
Degradation of Diclofenac in Urine by Electro-Permanganate Process Driven by Microbial Fuel Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Reactor and Operation
2.3. Analytical Methods
2.4. Electrochemical Measurements and Microbial Characterization
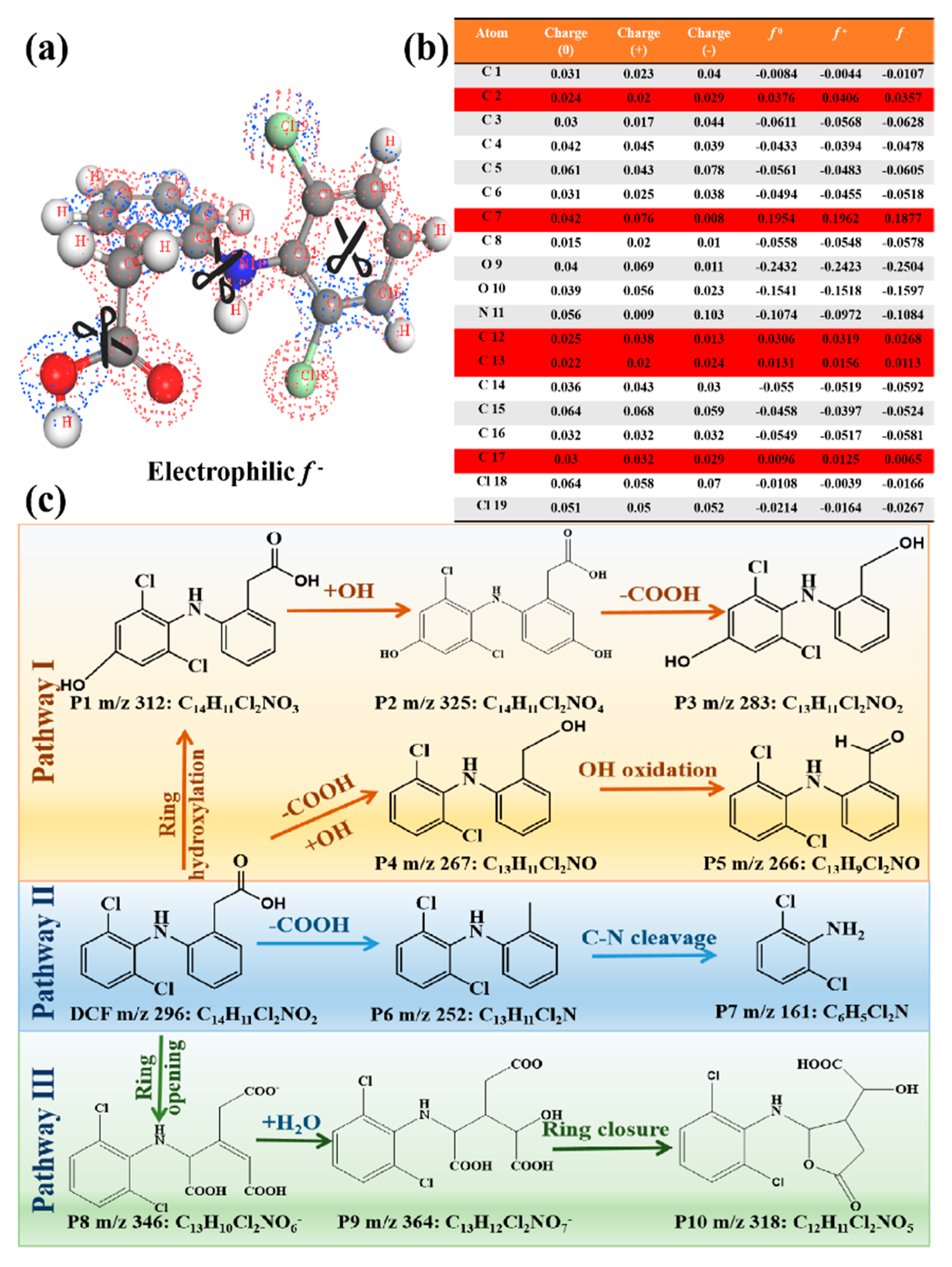
2.5. Model Construction and Density Functional Theory Calculations
3. Results
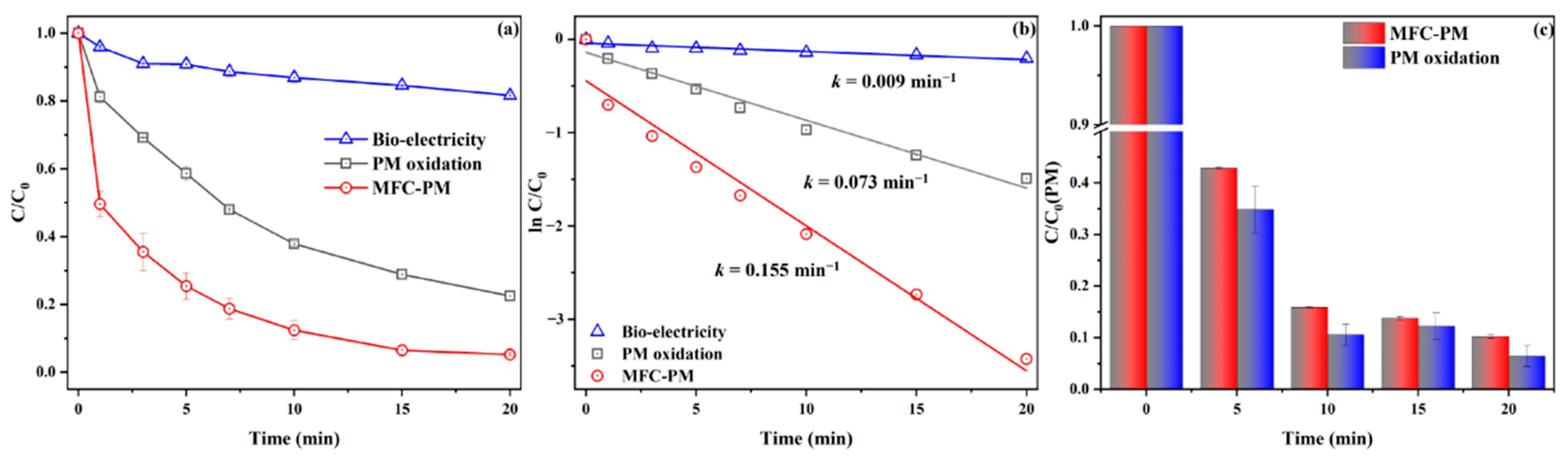
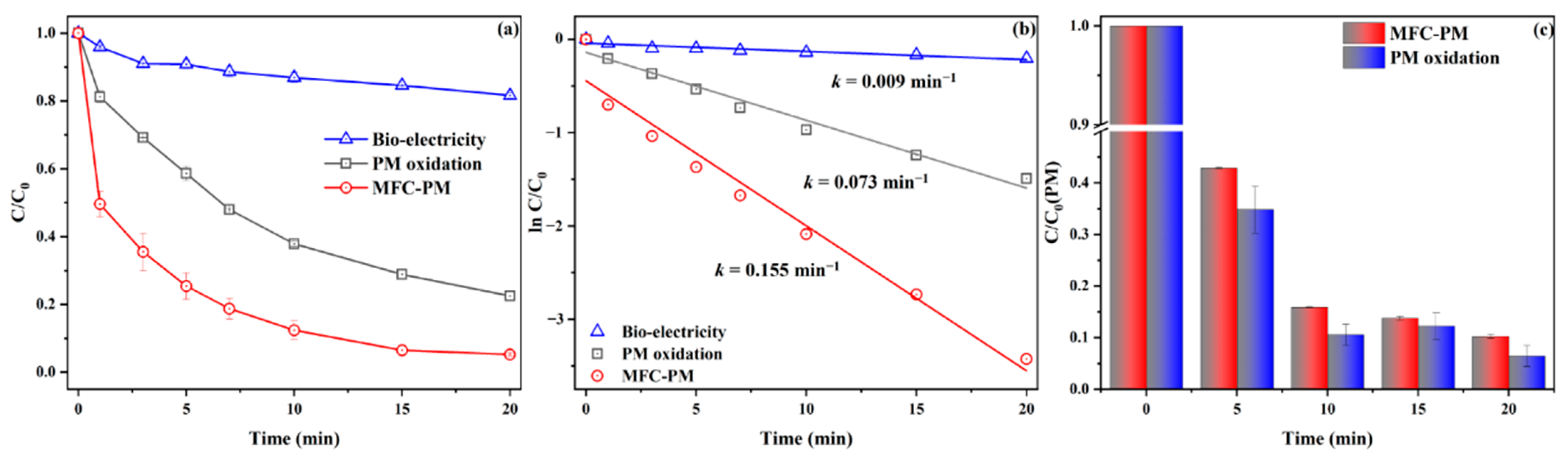
3.1. DCF Removal in the Cathode Chamber
3.2. Process Optimization
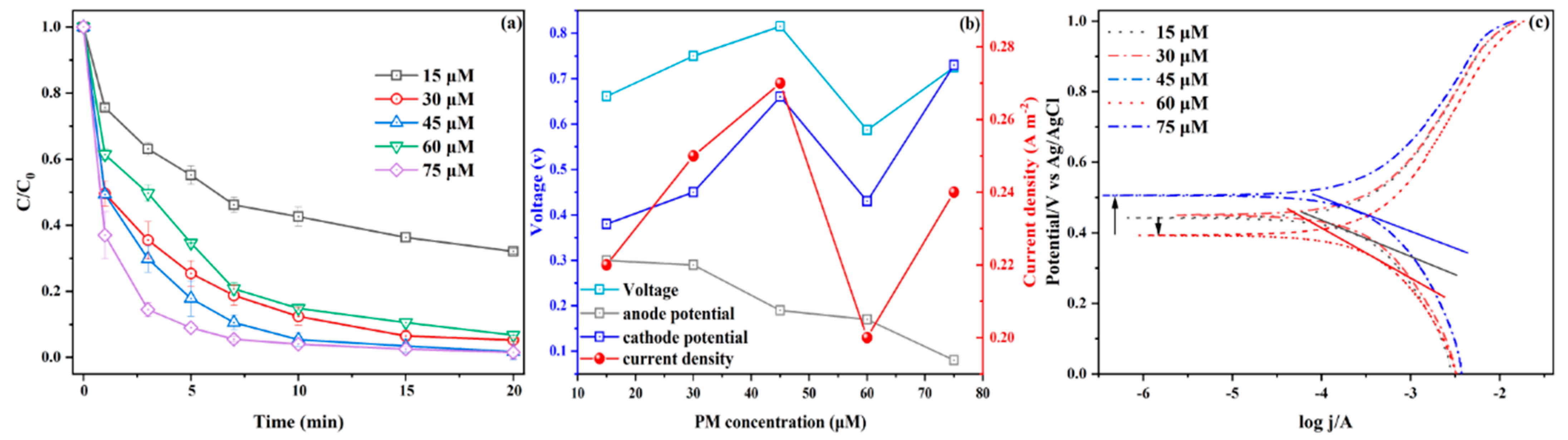
3.2.1. Effect of PM Dosages
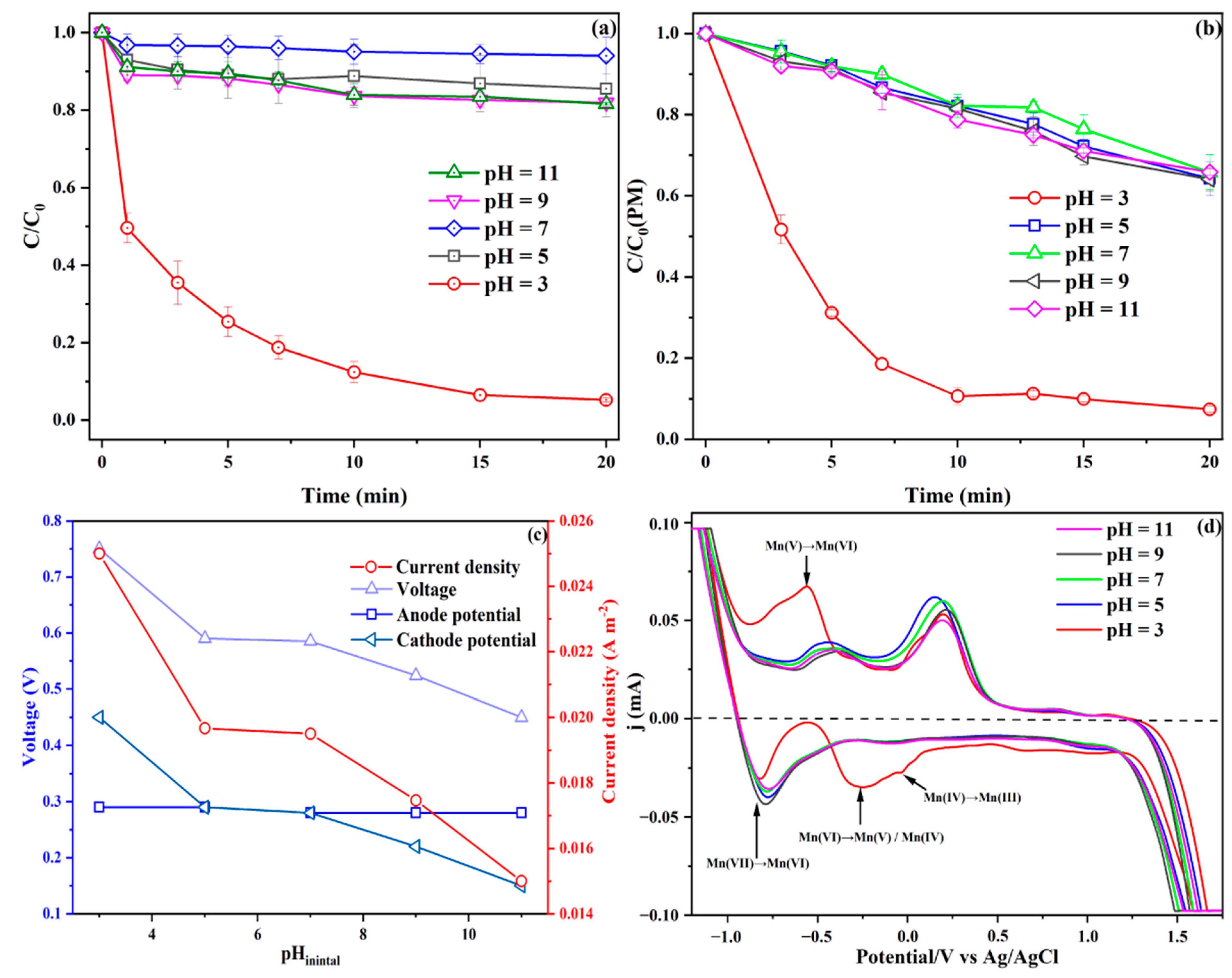
3.2.2. Effect of Catholyte pH
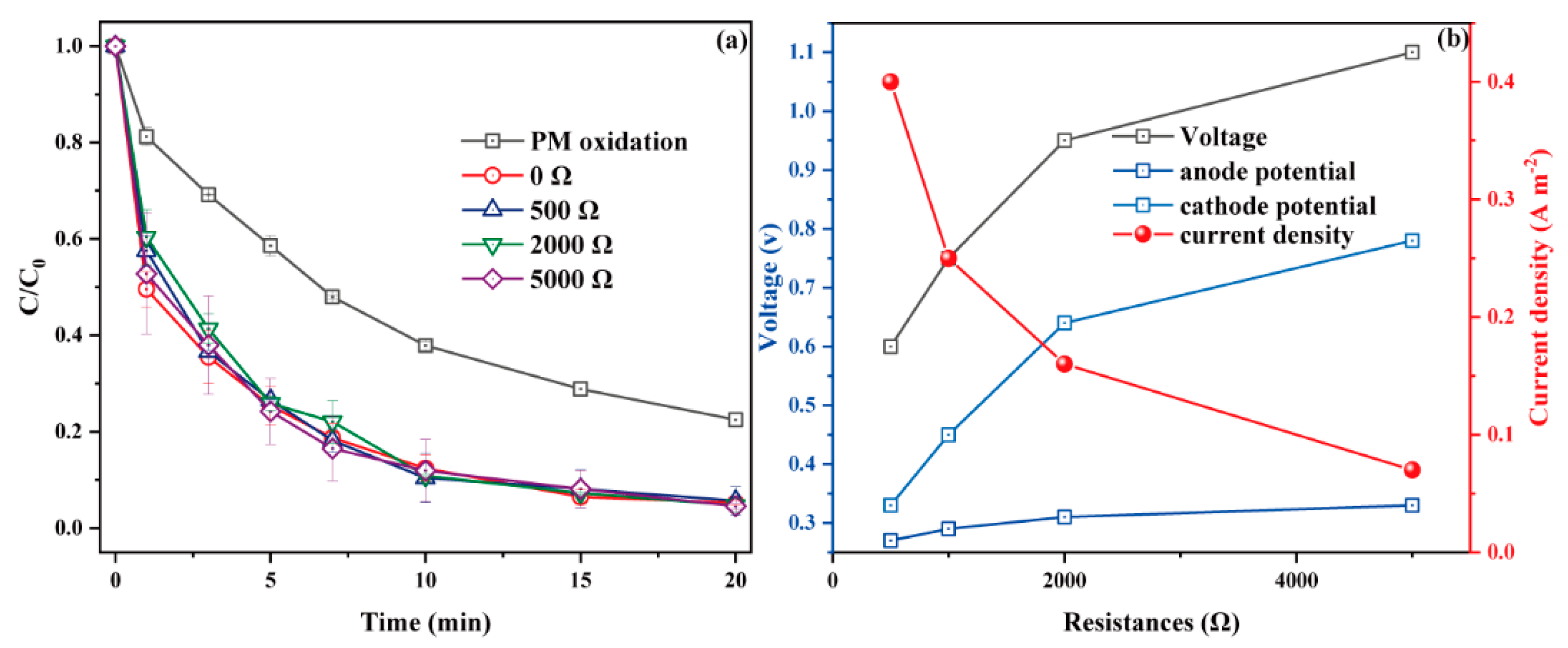
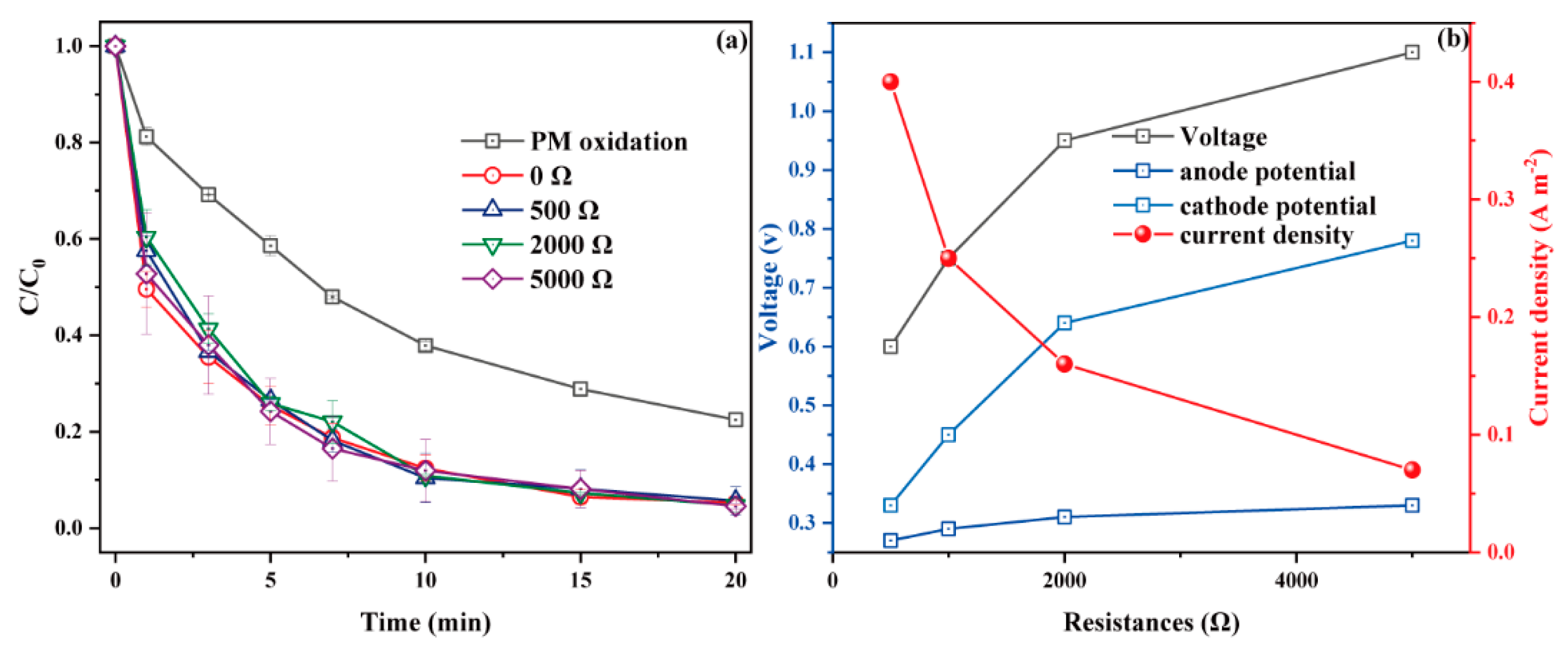
3.2.3. Effect of External Resistance
3.3. Proposed Electron-Transfer and DCF Removal Mechanism
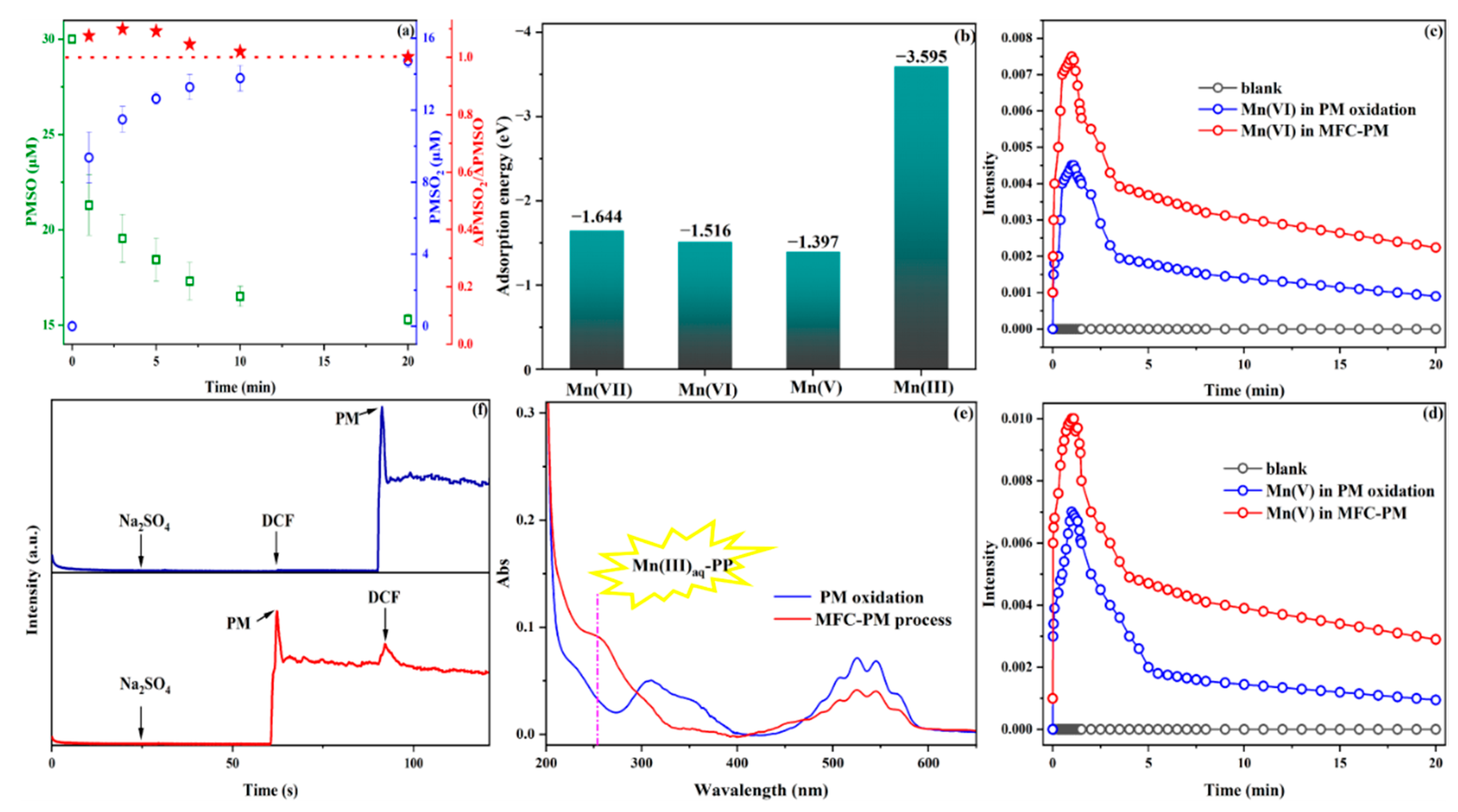
3.3.1. Identification of Reactive Species
3.3.2. Analysis of Electricity-Producing Communities
3.3.3. Reaction Mechanism
3.3.4. Degradation Pathways of DCF
3.4. Application of the MFC-PM Process
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Plakas, K.V.; Mantza, A.; Sklari, S.D.; Zaspalis, V.T.; Karabelas, A.J. Heterogeneous Fenton-like oxidation of pharmaceutical diclofenac by a catalytic iron-oxide ceramic microfiltration membrane. Chem. Eng. J. 2019, 373, 700–708. [Google Scholar] [CrossRef]
- De Oliveira, T.; Guegan, R.; Thiebault, T.; Le Milbeau, C.; Muller, F.; Teixeira, V.; Giovanela, M.; Boussafir, M. Adsorption of diclofenac onto organoclays: Effects of surfactant and environmental (pH and temperature) conditions. J. Hazard. Mater. 2017, 323, 558–566. [Google Scholar] [CrossRef]
- Vieno, N.; Sillanpaa, M. Fate of diclofenac in municipal wastewater treatment plant—A review. Environ. Int. 2014, 69, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.J.; Ongerth, J.E. Modelling of pharmaceutical residues in Australian sewage by quantities of use and fugacity calculations. Chemosphere 2004, 54, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Clara, M.; Strenn, B.; Gans, O.; Martinez, E.; Kreuzinger, N.; Kroiss, H. Removal of selected pharmaceuticals, fragrances and endocrine disrupting compounds in a membrane bioreactor and conventional wastewater treatment plants. Water Res. 2005, 39, 4797–4807. [Google Scholar] [CrossRef]
- Davies, N.M.; Anderson, K.E. Clinical pharmacokinetics of diclofenac—Therapeutic insights and pitfalls. Clin. Pharmacokinet. 1997, 33, 184–213. [Google Scholar] [CrossRef] [PubMed]
- Stierlin, H.; Faigle, J.W.; Sallmann, A.; Kung, W.; Richter, W.J.; Kriemler, H.P.; Alt, K.O.; Winkler, T. Biotransformation of Diclofenac Sodium (Voltaren) in Animals and in Man: 1. Isolation and Identification of Principal Metabolites. Xenobiotica 1979, 9, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhang, W.; Liu, W.; Li, Y.; Ye, J.; Liang, J.; Tong, M. Activation of sulfite by single-atom Fe deposited graphitic carbon nitride for diclofenac removal: The synergetic effect of transition metal and photocatalysis. Chem. Eng. J. 2021, 407, 127167. [Google Scholar] [CrossRef]
- Bae, S.; Kim, D.; Lee, W. Degradation of diclofenac by pyrite catalyzed Fenton oxidation. Appl. Catal. B Environ. 2013, 134, 93–102. [Google Scholar] [CrossRef]
- Dobrin, D.; Bradu, C.; Magureanu, M.; Mandache, N.B.; Parvulescu, V.I. Degradation of diclofenac in water using a pulsed corona discharge. Chem. Eng. J. 2013, 234, 389–396. [Google Scholar] [CrossRef]
- Perisic, D.J.; Gilja, V.; Stankov, M.N.; Katancic, Z.; Kusic, H.; Stangar, U.L.; Dionysiou, D.D.; Bozic, A.L. Removal of diclofenac from water by zeolite-assisted advanced oxidation processes. J. Photochem. Photobiol. A Chem. 2016, 321, 238–247. [Google Scholar] [CrossRef]
- Plakas, K.V.; Sklari, S.D.; Yiankakis, D.A.; Sideropoulos, G.T.; Zaspalis, V.T.; Karabelas, A.J. Removal of organic micropollutants from drinking water by a novel electro-Fenton filter: Pilot-scale studies. Water Res. 2016, 91, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Naddeo, V.; Belgiorno, V.; Ricco, D.; Kassinos, D. Degradation of diclofenac during sonolysis, ozonation and their simultaneous application. Ultrason. Sonochem. 2009, 16, 790–794. [Google Scholar] [CrossRef]
- Cheng, H.; Song, D.; Liu, H.; Qu, J. Permanganate oxidation of diclofenac: The pH-dependent reaction kinetics and a ring-opening mechanism. Chemosphere 2015, 136, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Shao, Y.; Gao, N.; Chen, J.; Zhang, Y.; Xiang, H.; Guo, Y. Degradation of diclofenac by UV-activated persulfate process: Kinetic studies, degradation pathways and toxicity assessments. Ecotoxicol. Environ. Saf. 2017, 141, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhou, P.; Zhou, H.; Liu, W.; Zhang, H.; Zhou, C.; Lai, L.; Ao, Z.; Su, S.; Lai, B. Insights into the Electron-Transfer Mechanism of Permanganate Activation by Graphite for Enhanced Oxidation of Sulfamethoxazole. Environ. Sci. Technol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhao, C.; Wang, T.; Kong, Z.; Zheng, L.; Ding, H.; Liu, Y.; Zheng, H. Simultaneously promoted reactive manganese species and hydroxyl radical generation by electro-permanganate with low additive ozone. Water Res. 2020, 189, 116623. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhou, Y.; Pang, S.; Jiang, J.; Yang, Z.; Shen, Y.; Wang, Z.; Wang, P.; Wang, L. New Insights into the Combination of Permanganate and Bisulfite as a Novel Advanced Oxidation Process: Importance of High Valent Manganese-Oxo Species and Sulfate Radical. Environ. Sci. Technol. 2019, 53, 3689–3696. [Google Scholar] [CrossRef]
- Chen, J.; Rao, D.; Dong, H.; Sun, B.; Shao, B.; Cao, G.; Guan, X. The role of active manganese species and free radicals in permanganate/bisulfite process. J. Hazard. Mater. 2020, 388, 121735. [Google Scholar] [CrossRef]
- Tian, S.; Wang, L.; Liu, Y.; Yang, T.; Huang, Z.; Wang, X.; He, H.; Jiang, J.; Ma, J. Enhanced Permanganate Oxidation of Sulfamethoxazole and Removal of Dissolved Organics with Biochar: Formation of Highly Oxidative Manganese Intermediate Species and in Situ Activation of Biochar. Environ. Sci. Technol. 2019, 53, 5282–5291. [Google Scholar] [CrossRef]
- Shi, Z.; Jin, C.; Zhang, J.; Zhu, L. Insight into mechanism of arsanilic acid degradation in permanganate-sulfite system: Role of reactive species. Chem. Eng. J. 2019, 359, 1463–1471. [Google Scholar] [CrossRef]
- Sun, B.; Li, D.; Linghu, W.; Guan, X. Degradation of ciprofloxacin by manganese(III) intermediate: Insight into the potential application of permanganate/bisulfite process. Chem. Eng. J. 2018, 339, 144–152. [Google Scholar] [CrossRef]
- Guo, K.; Zhang, J.; Li, A.; Xie, R.; Liang, Z.; Wang, A.; Ling, L.; Li, X.; Li, C.; Fang, J. Ultraviolet Irradiation of Permanganate Enhanced the Oxidation of Micropollutants by Producing HO• and Reactive Manganese Species. Environ. Sci. Technol. Lett. 2018, 5, 750–756. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Wang, X.; Zhang, J.; Ding, L.; Li, J.; Zheng, H.; Zhao, C. Generation of Active Mn(III)aq by a Novel Heterogeneous Electro-permanganate Process with Manganese(II) as Promoter and Stabilizer. Environ. Sci. Technol. 2019, 53, 9063–9072. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, C.; Liang, J.; Shang, R.; Zhu, X.; Ding, L.; Deng, H.; Zheng, H.; Strathmann, T.J. Rapid removal of diclofenac in aqueous solution by soluble Mn(III) (aq) generated in a novel Electro-activated carbon fiber-permanganate (E-ACF-PM) process. Water Res. 2019, 165, 114975. [Google Scholar] [CrossRef] [PubMed]
- Gude, V.G. Wastewater treatment in microbial fuel cells–an overview. J. Clean. Prod. 2016, 122, 287–307. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Z.; Chu, L.; Chen, R.; Zhang, C.; Toan, S.; Bagley, D.M.; Sun, J.; Dong, S.; Fan, M. Unified photoelectrocatalytic microbial fuel cell harnessing 3D binder-free photocathode for simultaneous power generation and dual pollutant removal. J. Power Sources 2021, 481, 229133. [Google Scholar] [CrossRef]
- Ahmadpour, T.; Aber, S.; Hosseini, M.G. Improved dye degradation and simultaneous electricity generation in a photoelectrocatalytic microbial fuel cell equipped with AgBr/CuO hybrid photocathode. J. Power Sources 2020, 474, 228589. [Google Scholar] [CrossRef]
- Xu, P.; Zheng, D.; He, Q.; Yu, J. The feasibility of ofloxacin degradation and electricity generation in photo-assisted microbial fuel cells with LiNbO3/CF photocatalytic cathode. Sep. Purif. Technol. 2020, 250, 117106. [Google Scholar] [CrossRef]
- Yu, X.; Fu, W.; Jiang, M.; Liu, G.; Zou, Y.; Chen, S. Automatic microbial electro-Fenton system driven by transpiration for degradation of acid orange 7. Sci. Total Environ. 2020, 725, 138508. [Google Scholar] [CrossRef]
- Zou, R.; Angelidaki, I.; Yang, X.; Tang, K.; Andersen, H.R.; Zhang, Y. Degradation of pharmaceuticals from wastewater in a 20-L continuous flow bio-electro-Fenton (BEF) system. Sci. Total Environ. 2020, 727, 138684. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, Q. Performance of electro-Fenton process coupling with microbial fuel cell for simultaneous removal of herbicide mesotrione. Bioresour. Technol. 2021, 319, 124244. [Google Scholar] [CrossRef]
- Yan, S.; Zhang, X.; Shi, Y.; Zhang, H. Natural Fe-bearing manganese ore facilitating bioelectro-activation of peroxymonosulfate for bisphenol A oxidation. Chem. Eng. J. 2018, 354, 1120–1131. [Google Scholar] [CrossRef]
- Long, X.; Wang, H.; Wang, C.; Cao, X.; Li, X. Enhancement of azo dye degradation and power generation in a photoelectrocatalytic microbial fuel cell by simple cathodic reduction on titania nanotube arrays electrode. J. Power Sources 2019, 415, 145–153. [Google Scholar] [CrossRef]
- Kageshima, Y.; Yoshimura, T.; Koh, S.; Mizuno, M.; Teshima, K.; Nishikiori, H. Photoelectrochemical Complete Decomposition of Cellulose for Electric Power Generation. Chemcatchem 2021, 13, 1530–1537. [Google Scholar] [CrossRef]
- Theerthagiri, J.; Lee, S.J.; Murthy, A.P.; Madhavan, J.; Choi, M.Y. Fundamental aspects and recent advances in transition metal nitrides as electrocatalysts for hydrogen evolution reaction: A review. Curr. Opin. Solid State Mater. Sci. 2020, 24, 100805. [Google Scholar] [CrossRef]
- Lee, S.J.; Theerthagiri, J.; Choi, M.Y. Time-resolved dynamics of laser-induced cavitation bubbles during production of Ni nanoparticles via pulsed laser ablation in different solvents and their electrocatalytic activity for determination of toxic nitroaromatics. Chem. Eng. J. 2021, 427, 130970. [Google Scholar] [CrossRef]
- Wen, Q.; Yang, T.; Wang, S.; Chen, Y.; Cong, L.; Qu, Y. Dechlorination of 4-chlorophenol to phenol in bioelectrochemical systems. J. Hazard. Mater. 2013, 244, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Chandraserkharan Meenu, P.; Sreelekshmy, B.R.; Basheer, R.; Sadasivan, S.M.; Vijayakumari Ramakrishnan, R.M.; Shibli, S.M.A. Development of a High-Performance Mediatorless Microbial Fuel Cell Comprising a Catalytic Steel Anode. ACS Appl. Bio Mater. 2018, 1, 1124–1133. [Google Scholar] [CrossRef]
- Kusmierek, E. Semiconductor Electrode Materials Applied in Photoelectrocatalytic Wastewater Treatment-an Overview. Catalysts 2020, 10, 439. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhou, S. Efficacy of Cu(II) as an electron-shuttle mediator for improved bioelectricity generation and Cr(VI) reduction in microbial fuel cells. Bioresour. Technol. 2019, 273, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, X.; Zhao, C.; Sun, Z.; Zheng, H.; Li, J.; Wang, Z. Optimization and mechanism of Acid Orange 7 removal by powdered activated carbon coupled with persulfate by response surface method. Water Sci. Technol. 2019, 79, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wu, X.; Luo, H.; Li, X.; Chen, W.; Chen, J.; Mo, Y.; Wang, W. CH4 control and associated microbial process from constructed wetland (CW) by microbial fuel cells (MFC). J. Environ. Manag. 2020, 260, 110071. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Kong, C. Enhanced removal of p-nitrophenol in a microbial fuel cell after long-term operation and the catabolic versatility of its microbial community. Chem. Eng. J. 2018, 339, 424–431. [Google Scholar] [CrossRef]
- Kim, Y.; Cha, C.; Engesser, K.; Kim, S. Degradation of various alkyl ethers by alkyl ether-degrading Actinobacteria isolated from activated sludge of a mixed wastewater treatment. Chemosphere 2008, 73, 1442–1447. [Google Scholar] [CrossRef]
- Liu, W.; Li, Y.; Liu, F.; Jiang, W.; Zhang, D.; Liang, J. Visible-light-driven photocatalytic degradation of diclofenac by carbon quantum dots modified porous g-C3N4: Mechanisms, degradation pathway and DFT calculation. Water Res. 2019, 151, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Tran, H.; Kim, D.; Oh, S.; Park, D.; Zhang, R.; Ahn, D. Simultaneous organics removal and bio-electrochemical denitrification in microbial fuel cells. Bioprocess Biosyst. Eng. 2008, 31, 315–321. [Google Scholar] [CrossRef]
- Xu, K.; Ben, W.; Ling, W.; Zhang, Y.; Qu, J.; Qiang, Z. Impact of humic acid on the degradation of levofloxacin by aqueous permanganate: Kinetics and mechanism. Water Res. 2017, 123, 67–74. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Wang, Y.; Zhang, J.; Duanmu, P.; Zheng, L.; Hasson, S.U.; Baldwin, A.; Wong, I.; Zhao, C. Degradation of Diclofenac in Urine by Electro-Permanganate Process Driven by Microbial Fuel Cells. Water 2021, 13, 2047. https://doi.org/10.3390/w13152047
Wang X, Wang Y, Zhang J, Duanmu P, Zheng L, Hasson SU, Baldwin A, Wong I, Zhao C. Degradation of Diclofenac in Urine by Electro-Permanganate Process Driven by Microbial Fuel Cells. Water. 2021; 13(15):2047. https://doi.org/10.3390/w13152047
Chicago/Turabian StyleWang, Xuxu, Ying Wang, Jian Zhang, Pengbo Duanmu, Liushi Zheng, Shabi UI Hasson, Andrew Baldwin, Irene Wong, and Chun Zhao. 2021. "Degradation of Diclofenac in Urine by Electro-Permanganate Process Driven by Microbial Fuel Cells" Water 13, no. 15: 2047. https://doi.org/10.3390/w13152047
APA StyleWang, X., Wang, Y., Zhang, J., Duanmu, P., Zheng, L., Hasson, S. U., Baldwin, A., Wong, I., & Zhao, C. (2021). Degradation of Diclofenac in Urine by Electro-Permanganate Process Driven by Microbial Fuel Cells. Water, 13(15), 2047. https://doi.org/10.3390/w13152047