
Key SARS-CoV-2 Mutations of Alpha, Gamma, and Eta Variants Detected in Urban Wastewaters in Italy by Long-Read Amplicon Sequencing Based on Nanopore Technology

 , , , ,
, , , ,
Abstract
:1. Introduction
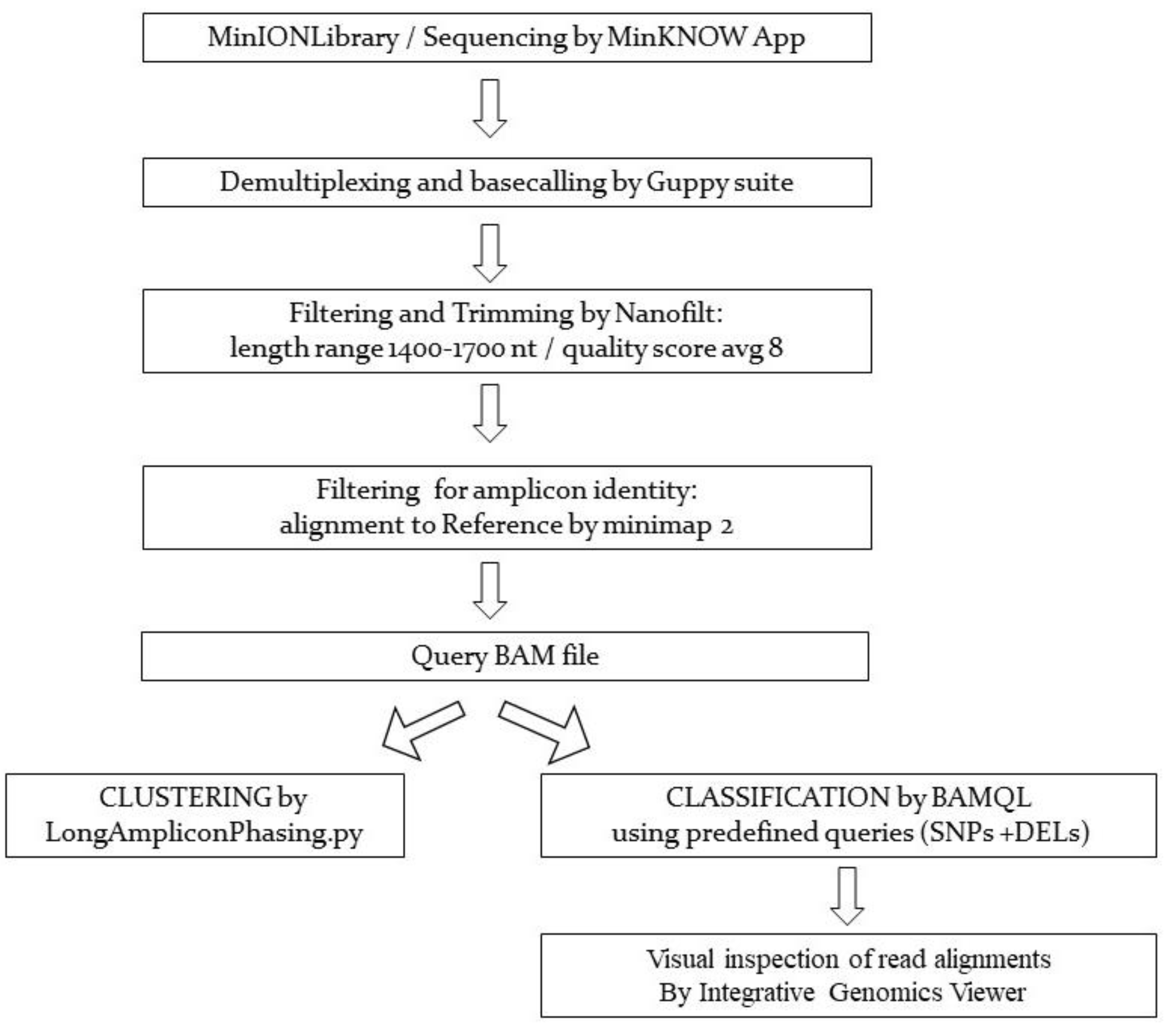
2. Materials and Methods
2.1. Sampling, Nucleic Acids Extraction, Real-Time RT-qPCR, RT-Nested-PCR, and Sanger Sequencing
2.2. Library Preparation of SARS-CoV-2 PCR Products for Nanopore Sequencing, Data Acquisition, Basecalling, and Bioinformatics Analyses
3. Results
3.1. Clustering Results
3.2. Classification Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Agrawal, S.; Orschler, L.; Lackner, S. Metatranscriptomic Analysis Reveals SARS-CoV-2 Mutations in Wastewater of the Frankfurt Metropolitan Area in Southern Germany. Microbiol. Resour. Announc. 2021, 10, e00280-21. [Google Scholar] [CrossRef]
- Bar-Or, I.; Weil, M.; Indenbaum, V.; Bucris, E.; Bar-Ilan, D.; Elul, M.; Levi, N.; Aguvaev, I.; Cohen, Z.; Shirazi, R.; et al. Detection of SARS-CoV-2 variants by genomic analysis of wastewater samples in Israel. Sci. Total Environ. 2021, 789, 148002. [Google Scholar] [CrossRef]
- Crits-Christoph, A.; Kantor, R.S.; Olm, M.R.; Whitney, O.N.; Al-Shayeb, B.; Lou, Y.C.; Flamholz, A.; Kennedy, L.C.; Greenwald, H.; Hinkle, A.; et al. Genome Sequencing of Sewage Detects Regionally Prevalent SARS-CoV-2 Variants. mBio 2021, 12, e02703-20. [Google Scholar] [CrossRef]
- Heijnen, L.; Elsinga, G.; de Graaf, M.; Molenkamp, R.; Koopmans, M.P.; Medema, G. Droplet digital RT-PCR to detect SARS-CoV-2 signature mutations of variants of concern in wastewater. Sci. Total Environ. 2021, 799, 149456. [Google Scholar] [CrossRef]
- Izquierdo-Lara, R.; Elsinga, G.; Heijnen, L.; Munnink, B.B.O.; Schapendonk, C.M.; Nieuwenhuijse, D.; Kon, M.; Lu, L.; Aarestrup, F.M.; Lycett, S.; et al. Monitoring SARS-CoV-2 Circulation and Diversity through Community Wastewater Sequencing, the Netherlands and Belgium. Emerg. Infect. Dis. 2021, 27, 1405–1415. [Google Scholar] [CrossRef]
- La Rosa, G.; Mancini, P.; Ferraro, G.B.; Veneri, C.; Iaconelli, M.; Lucentini, L.; Bonadonna, L.; Brusaferro, S.; Brandtner, D.; Fasanella, A.; et al. Rapid screening for SARS-CoV-2 variants of concern in clinical and environmental samples using nested RT-PCR assays targeting key mutations of the spike protein. Water Res. 2021, 197, 117104. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; Klapsa, D.; Wilton, T.; Zambon, M.; Bentley, E.; Bujaki, E.; Fritzsche, M.; Mate, R.; Majumdar, M. Tracking SARS-CoV-2 in Sewage: Evidence of Changes in Virus Variant Predominance during COVID-19 Pandemic. Viruses 2020, 12, 1144. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Feirer, N.; Brockman, M.; Preston, M.A.; Teter, S.J.; Ma, D.; Goueli, S.A.; Moorji, S.; Saul, B.; Cali, J.J. A direct capture method for purification and detection of viral nucleic acid enables epidemiological surveillance of SARS-CoV-2. Sci. Total Environ. 2021, 795, 148834. [Google Scholar] [CrossRef] [PubMed]
- Nemudryi, A.; Nemudraia, A.; Wiegand, T.; Surya, K.; Buyukyoruk, M.; Cicha, C.; Vanderwood, K.K.; Wilkinson, R.; Wiedenheft, B. Temporal Detection and Phylogenetic Assessment of SARS-CoV-2 in Municipal Wastewater. Cell Rep. Med. 2020, 1, 100098. [Google Scholar] [CrossRef]
- Prado, T.; Fumian, T.M.; Mannarino, C.F.; Resende, P.C.; Motta, F.C.; Eppinghaus, A.L.F.; Vale, V.H.C.D.; Braz, R.M.S.; Andrade, J.D.S.R.D.; Maranhão, A.G.; et al. Wastewater-based epidemiology as a useful tool to track SARS-CoV-2 and support public health policies at municipal level in Brazil. Water Res. 2021, 191, 116810. [Google Scholar] [CrossRef] [PubMed]
- Rimoldi, S.G.; Stefani, F.; Gigantiello, A.; Polesello, S.; Comandatore, F.; Mileto, D.; Maresca, M.; Longobardi, C.; Mancon, A.; Romeri, F.; et al. Presence and infectivity of SARS-CoV-2 virus in wastewaters and rivers. Sci. Total Environ. 2020, 744, 140911. [Google Scholar] [CrossRef]
- Rubio-Acero, R.; Beyerl, J.; Muenchhoff, M.; Roth, M.S.; Castelletti, N.; Paunovic, I.; Radon, K.; Springer, B.; Nagel, C.; Boehm, B.; et al. Spatially resolved qualified sewage spot sampling to track SARS-CoV-2 dynamics in Munich—One year of experience. Sci. Total Environ. 2021, 797, 149031. [Google Scholar] [CrossRef] [PubMed]
- Wilton, T.; Bujaki, E.; Klapsa, D.; Majumdar, M.; Zambon, M.; Fritzsche, M.; Mate, R.; Martin, J. Rapid Increase of SARS-CoV-2 Variant B.1.1.7 Detected in Sewage Samples from England between October 2020 and January 2021. mSystems 2021, 6, e0035321. [Google Scholar] [CrossRef]
- Mancini, P.; Ferraro, G.B.; Iaconelli, M.; Suffredini, E.; Valdazo-González, B.; Della Libera, S.; Divizia, M.; La Rosa, G. Molecular characterization of human Sapovirus in untreated sewage in Italy by amplicon-based Sanger and next-generation sequencing. J. Appl. Microbiol. 2019, 126, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Suffredini, E.; Iaconelli, M.; Equestre, M.; Valdazo-González, B.; Ciccaglione, A.R.; Marcantonio, C.; della Libera, S.; Bignami, F.; La Rosa, G. Genetic Diversity Among Genogroup II Noroviruses and Progressive Emergence of GII.17 in Wastewaters in Italy (2011–2016) Revealed by Next-Generation and Sanger Sequencing. Food Environ. Virol. 2018, 10, 141–150. [Google Scholar] [CrossRef]
- Iaconelli, M.; Valdazo-González, B.; Equestre, M.; Ciccaglione, A.R.; Marcantonio, C.; della Libera, S.; La Rosa, G. Molecular characterization of human adenoviruses in urban wastewaters using next generation and Sanger sequencing. Water Res. 2017, 121, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhang, J.; Xiao, A.; Gu, X.; Lee, W.L.; Armas, F.; Kauffman, K.; Hanage, W.; Matus, M.; Ghaeli, N.; et al. SARS-CoV-2 Titers in Wastewater Are Higher than Expected from Clinically Confirmed Cases. mSystems 2020, 5, e00614-20. [Google Scholar] [CrossRef]
- Costafreda, M.I.; Bosch, A.; Pintó, R.M. Development, Evaluation, and Standardization of a Real-Time TaqMan Reverse Transcription-PCR Assay for Quantification of Hepatitis A Virus in Clinical and Shellfish Samples. Appl. Environ. Microbiol. 2006, 72, 3846–3855. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, G.; Mancini, P.; Ferraro, G.B.; Veneri, C.; Iaconelli, M.; Bonadonna, L.; Lucentini, L.; Suffredini, E. SARS-CoV-2 has been circulating in northern Italy since December 2019: Evidence from environmental monitoring. Sci. Total Environ. 2021, 750, 141711. [Google Scholar] [CrossRef]
- Masella, A.P.; Lalansingh, C.M.; Sivasundaram, P.; Fraser, M.; Bristow, R.G.; Boutros, P.C. BAMQL: A query language for extracting reads from BAM files. BMC Bioinform. 2016, 17, 305. [Google Scholar] [CrossRef] [Green Version]
- Panchal, D.; Prakash, O.; Bobde, P.; Pal, S. SARS-CoV-2: Sewage surveillance as an early warning system and challenges in developing countries. Environ. Sci. Pollut. Res. Int. 2021, 28, 22221–22240. [Google Scholar] [CrossRef]
- Macku’ak, T.; Gál, M.; Špalková, V.; Fehér, M.; Briestenská, K.; Mikušová, M.; Tomčíková, K.; Tamáš, M.; Butor Škulcová, A. Wastewater-Based Epidemiology as an Early Warning System for the Spreading of SARS-CoV-2 and Its Mutations in the Population. Int. J. Environ. Res. Public Health 2021, 18, 5629. [Google Scholar] [CrossRef]
- Lundy, L.; Fatta-Kassinos, D.; Slobodnik, J.; Karaolia, P.; Cirka, L.; Kreuzinger, N.; Castiglioni, S.; Bijlsma, L.; Dulio, V.; Deviller, G.; et al. Making Waves: Collaboration in the time of SARS-CoV-2—Rapid development of an international co-operation and wastewater surveillance database to support public health decision-making. Water Res. 2021, 199, 117167. [Google Scholar] [CrossRef] [PubMed]
- Hamouda, M.; Mustafa, F.; Maraqa, M.; Rizvi, T.; Aly Hassan, A. Wastewater surveillance for SARS-CoV-2: Lessons learnt from recent studies to define future applications. Sci. Total Environ. 2021, 759, 143493. [Google Scholar] [CrossRef] [PubMed]
- Rooney, C.M.; Moura, I.B.; Wilcox, M.H. Tracking COVID-19 via sewage. Curr. Opin. Gastroenterol. 2021, 37, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Medema, G.; Been, F.; Heijnen, L.; Petterson, S. Implementation of environmental surveillance for SARS-CoV-2 virus to support public health decisions: Opportunities and challenges. Curr. Opin. Environ. Sci. Health 2020, 17, 49–71. [Google Scholar] [CrossRef]
- Ertöz, L.; Steinbach, M.; Kumar, V. Finding Clusters of Different Sizes, Shapes, and Densities in Noisy, High Dimensional Data. In Proceedings of the 2003 SIAM International Conference on Data Mining; Society for Industrial and Applied Mathematics, San Francisco, CA, USA, 1–3 May 2003; pp. 47–58. [Google Scholar]
- Liang, M.; Raley, C.; Zheng, X.; Kutty, G.; Gogineni, E.; Sherman, B.T.; Sun, Q.; Chen, X.; Skelly, T.; Jones, K.; et al. Distinguishing highly similar gene isoforms with a clustering-based bioinformatics analysis of PacBio single-molecule long reads. BioData Min. 2016, 9, 13. [Google Scholar] [CrossRef] [Green Version]
- Riaz, N.; Leung, P.; Barton, K.; Smith, M.A.; Carswell, S.; Bull, R.; Lloyd, A.R.; Rodrigo, C. Adaptation of Oxford Nanopore technology for hepatitis C whole genome sequencing and identification of within-host viral variants. BMC Genom. 2021, 22, 148. [Google Scholar] [CrossRef]
- Kono, N.; Arakawa, K. Nanopore sequencing: Review of potential applications in functional genomics. Dev. Growth Differ. 2019, 61, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.; Hui, J.; Mao, H. Nanopore Technology and Its Applications in Gene Sequencing. Biosensors 2021, 11, 214. [Google Scholar] [CrossRef]
- Ogden, R.; Vasiljevic, N.; Prost, S. Nanopore sequencing in non-human forensic genetics. Emerg. Top. Life Sci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, S.; Mangano, E.; Barizzone, N.; Bordoni, R.; Sorosina, M.; Clarelli, F.; Corrado, L.; Boneschi, F.M.; D’Alfonso, S.; De Bellis, G. Next Generation Sequencing of Pooled Samples: Guideline for Variants’ Filtering. Sci. Rep. 2016, 6, 33735. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Simpson, S.L.; Bertsch, P.M.; Bibby, K.; Bivins, A.; Blackall, L.L.; Bofill-Mas, S.; Bosch, A.; Brandão, J.; Choi, P.M.; et al. Minimizing errors in RT-PCR detection and quantification of SARS-CoV-2 RNA for wastewater surveillance. Sci. Total Environ. 2021, 149877. [Google Scholar] [CrossRef]


{kind=link}
| Alpha (B.1.1.7) | Beta (B.1.351) | Gamma (P.1) | Delta (B.1.617.2) | Eta (B.1.525) | Kappa (B.1.617.1) | Lambda (C.37) | Lota (B.1.526) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutation | % | Mutation | % | Mutation | % | Mutation | % | Mutation | % | Mutation | % | Mutation | % | Mutation | % |
| H69del | 94% | D80A | 87% | D138Y | 93% | K77T | 0.8% | A67V | 95% | T95I | 45% | G75V | 85% | T95I | 74% |
| V70del | 95% | D215G | 85% | R190 | 92% | T95I | 41% | H69- | 92% | G142D | 54% | T76I | 86% | D253G | 74% |
| Y144del | 94% | L241- | 77% | K417T | 95% | G142D | 54% | V70- | 92% | E154K | 67% | R246- | 76% | S477N | 29% |
| E484K | 0.2% | L242- | 78% | E484K | 96% | E156G | 88% | Y144- | 92% | L452R | 93% | S247- | 76% | E484K | 42% |
| S494P | 0.3% | L243- | 77% | N501Y | 96% | F157- | 89% | E484K | 97% | E484Q | 93% | Y248- | 77% | ||
| N501Y | 97% | P384L | 1.2% | R158- | 88% | L249- | 77% | ||||||||
| A570D | 98% | K417N | 88% | W258L | 0.2% | T250- | 77% | ||||||||
| E484K | 87% | K417N | 0.4% | P251- | 77% | ||||||||||
| N501Y | 87% | L452R | 97% | G252- | 77% | ||||||||||
| E516Q | 0.3% | T478K | 97% | D253N | 77% | ||||||||||
| L452Q | 90% | ||||||||||||||
| F490S | 90% | ||||||||||||||
| Sample ID | Sampling Location | Date | Sanger Sequencing | Nanopore Sequencing | |||
|---|---|---|---|---|---|---|---|
| Mutations Detected | Associated Variant | Pool Barcode | Clustering | Classification (BAMQL) | |||
| 4158 | Sabaudia (Belsito) | 28 April 2021 | D138Y, R190S, K417T, E484K, N501Y | Gamma | BC01 | Alpha + Gamma | Alpha + Gamma |
| 4159 | Sabaudia (Belsito) | 29 April 2021 | H69del, V70del, Y144del, N501Y, A570D | Alpha | |||
| 4162 | Sabaudia (Caprolace) | 28 April 2021 | S98F, H69del, V70del, Y144del, N501Y, A570D | Alpha | BC02 | Alpha | Alpha + Eta |
| 4163 | Sabaudia (Caprolace) | 29 April 2021 | H69del, V70del, Y144del, N501Y, A570D | Alpha | |||
| 4172 | Sabaudia (Caprolace) | 4 May 2021 | H69del, V70del, Y144del, N501Y, A570D | Alpha | BC03 | Alpha | Alpha |
| 4173 | Sabaudia (Caprolace) | 6 May 2021 | H69del, V70del, Y144del, N501Y, A570D | Alpha | |||
| Panel of Deletion and/or Amino Acid Substitutions in Linkage of Each Variant | Clustering | Classification (BAMQL) | ||||
|---|---|---|---|---|---|---|
| BC01 | BC02 | BC03 | BC01 | BC02 | BC03 | |
| Alpha (H69del, V70del, Y144del, N501Y, A570D) | 11,055 | 70,714 | 85,971 | 54,489 | 85,191 | 105,315 |
| Alpha + S98F (C21855T) | / | 8604 | / | / | 38,460 | / |
| Alpha + E484K (G23012A) | / | / | / | / | 761 | / |
| Alpha + S98F + E484K (C21855T + G23012A) | / | / | / | / | 931 | / |
| Gamma (D138Y, R190S, K417T, E484K, N501Y) | 8443 | / | / | 69,980 | / | / |
| Eta (A67V, H69del, V70del, Y144del, E484K) | / | / | / | / | 4325 | / |
| Eta + S151I (G22014T) | / | / | / | / | 2036 | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Rosa, G.; Brandtner, D.; Mancini, P.; Veneri, C.; Bonanno Ferraro, G.; Bonadonna, L.; Lucentini, L.; Suffredini, E. Key SARS-CoV-2 Mutations of Alpha, Gamma, and Eta Variants Detected in Urban Wastewaters in Italy by Long-Read Amplicon Sequencing Based on Nanopore Technology. Water 2021, 13, 2503. https://doi.org/10.3390/w13182503
La Rosa G, Brandtner D, Mancini P, Veneri C, Bonanno Ferraro G, Bonadonna L, Lucentini L, Suffredini E. Key SARS-CoV-2 Mutations of Alpha, Gamma, and Eta Variants Detected in Urban Wastewaters in Italy by Long-Read Amplicon Sequencing Based on Nanopore Technology. Water. 2021; 13(18):2503. https://doi.org/10.3390/w13182503
Chicago/Turabian StyleLa Rosa, Giuseppina, David Brandtner, Pamela Mancini, Carolina Veneri, Giusy Bonanno Ferraro, Lucia Bonadonna, Luca Lucentini, and Elisabetta Suffredini. 2021. "Key SARS-CoV-2 Mutations of Alpha, Gamma, and Eta Variants Detected in Urban Wastewaters in Italy by Long-Read Amplicon Sequencing Based on Nanopore Technology" Water 13, no. 18: 2503. https://doi.org/10.3390/w13182503