
Quantitative Analysis of Malachite Green in Environmental Samples Using Liquid Chromatography-Mass Spectrometry

 ,
,  , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Instrumentation
2.3. Sample Collection
3. Experimental
3.1. Preparation of Standard Solution
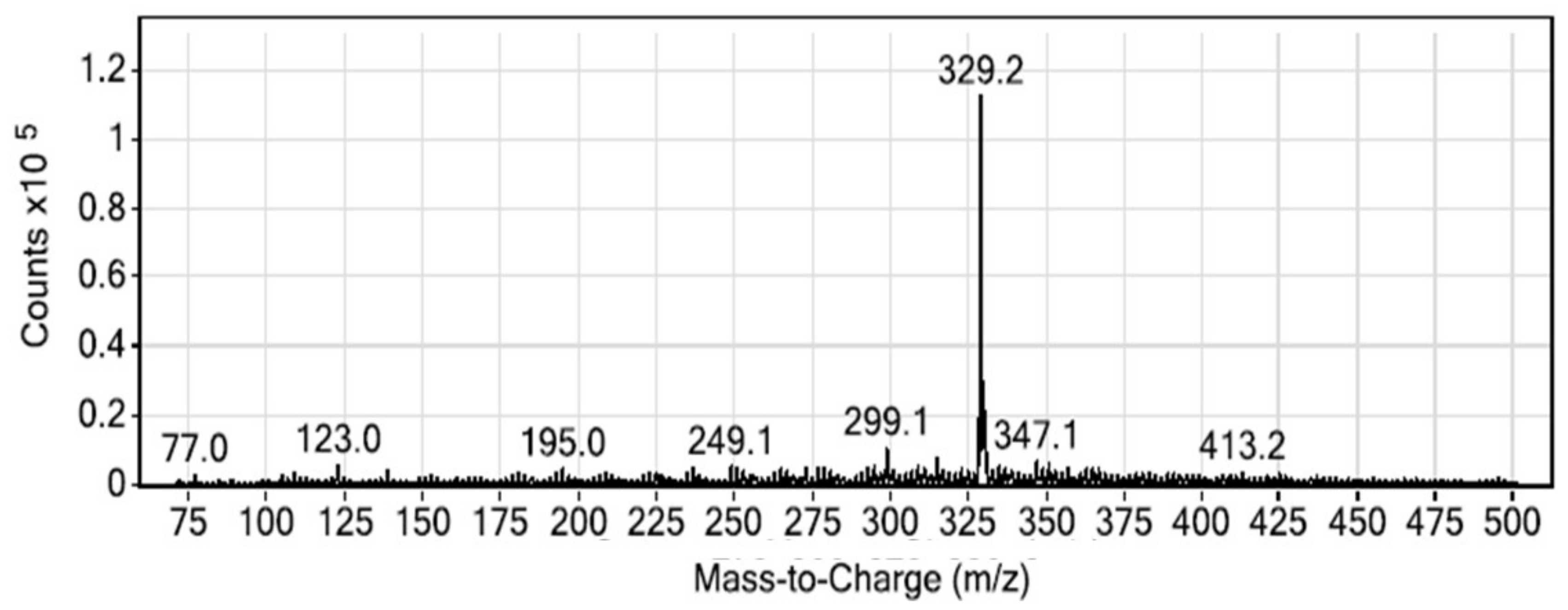
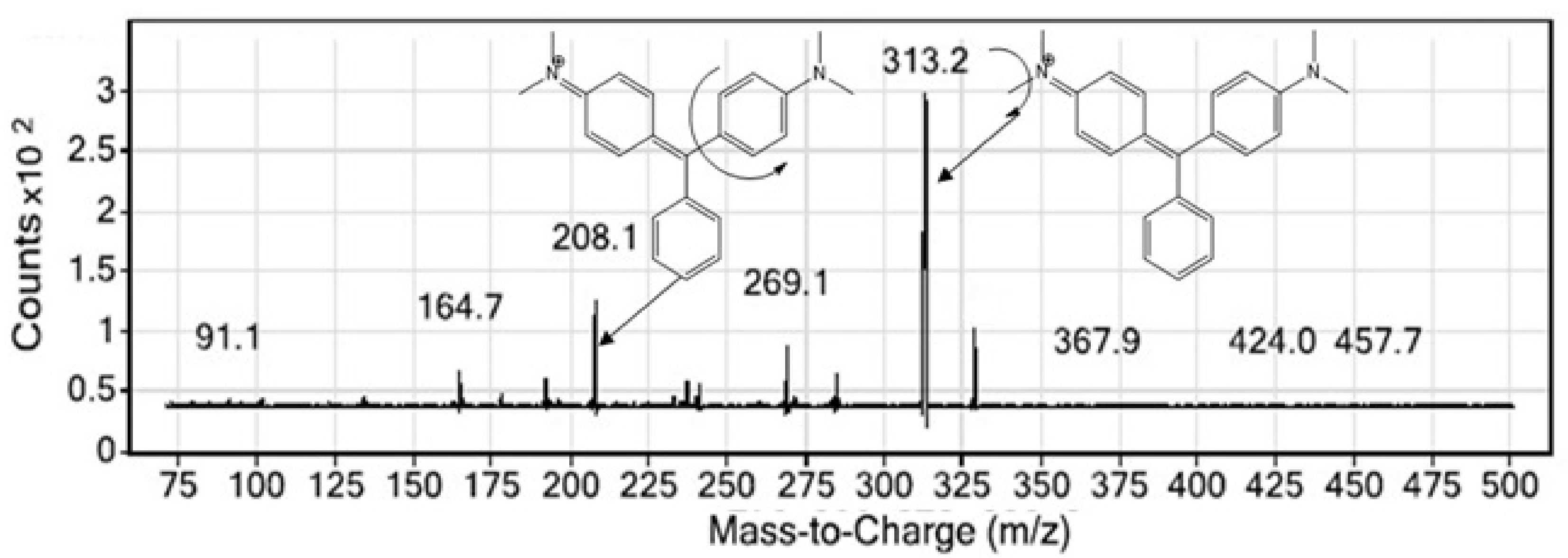
3.2. LC-MS/MS Investigation of MG
Liquid Chromatographic Conditions
3.3. MS/MS Conditions
3.4. Synthesis of Adsorbent Materials
3.5. Sample Extraction Procedure
4. Results and Discussion
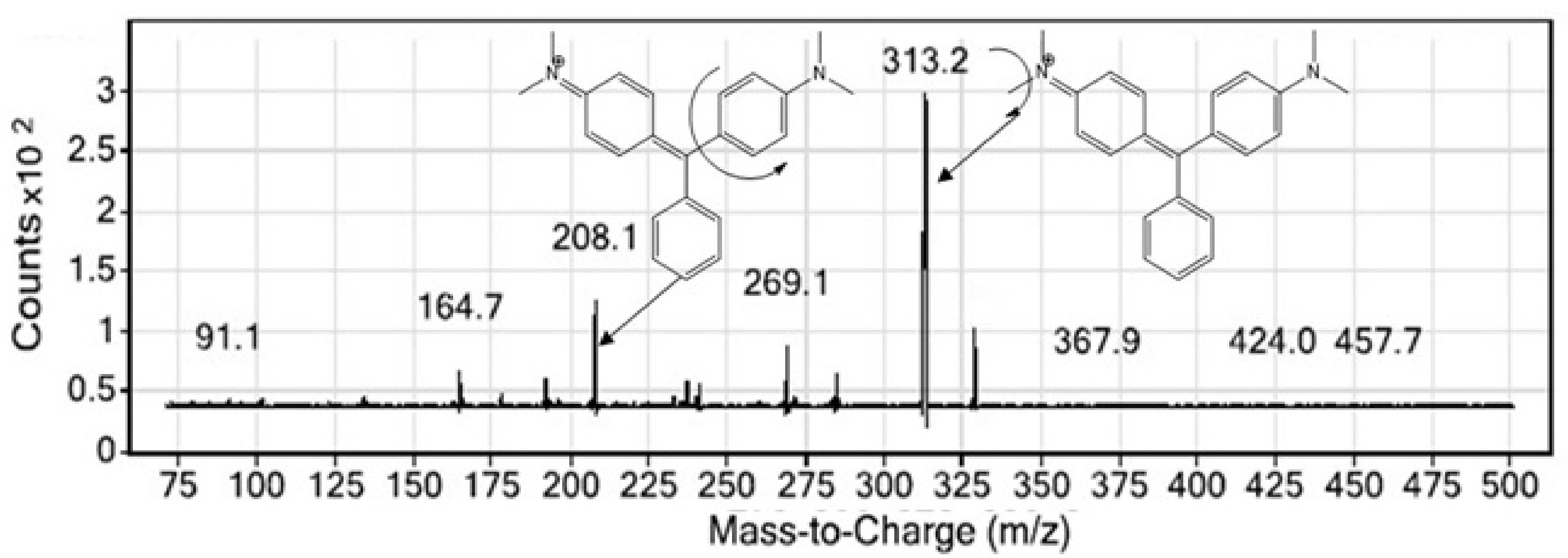
4.1. Method Development and Optimization
4.2. Method Validation for Liquid Chromatography
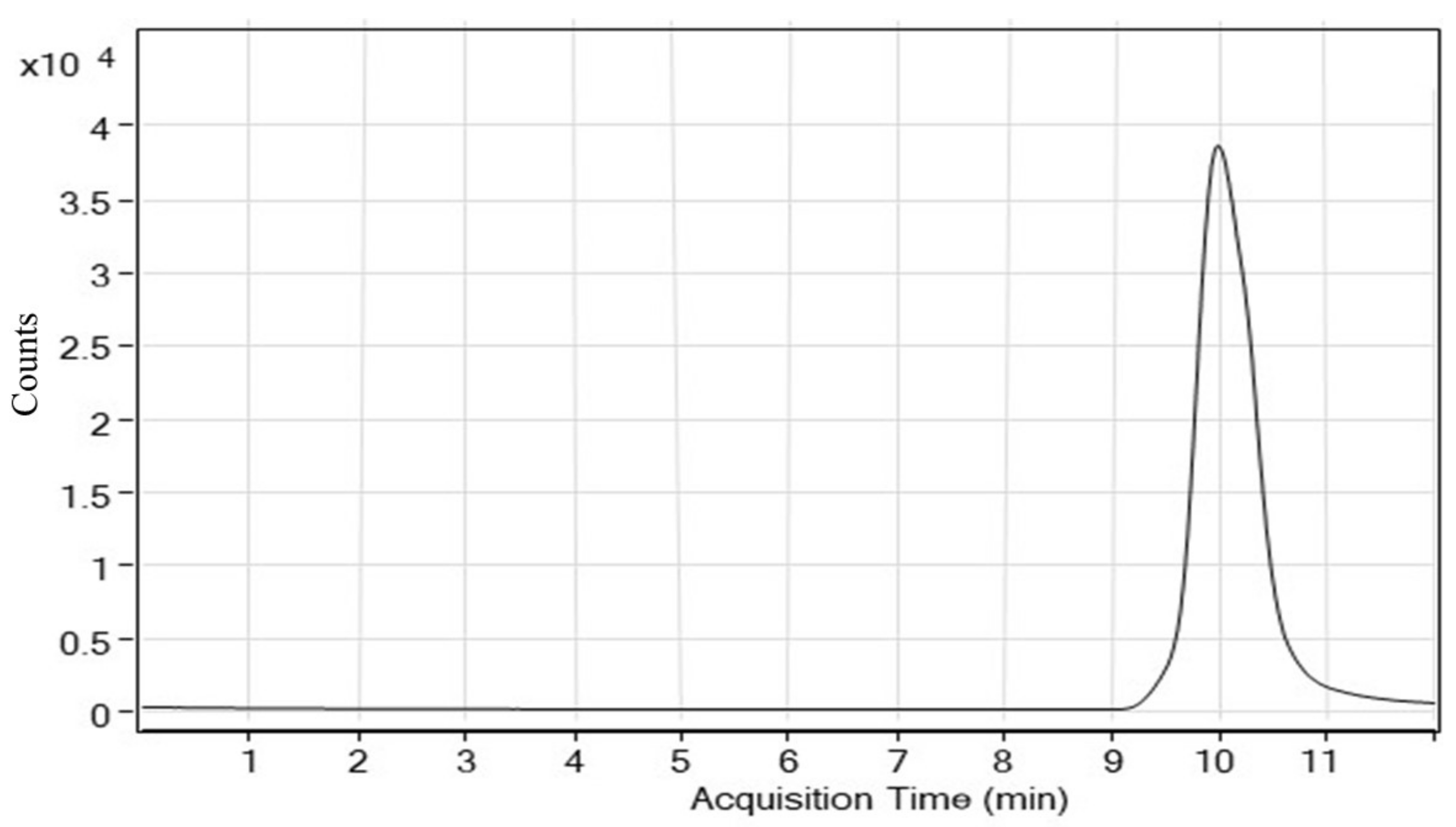
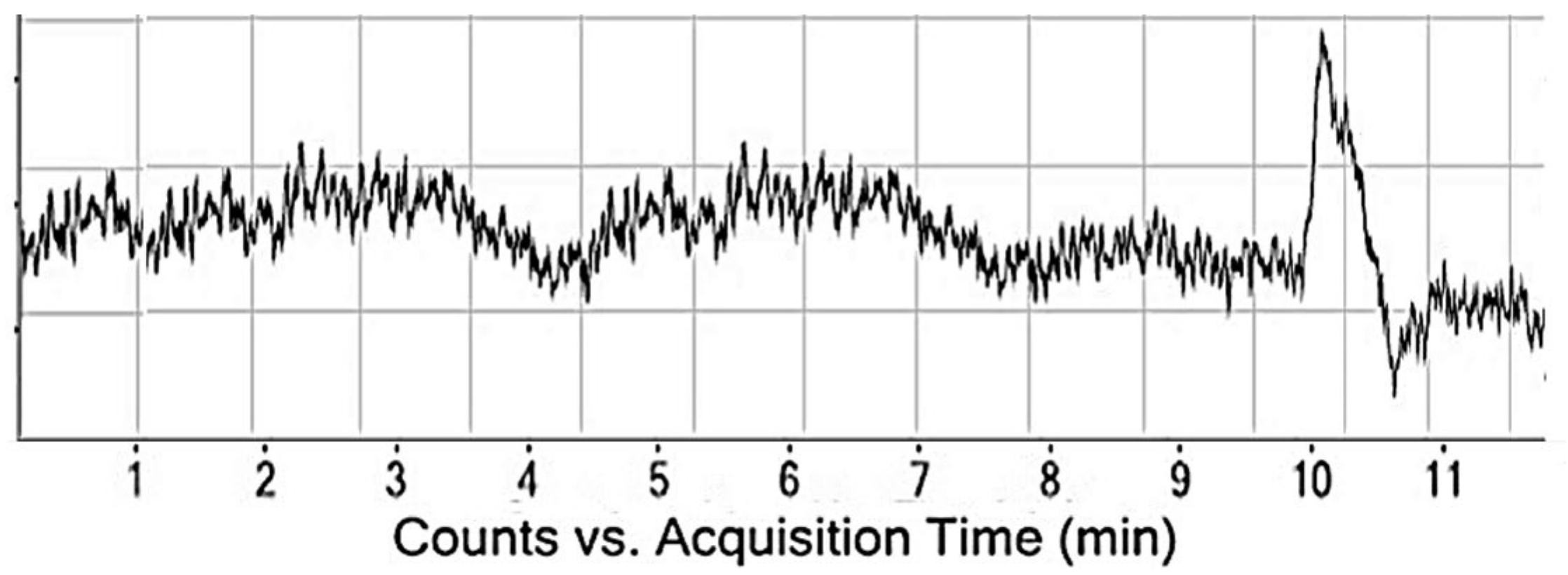
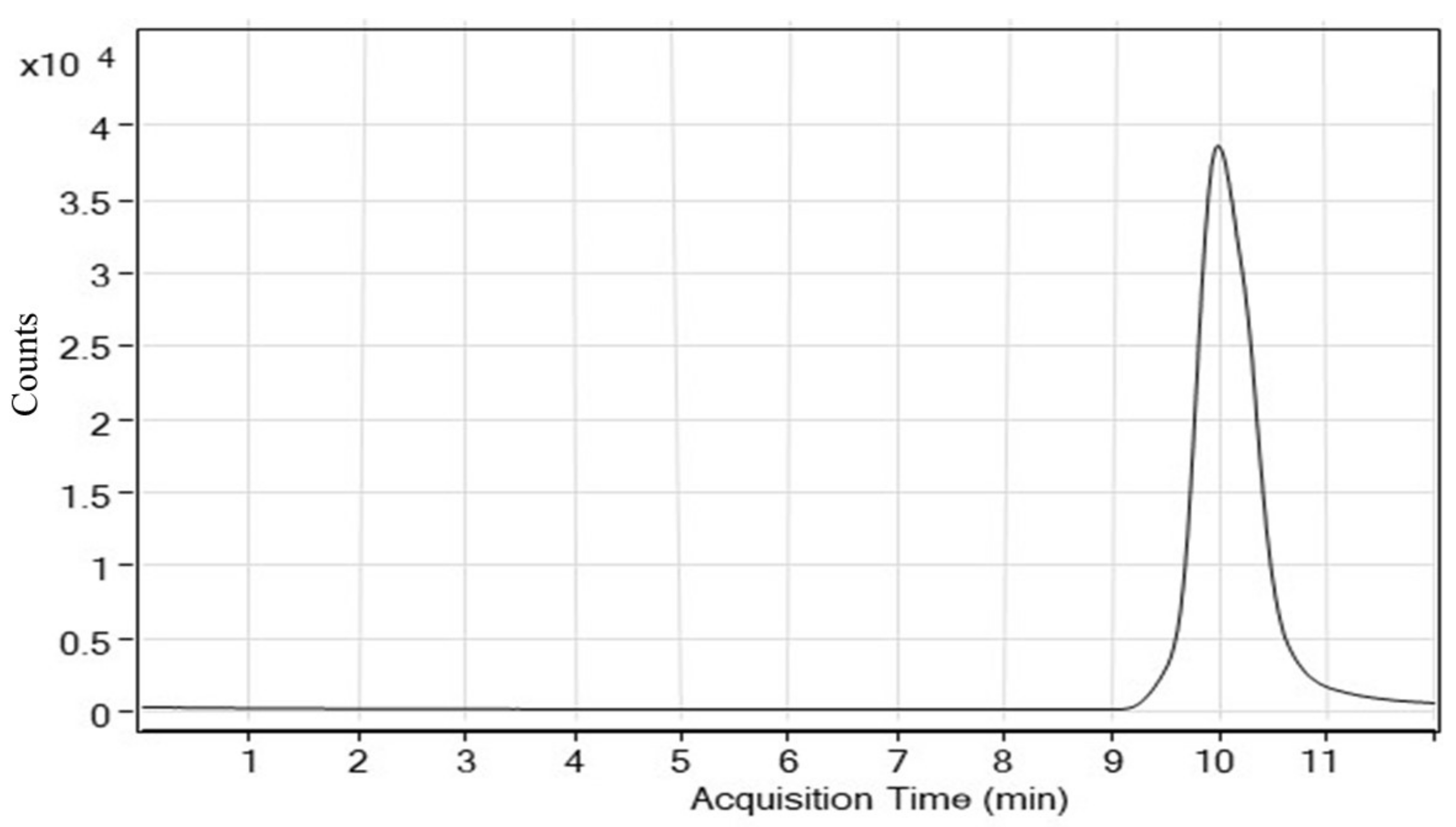
4.2.1. System Suitability
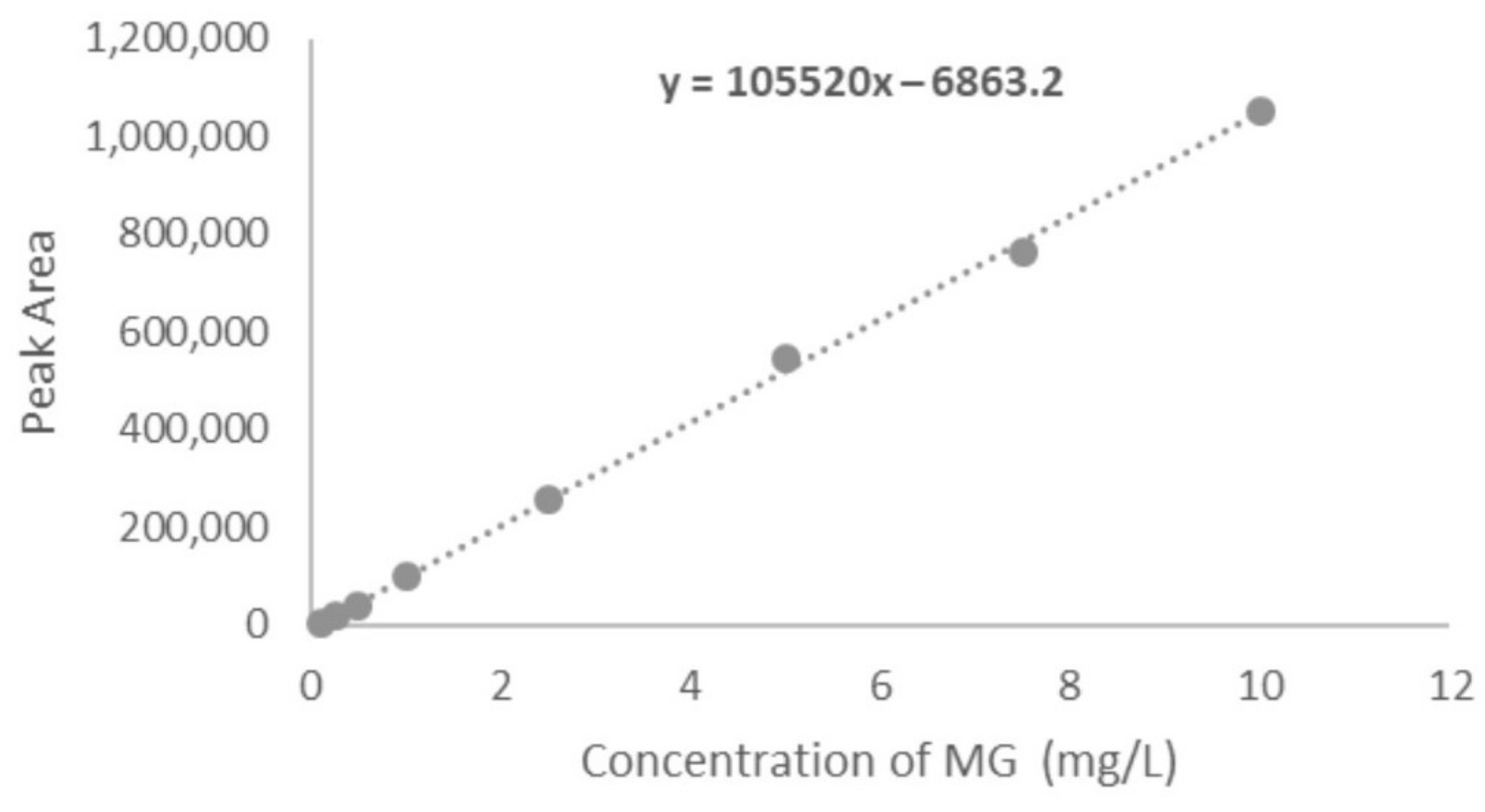
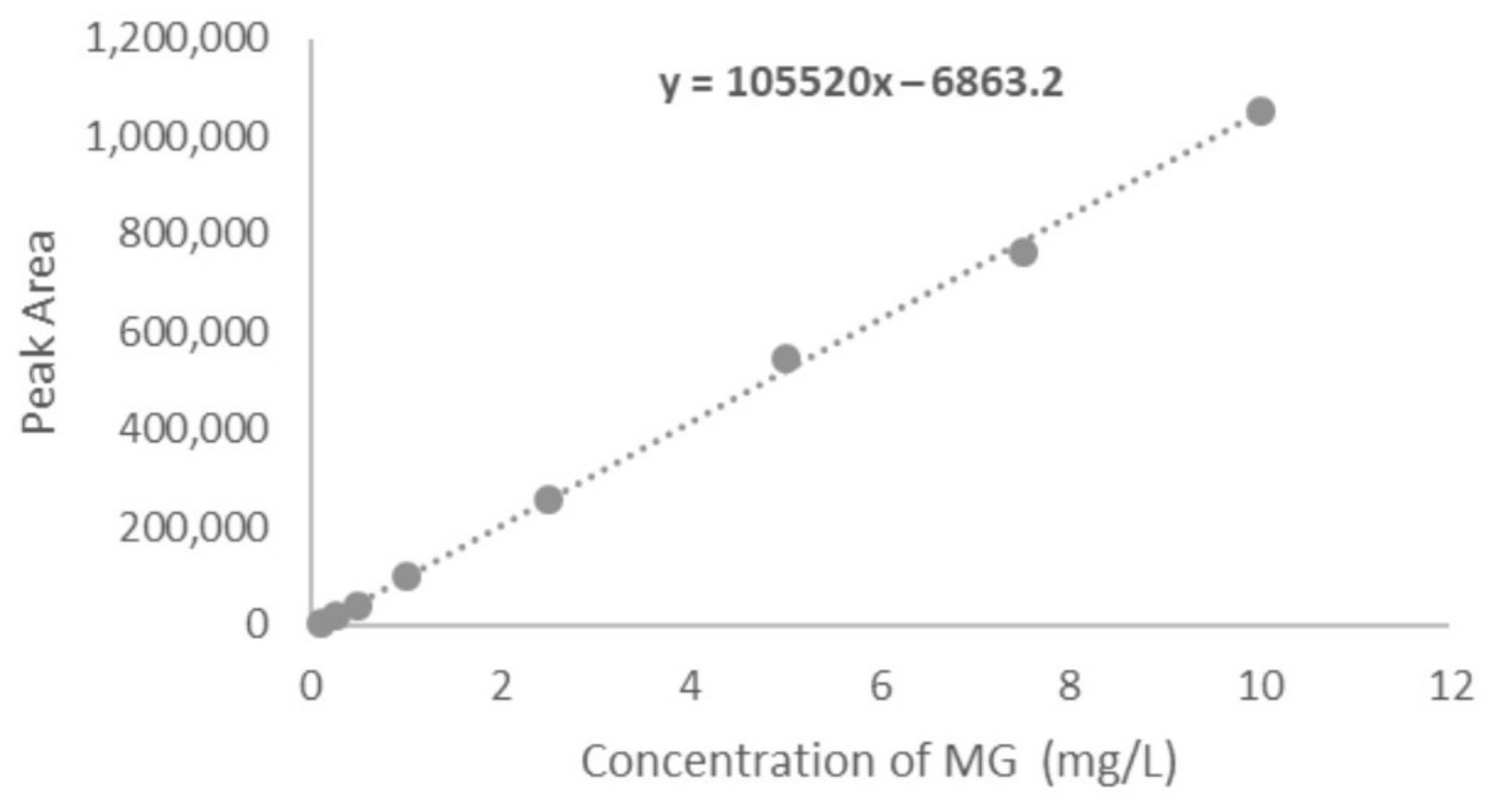
4.2.2. Linearity and Range
4.2.3. Precision and Accuracy
4.2.4. Limit of Detection (LOD) and Limit of Quantitation (LOQ)
4.2.5. Recovery Parameter for Determination of MG
4.2.6. Stability Studies
4.2.7. Application of Sorbent for Uptake of Dyes
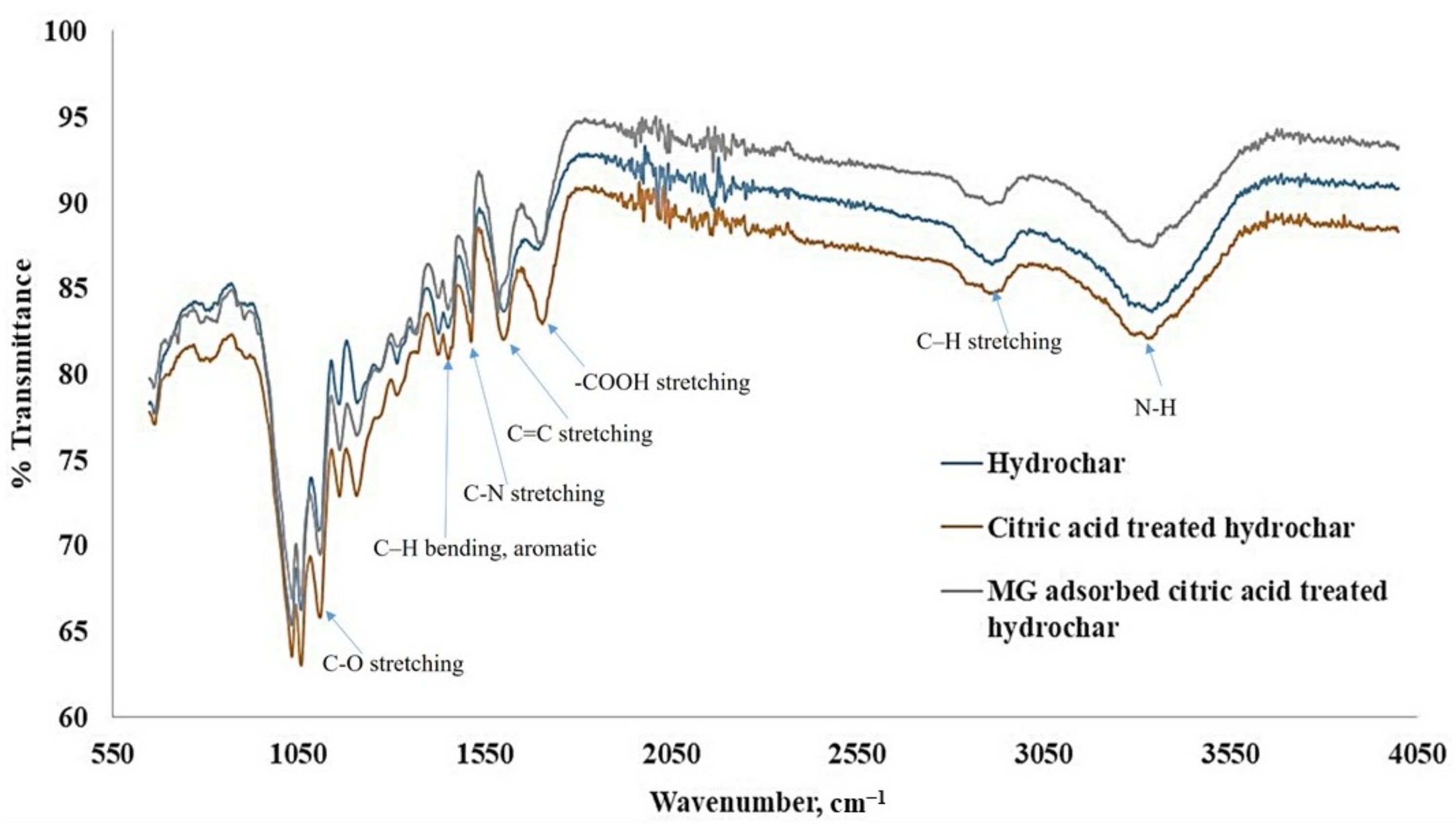
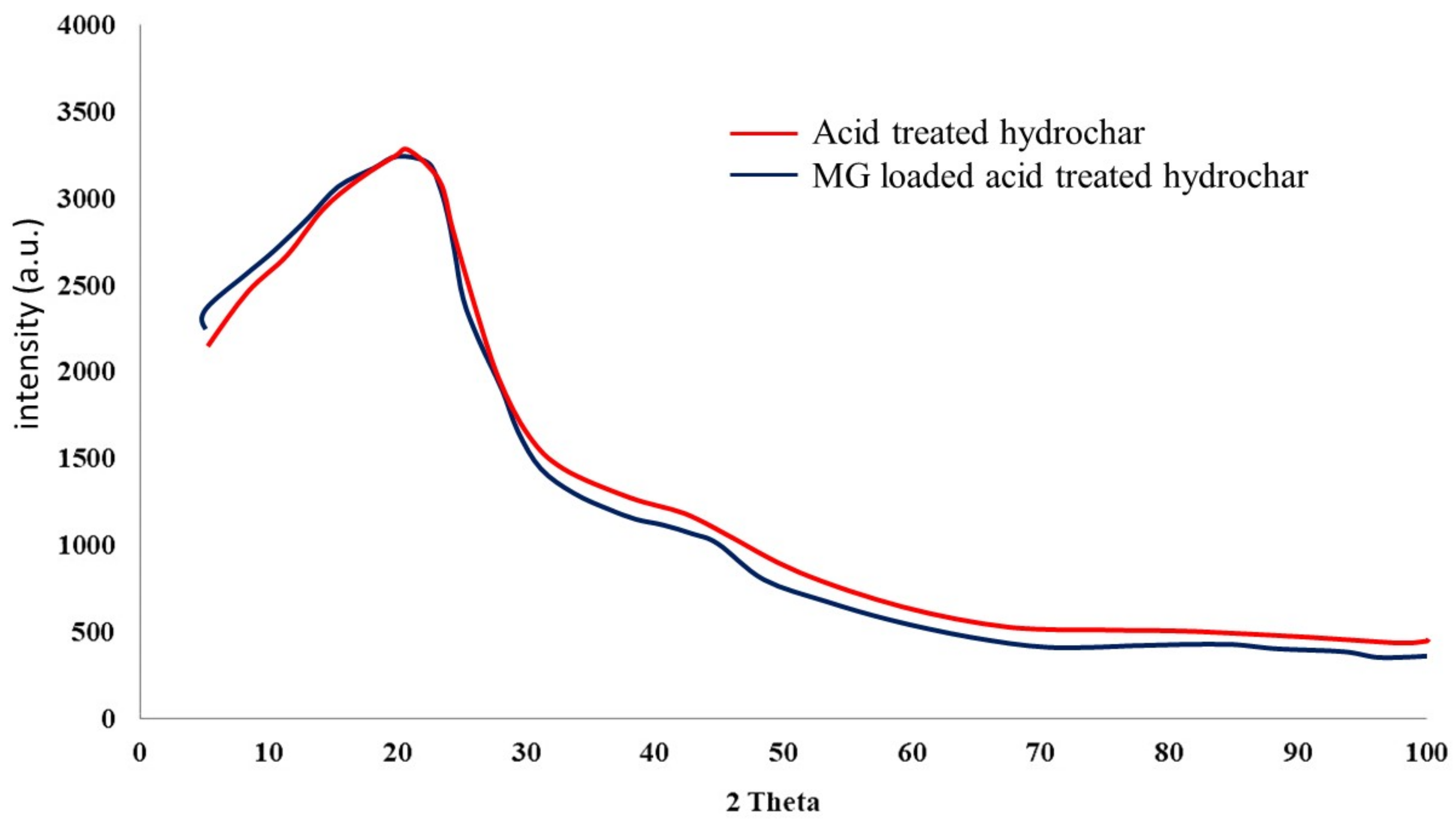
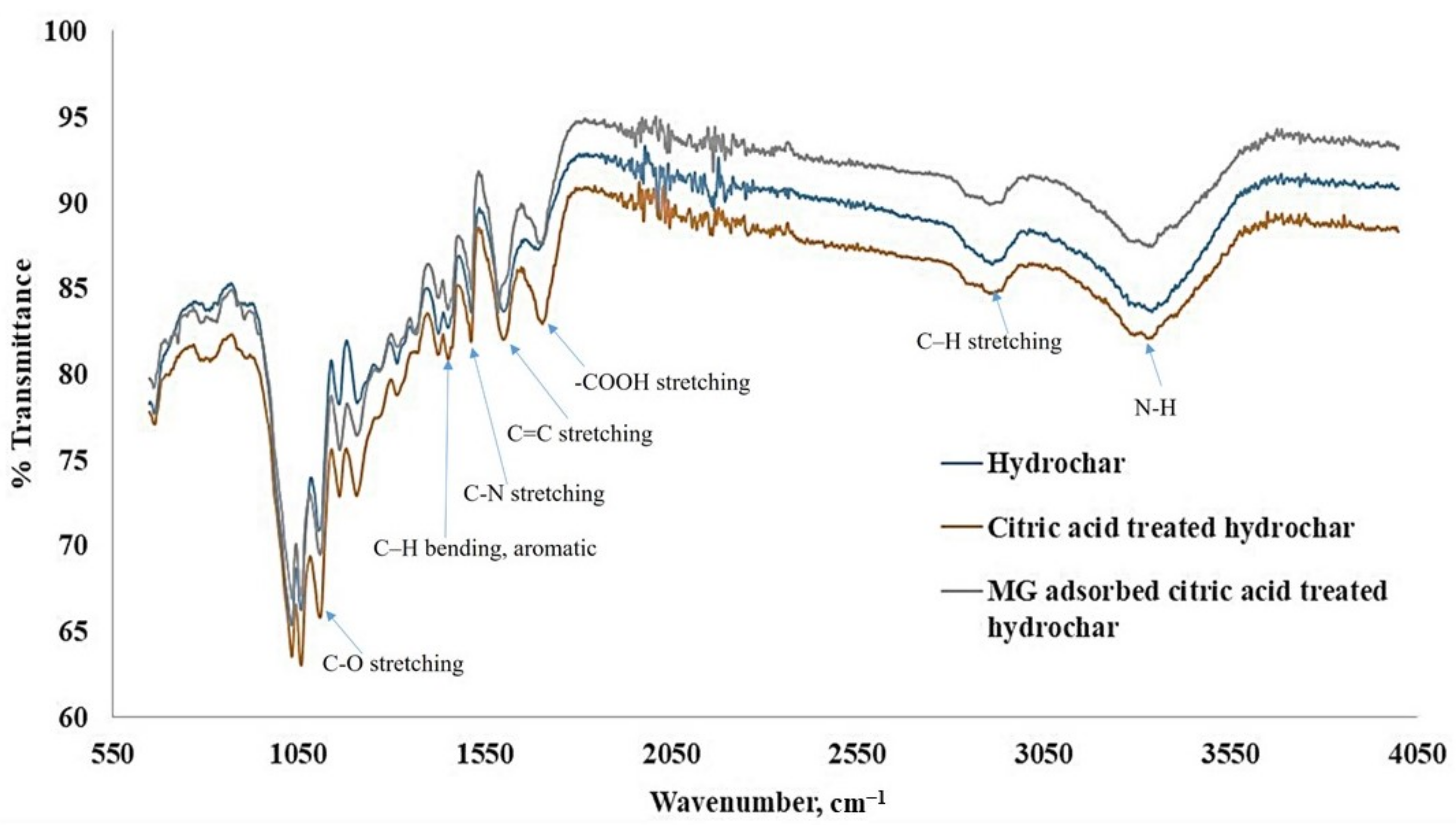
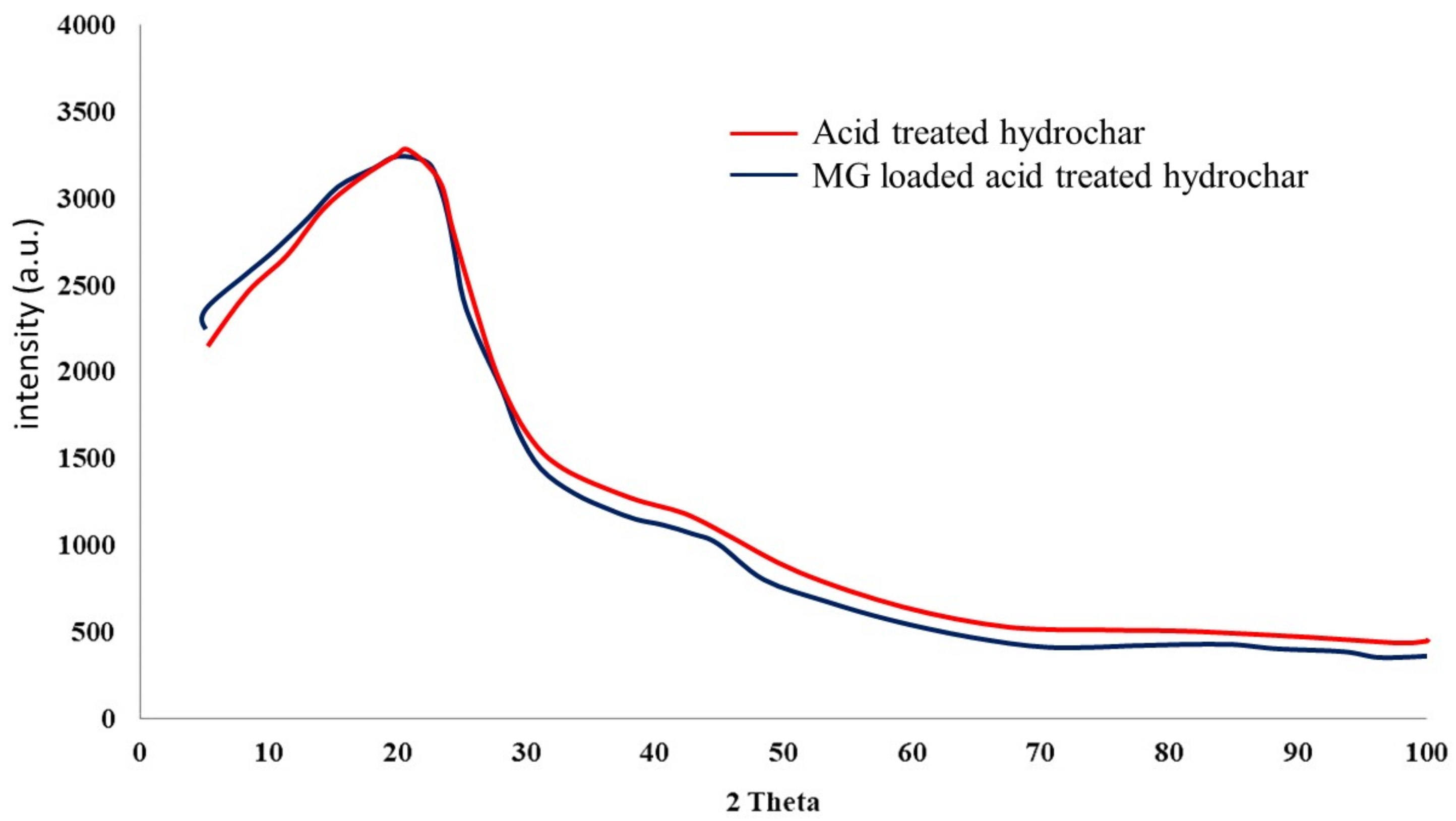
4.3. Sorbent Material Characterization
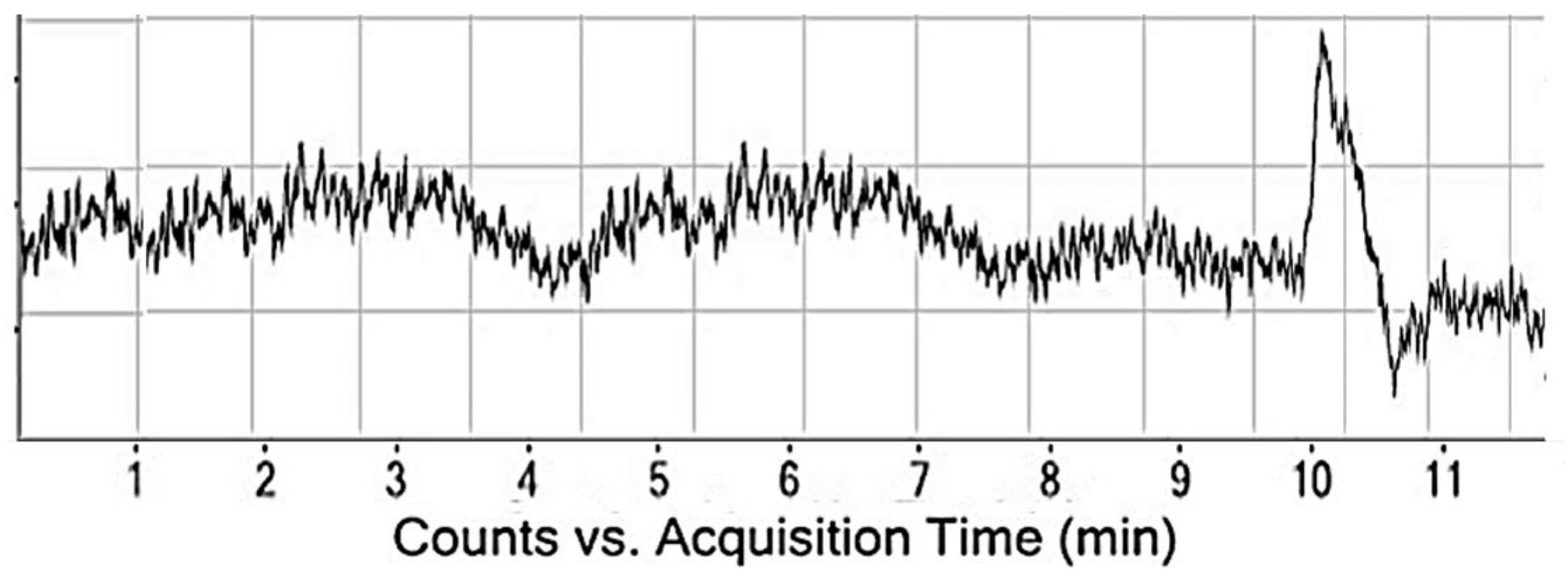
4.4. Application of Developed Method
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hashimoto, J.C.; Paschoal, J.A.R.; de Queiroz, J.F.; Reyes, F.G.R. Considerations on the Use of Malachite Green in Aquaculture and Analytical Aspects of Determining the Residues in Fish: A Review. J. Aquat. Food Prod. Technol. 2014, 20, 273–294. [Google Scholar] [CrossRef]
- Raval, N.P.; Shah, P.U.; Shah, N.K. Nanoparticles Loaded Biopolymer as Effective Adsorbent for Adsorptive Removal of Malachite Green from Aqueous Solution. Water Conserv. Sci. Eng. 2016, 1, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Raval, N.P.; Shah, P.U.; Shah, N.K. Malachite green “acataionic dye and its removal from aqueous by adsorption”. Appl. Water Sci. 2017, 7, 3407–3445. [Google Scholar] [CrossRef] [Green Version]
- Farhadi, K.; Maleki, R.; Nezhad, N.M.; Samadi, N. Spectrophotometric Determination of Malachite Green Residue in Water Samples After Preconcentration on Surfactant-Coated Alumina. Spectrosc. Lett. 2010, 43, 101–107. [Google Scholar] [CrossRef]
- An, L.; Deng, J.; Zhou, L.; Li, H.; Chen, F.; Wang, H.; Liu, Y. Simultaneous spectrophotometric determination of trace amount of malachite green and crystal violet in water after cloud point extraction using partial least squares regression. J. Hazard. Mater. 2010, 175, 883–888. [Google Scholar] [CrossRef]
- Afkhami, A.; Moosavi, R.; Madrakian, T. Preconcentration and spectrophotometric determination of low concentrations of malachite green and leuco-malachite green in water samples by high performance solid phase extraction using maghemite nanoparticles. Talanta 2010, 82, 785–789. [Google Scholar] [CrossRef]
- Pourreza, N.; Elhami, S. Spectrophtometric determination of malachite green in fish farming water samples after cloud point extraction using nonionic surfactant Triton X-100. Anal. Chim. Acta 2017, 596, 62–65. [Google Scholar] [CrossRef]
- Zhu, D.; Li, Q.; Honeychurch, K.C.; Piano, M.; Chen, G. Determination of Malachite Green in Aquaculture Water by Adsorptive Stripping Voltammetry. Anal. Lett. 2016, 49, 1436–1451. [Google Scholar] [CrossRef]
- Huang, W.; Yang, C.; Qu, W.; Zhang, S. Voltammetric determination of malachite green in fish samples based on the enhancement effect of anionic surfactant. Russ. J. Electrochem. 2008, 44, 946–951. [Google Scholar] [CrossRef]
- Yi, H.; Qu, W.; Huang, W. Electrochemical determination of malachite green using a multi-wall carbon nanotube modified glassy carbon electrode. Microchim. Acta 2008, 160, 291–296. [Google Scholar] [CrossRef]
- NurulHidayah, A.P.; Faridah, S.; Azura, M.S.N.; Gayah, A.R.; Othman, M.; Fatimah, A.B. Malachite Green and Leuco-Malachite Green Detection in Fish Using Modified Enzyme Biosensor. Procedia Chem. 2016, 20, 85–89. [Google Scholar]
- Mitrowska, K.; Zmudzki, A.P.J. Determination of malachite green and leucomalachite green in carpmuscle by liquid chromatography with visible and fluorescence detection. J. Chromatogr. A 2005, 1089, 187–192. [Google Scholar] [CrossRef]
- Bajc, Z.; Doganoc, D.Z.; Gačnik, K.S. Determination of malachite green and leucomalachite green in trout and carp muscle by liquid chromatography with visible and fluorescence detection. Slov. Vet. Res. 2007, 44, 81–90. [Google Scholar]
- Chen, G.; Miao, S. HPLC Determination and MS Confirmation of Malachite Green, Gentian Violet, and Their Leuco Metabolite Residues in Channel Catfish Muscle. J. Agric. Food Chem. 2010, 58, 7109–7114. [Google Scholar] [CrossRef]
- Xie, J.; Peng, T.; Chen, D.-D.; Zhang, Q.-J.; Wang, G.-M.; Wang, X.; Guo, Q.; Jiang, F.; Chen, D.; Deng, J. Determination of malachite green, crystal violet and their leuco-metabolites in fish by HPLC–VIS detection after immunoaffinity column clean-up. J. Chromatogr. B 2013, 913–914, 123–128. [Google Scholar] [CrossRef]
- Zeng, S.; Ye, J.; Lin, L.; Chen, W.; Yang, C. Application of UPLC-MS/MS for Simultaneous Determination of Malachite Green and Metabolites Residues in Tilapia. IOP Conf. Ser. Mat. Sci. Eng. 2019, 563, 052062. [Google Scholar] [CrossRef]
- Ascari, J.; Dracz, S.; Santos, F.A.; Lima, J.A.; Diniz, M.H.G.; Vargas, E.A. Validation of an LC-MS/MS method for malachite green (MG), leucomalachite green (LMG), crystal violet (CV) and leucocrystal violet (LCV) residues in fish and shrimp. Food Addit. Contam. A 2012, 29, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Hakami, A.A.H.; Wabaidur, S.M.; Khan, M.A.; AlOthman, Z.A.; Siddiqui, M.R. Extraction Procedures and Analytical Methods for the Determination of Methylene Blue, Rhodamine B and Crystal Violet—An Overview. Curr. Anal. Chem. 2021, 17, 708–728. [Google Scholar] [CrossRef]
- Bilal, M.; Ihsanullah, I.; Younas, M.; Shah, M.U.H. Recent advances in applications of low-cost adsorbents for the removal of heavy metals from water: A critical review. Sep. Purif. Technol. 2022, 278, 119510. [Google Scholar] [CrossRef]
- Ferrentino, B.; Ceccato, R.; Marchetti, V.; Andreottola, G.; Fiori, L. Sewage Sludge Hydrochar: An Option for Removal of Methylene Blue from Wastewater. Appl. Sci. 2020, 10, 3445. [Google Scholar] [CrossRef]
- Hamouda, R.A.; El-Naggar, N.E.; Doleib, N.M.; Saddiq, A.A. Bioprocessing strategies for cost-effective simultaneous removal of chromium and malachite green by marine alga Enteromorpha intestinalis. Sci. Rep. 2020, 10, 13479. [Google Scholar] [CrossRef]
- Iqbal, M. UHPLC-MS/MS assay using environment friendly organic solvents: A green approach for fast determination of quetiapine in rat plasma. Arab. J. Chem. 2019, 12, 1774–1782. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.R.; Wabaidur, S.M.; Busquets, R.; Khan, M.A.; Siddiqui, M.R.; Azam, M. Identification of malachite green in industrial wastewater using lignocellulose biomass composite bio-sorbent and UPLC-MS/MS: A green environmental approach. Process Saf. Environ. 2019, 126, 106–166. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Gai, P.; Hao, T.; Duan, J.; Wang, S. Determination of Malachite Green Residues in Fish Using a Highly Sensitive Electrochemiluminescence Method Combined with Molecularly Imprinted Solid Phase Extraction. J. Agric. Food Chem. 2011, 59, 5257–5262. [Google Scholar] [CrossRef]
- Javad, S.; Rahim, M.; Rezaei, M.; Razmi, H. Magnetic solid-phase extraction of malachite green using soluble eggshell membrane protein doped with magnetic graphene oxide nanocomposite. Int. J. Environ. Anal. Chem. 2018, 98, 1242–1252. [Google Scholar]
- Hammud, H.H.; Shmait, A.; Hourani, N. Removal of Malachite Green from water using hydrothermally carbonized pine needles. RSC Adv. 2015, 5, 7909–7920. [Google Scholar] [CrossRef]
- Ribeiro, R.L.V.; Bottoli, C.B.G.; Collins, K.E.; Collins, C.H. Reevaluation of Ethanol as Organic Modifier for Use in HPLC-RP Mobile Phases. J. Braz. Chem. Soc. 2004, 15, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Surekha, M.; Reddy, S.M. PRESERVATIVES|Classification and Properties. In Encyclopedia of Food Microbiology, 2nd ed.; Academic Press: Cambridge, MA, USA, 2014; pp. 69–75. [Google Scholar]
- Shukla, A.K.; Sampath, S.; Rajan, A.S. Graphite-Grafted Alkaline Iron Electrode. Indian Patent 2016/110862 A1, 14 July 2016. [Google Scholar]
- Cerny, J.; Jindra, J.; Micka, K. Comparative study of porous iron electrodes. J. Power Sources 1993, 45, 267–279. [Google Scholar] [CrossRef]
- Inagaki, M.; Okada, Y.; Miura, H.; Konno, H. Preparation of carbon-coated transition metal particles from mixtures of metal oxide and polyvinylchloride. Carbon 1999, 37, 329–334. [Google Scholar] [CrossRef]
- Rajan, A.S.; Sampath, S.; Shukla, A.K. An in situ carbon-grafted alkaline iron electrode for iron-based accumulators. Energy Environ. Sci. 2014, 7, 1110–1116. [Google Scholar] [CrossRef]
- Khan, M.R.; Khan, M.A.; AlOthman, Z.A.; AlSohaimi, I.H.; Naushad, M.; Shalaan, N.H. Quantitative determination of methylene blue in environmental samples by solid-phase extraction and ultra-performance liquid chromatographytandem mass spectrometry: A green approach. RSC Adv. 2014, 4, 34037–34044. [Google Scholar] [CrossRef]
- Hakami, A.A.H.; Wabaidur, S.M.; Khan, M.A.; AlOthman, Z.A.; Rafatullah, M.; Siddiqui, M.R. Development of Ultra-Performance Liquid Chromatography–Mass Spectrometry Method for Simultaneous Determination of Three Cationic Dyes in Environmental Samples. Molecules 2020, 25, 4564. [Google Scholar] [CrossRef] [PubMed]
- AlAmmari, A.M.; Khan, M.R.; Aqel, A. Trace identification of endocrine-disrupting bisphenol A in drinking water by solid-phase extraction and ultra-performance liquid chromatography-tandem mass spectrometry. J. King Saud Univ. Sci. 2020, 32, 1634–1640. [Google Scholar] [CrossRef]
- Zocolo, G.J.; dos Sontos, G.P.; Vendemiatti, J.; Vacchi, F.I.; de Aragão Umbuzeiro, G.; Zanoni, M.V.B. Using SPE-LC-ESI-MS/MS Analysis to Assess Disperse Dyes in Environmental Water Samples. J. Chromatogr. Sci. 2015, 53, 1257–1264. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Values |
|---|---|
| Gas Temperature | 350 °C |
| Gas Flow | 11 L/min |
| Nebulizer | 35 psi |
| Capillary | 4000 V |
| Collision Energy | 40 eV |
| Fragmentor voltage | 150 V |
| S. No | Retention Time | Concentration of MG (mg L−1) from Corresponding Peak Area |
|---|---|---|
| 1 | 9.988 | 9.99 |
| 2 | 9.998 | 9.82 |
| 3 | 9.999 | 9.91 |
| 4 | 9.998 | 9.97 |
| 5 | 9.998 | 9.91 |
| 6 | 9.999 | 9.87 |
| Mean | 9.998 | 9.92 |
| % RSD | 0.05 | 0.500 |
| Intra-Day Precision | Inter-Day Precision | |||||||
|---|---|---|---|---|---|---|---|---|
| Taken | Found | RSD (%) | SAE | Recovery (%) | Found | RSD (%) | SAE * | Recovery (%) |
| 0.1 | 0.0991 | 0.709 | 0.0016 | 99.10 | 0.0989 | 1.155 | 0.002 | 98.86 |
| 0.25 | 0.2488 | 1.748 | 0.0097 | 99.53 | 0.247 | 1.893 | 0.010 | 98.92 |
| 5.0 | 5.069 | 0.844 | 0.0957 | 101.39 | 5.003 | 1.406 | 0.157 | 100.06 |
| 10.0 | 9.997 | 0.952 | 0.2127 | 99.97 | 9.995 | 1.161 | 0.259 | 99.95 |
| Spiked (mg/L) | Found (mg/L) | RSD (%) | SAE * | Recovery (%) |
|---|---|---|---|---|
| 0.1 | 0.098 | 1. 71 | 0.001 | 97.98 |
| 0.5 | 0.486 | 3.83 | 0.04 | 97.28 |
| 1.0 | 0.983 | 1.62 | 0.03 | 98.30 |
| 7.5 | 7.41 | 2.01 | 0.33 | 98.75 |
| 10 | 9.86 | 1.87 | 0.41 | 98.62 |
| Sample | Type of Sample | Concentration of MG Found, mg/L |
|---|---|---|
| 01 | Laundry Waste | 0.39 |
| 02 | Lake Water | 0.43 |
| 03 | Fish Storage water | 2.02 |
| 04 | Leather Industry waste | 2.56 |
| 05 | University Tap water | ND * |
| 06 | Bottled drinking water | ND * |
| 07 | Irrigation supply water | ND * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain Hakami, A.A.; Ahmed, M.A.; Khan, M.A.; AlOthman, Z.A.; Rafatullah, M.; Islam, M.A.; Siddiqui, M.R. Quantitative Analysis of Malachite Green in Environmental Samples Using Liquid Chromatography-Mass Spectrometry. Water 2021, 13, 2864. https://doi.org/10.3390/w13202864
Hussain Hakami AA, Ahmed MA, Khan MA, AlOthman ZA, Rafatullah M, Islam MA, Siddiqui MR. Quantitative Analysis of Malachite Green in Environmental Samples Using Liquid Chromatography-Mass Spectrometry. Water. 2021; 13(20):2864. https://doi.org/10.3390/w13202864
Chicago/Turabian StyleHussain Hakami, Afnan Ali, Mohammed Asif Ahmed, Moonis Ali Khan, Zeid A. AlOthman, Mohd Rafatullah, Md. Ataul Islam, and Masoom Raza Siddiqui. 2021. "Quantitative Analysis of Malachite Green in Environmental Samples Using Liquid Chromatography-Mass Spectrometry" Water 13, no. 20: 2864. https://doi.org/10.3390/w13202864
APA StyleHussain Hakami, A. A., Ahmed, M. A., Khan, M. A., AlOthman, Z. A., Rafatullah, M., Islam, M. A., & Siddiqui, M. R. (2021). Quantitative Analysis of Malachite Green in Environmental Samples Using Liquid Chromatography-Mass Spectrometry. Water, 13(20), 2864. https://doi.org/10.3390/w13202864