phytol. 2007, 176, 590–599. [Google Scholar] [CrossRef]
- Su, Y.H.; McGrath, S.P.; Zhu, Y.G.; Zhao, F.J. Highly efficient xylem transport of arsenite in the arsenic hyperaccumulator Pteris vittata. New Phytol. 2008, 180, 434–441. [Google Scholar] [CrossRef]
- Gao, Y.; Mucci, A. Acid base reactions, phosphate and arsenate complexation, and their competitive adsorption at the surface of goethite in 0.7 M NaCl solution. Geochim. Cosmochim. Acta 2001, 65, 2361–2378. [Google Scholar] [CrossRef]
- Tu, S.; Ma, L.Q. Interactive effects of pH, arsenic and phosphorus on uptake of As and P and growth of the arsenic hyperaccumulator Pteris vittata L. under hydroponic conditions. Environ. Exp. Bot. 2003, 50, 243–251. [Google Scholar] [CrossRef]
- Burlo, F.; Guijarro, I.; Carbonell-Barrachina, A.A.; Vlaero, D.; Martinez-Sanchez, F. Arsenic species: Effects on and accumulation by tomato plants. J. Agric. Food Chem. 1999, 47, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Marschner, H. Mineral Nutrition of Higher Plants, 2nd ed.; Academic Press Ltd.: London, UK, 1995. [Google Scholar]
- Shaibur, M.R.; Kitajima, N.; Huq, S.I.; Kawai, S. Arsenic–iron interaction: Effect of additional iron on arsenic-induced chlorosis in barley grown in water culture. J. Soil Sci. Plant Nutr. 2009, 55, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Ma, L.Q.; Saha, U.; Mathews, S.; Sundaram, S.; Rathinasabapathi, B.; Zhou, Q. Sulfate and glutathione enhanced arsenic accumulation by arsenic hyperaccumulator Pteris vittata L. Environ. Pollut. 2010, 158, 1530–1535. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.F.; Yamaji, N.; Mitani, N.; Xu, X.Y.; Su, Y.H.; McGrath, S.P.; Zhao, F.J. Transporters of arsenite in rice and their role in arsenic accumulation in rice grain. Proc. Natl. Acad. Sci. USA 2008, 105, 9931–9935. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ma, L.Q.; Rathinasabapathi, B.; Liu, Y.; Zeng, G. Uptake and translocation of arsenite and arsenate by Pteris vittata L.: Effects of silicon, boron and mercury. Environ. Exp. Bot. 2010, 68, 222–229. [Google Scholar] [CrossRef]
- Farnese, F.S.; Oliveira, J.A.; Farnese, M.S.; Gusman, G.S.; Silveira, N.M.; Siman, L.I. Uptake arsenic by plants: Effects on mineral nutrition, growth and antioxidant capacity. Idesia 2014, 32, 96–106. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Instrumental Parameters | |
|---|---|
| Plasma gas flow | 10 L min−1 |
| Auxiliary gas flow | 0.2 L min−1 |
| Nebulizer gas flow | 0.55 L min−1 |
| RF power | 1450 watts |
| Viewing height | 15 mm |
| Plasma view | Axial |
| Read parameters | Auto |
| Peristaltic pump flow rate | 1.5 mL min−1 |
| Processing peak | Height |
| Calibration | Linear calculated intercept |
| Injector Alumina | 2.0 mm i.d. |
| Quartztorch | 1 slot |
| T0 | µXRF | ICP-OES | T30 | µXRF | ICP-OES | T60 | µXRF | ICP |
|---|---|---|---|---|---|---|---|---|
| Fe | 222.5 ± 32 | 182.9 ± 17 | Fe | 173.0 ± 17 | 174.7 ± 48 | Fe | 122.3 ± 26 | 124.6 ± 5 |
| Ca | 7186.6 ± 620 | 6256.4 ± 863 | Ca | 4700.1 ± 527 | 4440.5 ± 429 | Ca | 3752.6 ± 544 | 3524.4 ± 150 |
| P | 1130.6 ± 139 | 1394.7 ± 265 | P | 1407.9 ± 193 | 1433.6 ± 43 | P | 1482.3 ± 205 | 1505.8 ± 61 |
| K | 4960.0 ± 594 | 5628.4 ± 603 | K | 7173.6 ± 407 | 6980.1 ± 179 | K | 9833.2 ± 728 | 9663.0 ± 2803 |
| Mn | 96.4 ± 12 | 106.3 ± 9 | Mn | 50.9 ± 7 | 53.0 ± 3 | Mn | 31.5 ± 2 | 33.8 ± 1 |
| Cu | 5.5 ± 1 | 24.9 ± 11 | Cu | 88.9 ± 23 | 91.4 ± 5 | Cu | 78.4 ± 16 | 88.6 ± 7 |
| Zn | 49.0 ± 5 | 41,6 ± 3 | Zn | 69.3 ± 4 | 73.2 ± 16 | Zn | 25.4 ± 1 | 27,5 ± 4 |
| S | 4823.4 ± 535 | 5568.6 ± 288 | S | 6715.3 ± 871 | 6727.1 ± 392 | S | 4809.4 ± 635 | 4785.6 ± 55 |
| Al | 45.4 ± 13 | 86.9 ± 21 | Al | 68.7 ± 9 | 76.3 ± 14 | Al | 38.7 ± 8 | 56.6 ± 2 |
| As | - | 3.0 ± 1 | As | 105.7 ± 5 | 108.4 ± 25 | As | 466.9 ± 39 | 468.5 ± 112 |
| Si | 1517.6 ± 551 | 1165.5 ± 231 | Si | 2570.2 ± 661 | 2579.2 ± 400 | Si | 1541.5 ± 204 | 1532.5 ± 491 |
| T0 | µXRF | ICP-OES | T30 | µXRF | ICP-OES | T45 | µXRF | ICP |
|---|---|---|---|---|---|---|---|---|
| Fe | 175.1 ± 42 | 186.3 ± 13 | Fe | 142.5 ± 29 | 144.2 ± 18 | Fe | 94.3 ± 23 | 96.6 ± 8 |
| Ca | 6222.8 ± 705 | 5095.5 ± 897 | Ca | 5521.1 ± 543 | 5221.6 ± 110 | Ca | 5138.3 ± 518 | 4896.8 ± 62 |
| P | 1370.8 ± 122 | 1184.4 ± 222 | P | 1077.2 ± 115 | 1101.7 ± 28 | P | 909.6 ± 125 | 951.9 ± 42 |
| K | 4774.0 ± 278 | 5228.7 ± 441 | K | 7726.4 ± 798 | 7524.7 ± 427 | K | 6454.6 ± 554 | 6328.7 ± 158 |
| Mn | 100.8 ± 18 | 130.8 ± 18 | Mn | 78.8 ± 13 | 80.3 ± 10 | Mn | 51.6 ± 10 | 53.6 ± 1 |
| Cu | 16.5 ± 3 | 20.5 ± 3 | Cu | 19.4 ± 3 | 25.0 ± 2 | Cu | 14.7 ± 3 | 24.4 ± 1 |
| Zn | 38.8 ± 4 | 41.7 ± 2 | Zn | 44.0 ± 2 | 46.1 ± 2 | Zn | 49.4 ± 5 | 51.9 ± 3 |
| S | 5481.6 ± 394 | 5417.9 ± 234 | S | 7350.0 ± 795 | 7250.5 ± 229 | S | 5303.7 ± 693 | 5311.6 ± 111 |
| Al | 51.9 ± 9 | 83.7 ± 18 | Al | 49.0 ± 10 | 59.0 ± 20 | Al | 14.7 ± 4 | 27.1 ± 5 |
| As | - | 5.8 ± 2 | As | 17169 ± 139 | 1651.7 ± 125 | As | 3357.2 ± 302 | 3240.5 ± 245 |
| Si | 1153.4 ± 224 | 945.4 ± 225 | Si | 2745.2 ± 816 | 2725.2 ± 811 | Si | 1935.1 ± 367 | 1940.5 ± 257 |
| Macro- and Micronutrient | Quantity in S1 Samples | Quantity in S2 Samples |
|---|---|---|
| As | 319.7 ± 22 | 288.4 ± 45 |
| Fe | 227.5 ± 40 | 228.4 ± 49 |
| Ca | 4296.6 ± 289 | 5369.2 ± 554 |
| P | 1047.9 ± 125 | 1001.9 ± 113 |
| K | 8595.6 ± 619 | 6413.7 ± 485 |
| Mn | 100.86 ± 14 | 104.5 ± 19 |
| S | 3975.13 ± 421 | 8491.7 ± 434 |
| Cu | 93.8 ± 26 | 12.7 ± 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capobianco, G.; Bonifazi, G.; Serranti, S.; Marabottini, R.; Antenozio, M.L.; Cardarelli, M.; Brunetti, P.; Stazi, S.R. A Green Approach Based on Micro-X-ray Fluorescence for Arsenic, Micro- and Macronutrients Detection in Pteris vittata. Water 2022, 14, 2202. https://doi.org/10.3390/w14142202
Capobianco G, Bonifazi G, Serranti S, Marabottini R, Antenozio ML, Cardarelli M, Brunetti P, Stazi SR. A Green Approach Based on Micro-X-ray Fluorescence for Arsenic, Micro- and Macronutrients Detection in Pteris vittata. Water. 2022; 14(14):2202. https://doi.org/10.3390/w14142202
Chicago/Turabian StyleCapobianco, Giuseppe, Giuseppe Bonifazi, Silvia Serranti, Rosita Marabottini, Maria Luisa Antenozio, Maura Cardarelli, Patrizia Brunetti, and Silvia Rita Stazi. 2022. "A Green Approach Based on Micro-X-ray Fluorescence for Arsenic, Micro- and Macronutrients Detection in Pteris vittata" Water 14, no. 14: 2202. https://doi.org/10.3390/w14142202