
Methylobacterium oryzae Influences Isoepoxydon Dehydrogenase Gene Expression and Patulin Production by Penicillium expansum




Abstract
1. Introduction
2. Materials and Methods
2.1. Microorganisms and Culture Conditions
2.2. Stock Solution of Fungal Spores
2.3. Apple Puree Agar Medium (APAM) and Apple Juice (APJ) Preparation
2.4. Biofilm Formation
2.5. Biofilm Mass Quantification by Crystal Violet
2.6. Number of Bacteria in Single and Inter-Kingdom Biofilms
2.7. RT-qPCR Method to Measure Idh Gene Expression of P. expansum
2.7.1. RNA Extraction and DNase Treatment
2.7.2. The cDNA Synthesis
2.7.3. Quantitative Real-Time PCR (qPCR) Assay
2.8. Patulin Extraction and Quantification by UHPLC
2.8.1. Reagents and Chemicals
2.8.2. Patulin Extraction
2.8.3. UHPLC Apparatus and Conditions
2.9. Statistical Analysis
3. Results
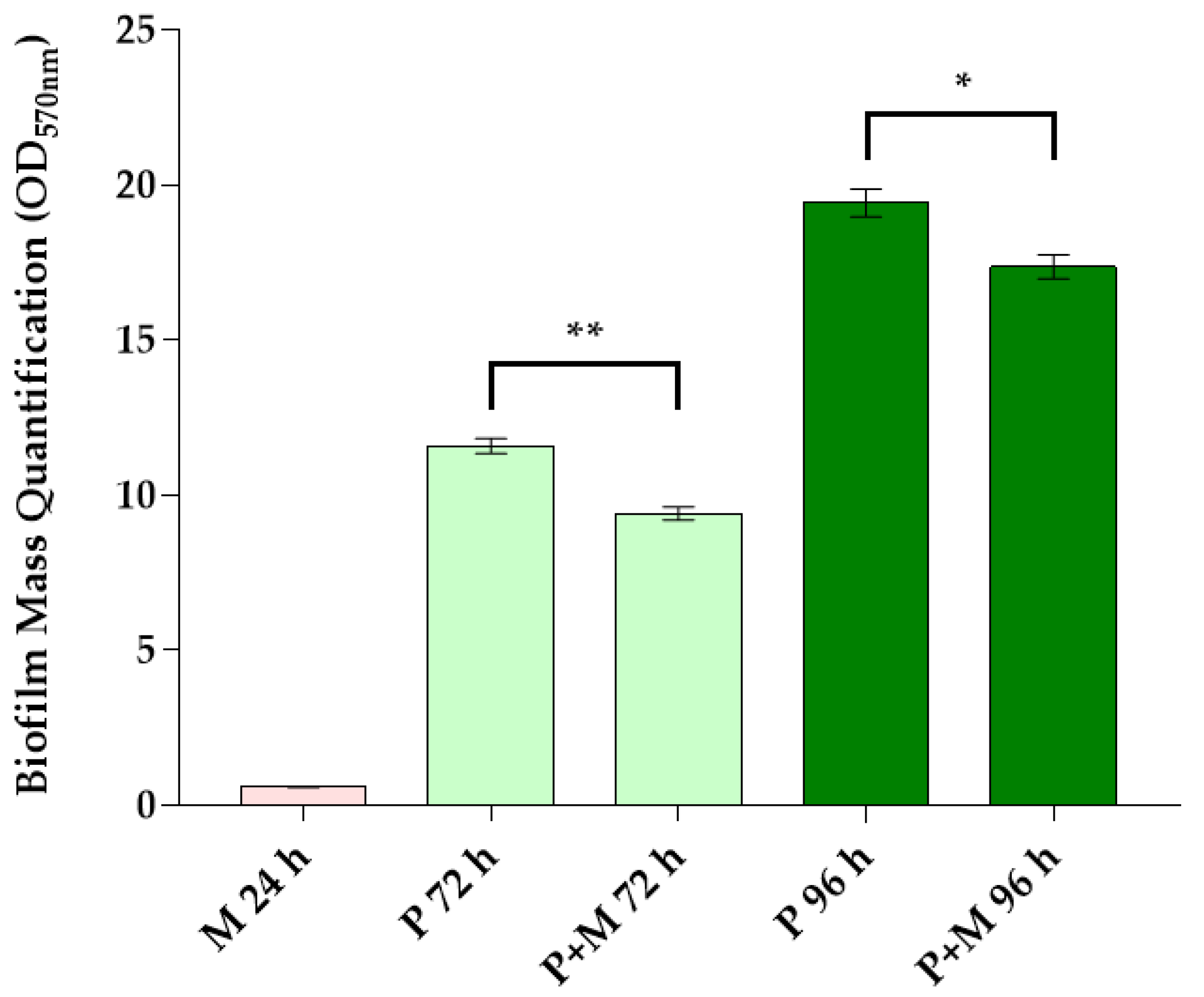
3.1. Biofilm Mass Quantification
3.2. Bacterial Cell Density in Single and Inter-Kingdom Biofilms
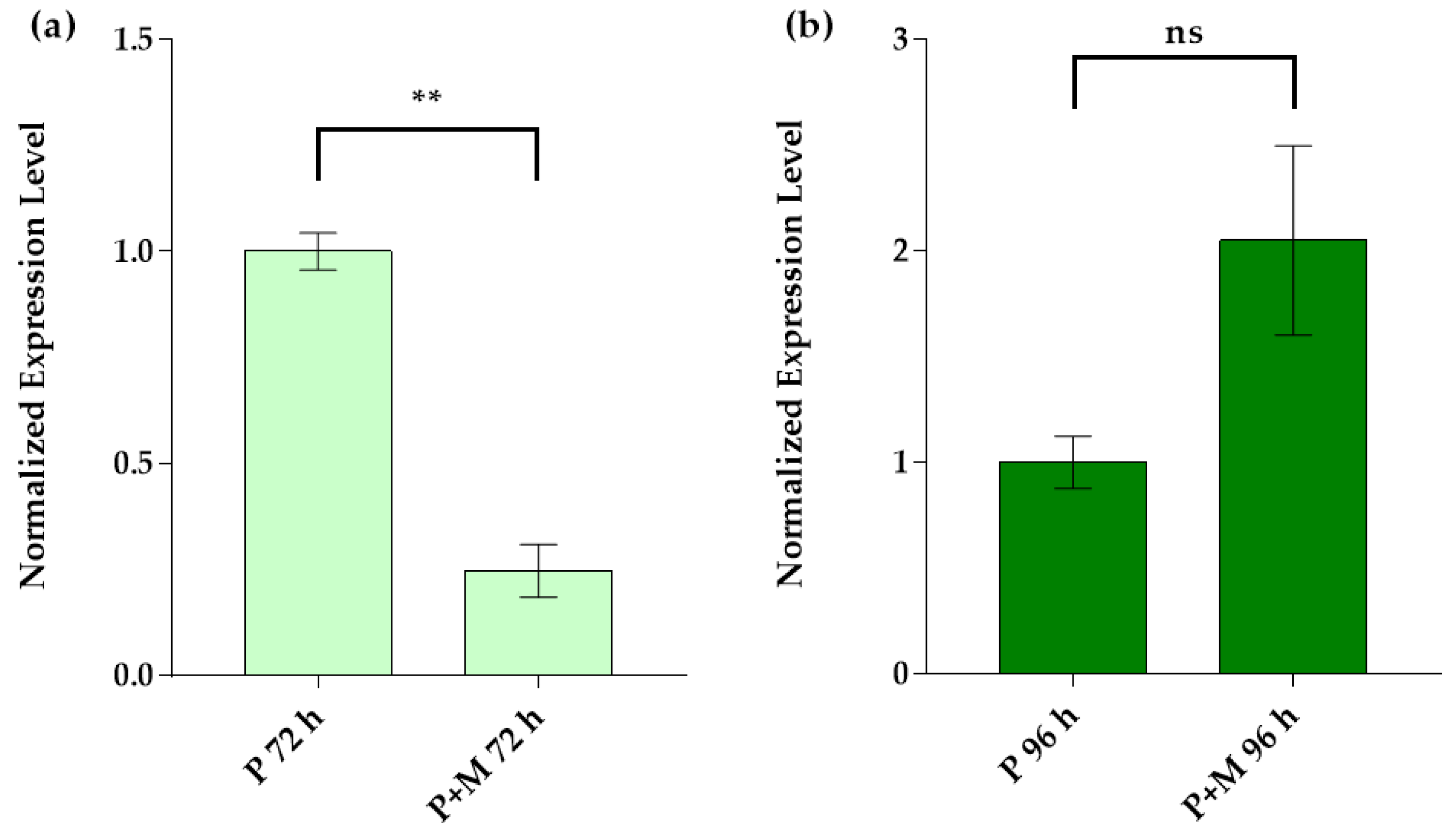
3.3. Idh Gene Expression in Single and Inter-Kingdom Biofilms
3.4. Patulin Production in Single and Inter-Kingdom Biofilms
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Flemming, H.C.; Percival, S.L.; Walker, J.T. Contamination potential of biofilms in water distribution systems. Water Sci. Technol. Water Supply 2002, 2, 271–280. [Google Scholar] [CrossRef]
- Siqueira, V.M.; Oliveira, H.M.B.; Santos, C.; Paterson, R.R.M.; Gusmão, N.B.; Lima, N. Biofilms from a Brazilian water distribution system include filamentous fungi. Can. J. Microbiol. 2013, 59, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Paterson, R.R.; Lima, N. Fungal contamination of drinking water III. In Water Encyclopedia; Lehr, J., Keeley, J., Lehr, J., Kingery, T.B., Eds.; John Wiley & Sons: New York, NY, USA, 2005. [Google Scholar]
- Feazel, L.M.; Baumgartner, L.K.; Peterson, K.L.; Frank, D.N.; Harris, J.K.; Pace, N.R. Opportunistic pathogens enriched in showerhead biofilms. Proc. Natl. Acad. Sci. USA 2009, 106, 16393–16398. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H.; Hu, C.; Yang, M.; Hu, H.; Niu, J. Characteristics of biofilms and iron corrosion scales with ground and surface waters in drinking water distribution systems. Corros. Sci. 2015, 90, 331–339. [Google Scholar] [CrossRef]
- Wang, H.; Zhu, Y.; Hu, C. Impacts of bacteria and corrosion on removal of natural organic matter and disinfection byproducts in different drinking water distribution systems. Int. Biodeterior. Biodegrad. 2017, 117, 52–59. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, K.; Zhang, T.; Li, C.; Mao, X. An ignored and potential source of taste and odor (T&O) issues—biofilms in drinking water distribution system (DWDS). Appl. Microbiol. Biotechnol. 2017, 101, 3537–3550. [Google Scholar] [CrossRef]
- Douterelo, I.; Jackson, M.; Solomon, C.; Boxall, J. Microbial analysis of in situ biofilm formation in drinking water distribution systems: Implications for monitoring and control of drinking water quality. Appl. Microbiol. Biotechnol. 2016, 100, 3301–3311. [Google Scholar] [CrossRef]
- Douterelo, I.; Calero-Preciado, C.; Soria-Carrasco, V.; Boxall, J.B. Whole metagenome sequencing of chlorinated drinking water distribution systems. Environ. Sci. Water Res. Technol. 2018, 4, 2080–2091. [Google Scholar] [CrossRef]
- Douterelo, I.; Fish, K.E.; Boxall, J.B. Succession of bacterial and fungal communities within biofilms of a chlorinated drinking water distribution system. Water Res. 2018, 141, 74–85. [Google Scholar] [CrossRef]
- Bryers, J.D.; Ratner, B.D. Bioinspired implant materials befuddle bacteria. ASM News 2004, 70, 232–237. [Google Scholar]
- Afonso, T.B.; Simões, L.C.; Lima, N. In vitro assessment of inter-kingdom biofilm formation by bacteria and filamentous fungi isolated from a drinking water distribution system. Biofouling 2019, 35, 1041–1054. [Google Scholar] [CrossRef] [PubMed]
- Afonso, T.B.; Simões, L.C.; Lima, N. Effect of quorum sensing and quenching molecules on inter-kingdom biofilm formation by Penicillium expansum and bacteria. Biofouling 2020, 36, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.B.; Givskov, M. Quorum-sensing inhibitors as anti-pathogenic drugs. Int. J. Med. Microbiol. 2006, 296, 149–161. [Google Scholar] [CrossRef]
- Antunes, L.C.M.; Ferreira, R.B.R.; Buckner, M.M.C.; Finlay, B.B. Quorum sensing in bacterial virulence. Microbiology 2010, 156, 2271–2282. [Google Scholar] [CrossRef] [PubMed]
- Shrout, J.D.; Tolker-Nielsen, T.; Givskov, M.; Parsek, M.R. The contribution of cell-cell signaling and motility to bacterial biofilm formation. MRS Bull. 2011, 36, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, N.; Keller, N.P. Mycotoxins in conversation with bacteria and fungi. Front. Microbiol. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- González, J.E.; Keshavan, N.D. Messing with Bacterial Quorum Sensing. Microbiol. Mol. Biol. Rev. 2006, 70, 859–875. [Google Scholar] [CrossRef]
- Paterson, R.R.M.; Kelley, J.; Gallagher, M. Natural occurrence of aflatoxins and Aspergillus flavus (Link) in water. Lett. Appl. Microbiol. 1997, 25, 435–436. [Google Scholar] [CrossRef]
- Russell, R.; Paterson, M. Zearalenone production and growth in drinking water inoculated with Fusarium graminearum. Mycol. Prog. 2007, 6, 109–113. [Google Scholar] [CrossRef]
- Martín-Rodríguez, A.J.; Reyes, F.; Martín, J.; Pérez-Yépez, J.; León-Barrios, M.; Couttolenc, A.; Espinoza, C.; Trigos, Á.; Martín, V.S.; Norte, M.; et al. Inhibition of bacterial quorum sensing by extracts from aquatic fungi: First report from marine endophytes. Mar. Drugs 2014, 12, 5503–5526. [Google Scholar] [CrossRef]
- van Rij, E.T.; Girard, G.; Lugtenberg, B.J.J.; Bloemberg, G.V. Influence of fusaric acid on phenazine-1-carboxamide synthesis and gene expression of Pseudomonas chlororaphis strain PCL1391. Microbiology 2005, 151, 2805–2814. [Google Scholar] [CrossRef] [PubMed]
- Bacon, C.W.; Hinton, D.M.; Mitchell, T.R. Is Quorum Signaling by Mycotoxins a New Risk-Mitigating Strategy for Bacterial Biocontrol of Fusarium verticillioides and Other Endophytic Fungal Species? J. Agric. Food Chem. 2017, 65, 7071–7080. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.B.; Skindersoe, M.E.; Bjarnsholt, T.; Phipps, R.K.; Christensen, K.B.; Jensen, P.O.; Andersen, J.B.; Koch, B.; Larsen, T.O.; Hentzer, M.; et al. Identity and effects of quorum-sensing inhibitors produced by Penicillium species. Microbiology 2005, 151, 1325–1340. [Google Scholar] [CrossRef] [PubMed]
- Liaqat, I.; Bachmann, R.T.; Sabri, A.N.; Edyvean, R.G.J. Isolate-specific effects of patulin, penicillic acid and EDTA on biofilm formation and growth of dental unit water line biofilm isolates. Curr. Microbiol. 2010, 61, 148–156. [Google Scholar] [CrossRef]
- Liaqat, I.; Bachmann, R.T.; Edyvean, R.G.J. Type 2 quorum sensing monitoring, inhibition and biofilm formation in marine microrganisms. Curr. Microbiol. 2014, 68, 342–351. [Google Scholar] [CrossRef]
- Puel, O.; Galtier, P.; Oswald, I.P. Biosynthesis and toxicological effects of patulin. Toxins 2010, 2, 613–631. [Google Scholar] [CrossRef]
- Li, B.; Chen, Y.; Zong, Y.; Shang, Y.; Zhang, Z.; Xu, X.; Wang, X.; Long, M.; Tian, S. Dissection of patulin biosynthesis, spatial control and regulation mechanism in Penicillium expansum. Environ. Microbiol. 2019, 21, 1124–1139. [Google Scholar] [CrossRef]
- White, S.; O’Callaghan, J.; Dobson, A.D.W. Cloning and molecular characterization of Penicillium expansum genes upregulated under conditions permissive for patulin biosynthesis. FEMS Microbiol. Lett. 2006, 255, 17–26. [Google Scholar] [CrossRef]
- Bustin, S.A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 2000, 25, 155–166. [Google Scholar] [CrossRef]
- Gonçalves, A.B.; Paterson, R.R.M.; Lima, N. Survey and significance of filamentous fungi from tap water. Int. J. Hyg. Environ. Health 2006, 209, 257–264. [Google Scholar] [CrossRef]
- Simões, L.C.; Simões, M.; Lima, N. Kinetics of biofilm formation by drinking water isolated Penicillium expansum. Biofouling 2015, 31, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Simões, L.C.; Simõoes, M.; Oliveira, R.; Vieira, M.J. Potential of the adhesion of bacteria isolated from drinking water to materials. J. Basic Microbiol. 2007, 47, 174–183. [Google Scholar] [CrossRef]
- Simões, L.C.; Simões, M.; Vieira, M.J. Influence of the diversity of bacterial isolates from drinking water on resistance of biofilms to disinfection. Appl. Environ. Microbiol. 2010, 76, 6673–6679. [Google Scholar] [CrossRef] [PubMed]
- Baert, K.; Devlieghere, F.; Flyps, H.; Oosterlinck, M.; Ahmed, M.M.; Rajković, A.; Verlinden, B.; Nicolaï, B.; Debevere, J.; De Meulenaer, B. Influence of storage conditions of apples on growth and patulin production by Penicillium expansum. Int. J. Food Microbiol. 2007, 119, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Stepanović, S.; Vuković, D.; Dakić, I.; Savić, B.; Švabić-Vlahović, M. A modified microtiter-plate test for quantification of staphylococcal biofilm formation. J. Microbiol. Methods 2000, 40, 175–179. [Google Scholar] [CrossRef]
- De Clercq, N.; Vlaemynck, G.; Van Pamel, E.; Van Weyenberg, S.; Herman, L.; Devlieghere, F.; De Meulenaer, B.; Van Coillie, E. Isoepoxydon dehydrogenase (idh) gene expression in relation to patulin production by Penicillium expansum under different temperature and atmosphere. Int. J. Food Microbiol. 2016, 220, 50–57. [Google Scholar] [CrossRef]
- Werbrouck, H.; Botteldoorn, N.; Uyttendaele, M.; Herman, L.; Van Coillie, E. Quantification of gene expression of Listeria monocytogenes by real-time reverse transcription PCR: Optimization, evaluation and pitfalls. J. Microbiol. Methods 2007, 69, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, 2002–2007. [Google Scholar] [CrossRef]
- Sargenti, S.R.; Almeida, C.A.A. Determination of patulin in apple juice by hplc using a simple and fast sample preparation method. Eclet. Quim. 2010, 35, 15–21. [Google Scholar] [CrossRef]
- Santos, I.M.; Abrunhosa, L.; Venâncio, A.; Lima, N. The effect of culture preservation techniques on patulin and citrinin production by Penicillium expansum Link. Lett. Appl. Microbiol. 2002, 35, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.R.N.; Spadaro, D.; Lore, A.; Gullino, M.L.; Garibaldi, A. Potential of patulin production by Penicillium expansum strains on various fruits. Mycotoxin Res. 2010, 26, 257–265. [Google Scholar] [CrossRef]
- Mowat, E.; Rajendran, R.; Williams, C.; McCulloch, E.; Jones, B.; Lang, S.; Ramage, G. Pseudomonas aeruginosa and their small diffusible extracellular molecules inhibit Aspergillus fumigatus biofilm formation. FEMS Microbiol. Lett. 2010, 313, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Kousser, C.; Clark, C.; Sherrington, S.; Voelz, K.; Hall, R.A. Pseudomonas aeruginosa inhibits Rhizopus microsporus germination through sequestration of free environmental iron. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Schena, L.; Nigro, F.; Ippolito, A.; Gallitelli, D. Real-time quantitative PCR: A new technology to detect and study phytopathogenic and antagonistic fungi. Eur. J. Plant Pathol. 2004, 110, 893–908. [Google Scholar] [CrossRef]
- Paterson, R.R.M. Internal amplification controls have not been employed in fungal PCR hence potential false negative results. J. Appl. Microbiol. 2007, 102, 1–10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schrader, C.; Schielke, A.; Ellerbroek, L.; Johne, R. PCR inhibitors—Occurrence, properties and removal. J. Appl. Microbiol. 2012, 113, 1014–1026. [Google Scholar] [CrossRef]
- Sweeney, M.J.; Pàmies, P.; Dobson, A.D.W. The use of reverse transcription-polymerase chain reaction (RT-PCR) for monitoring aflatoxin production in Aspergillus parasiticus 439. Int. J. Food Microbiol. 2000, 56, 97–103. [Google Scholar] [CrossRef]
- Mayer, Z.; Färber, P.; Geisen, R. Monitoring the production of aflatoxin B1 in wheat by measuring the concentration of nor-1 mRNA. Appl. Environ. Microbiol. 2003, 69, 1154–1158. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jiao, F.; Kawakami, A.; Nakajima, T. Effects of different carbon sources on trichothecene production and Tri gene expression by Fusarium graminearum in liquid culture. FEMS Microbiol. Lett. 2008, 285, 212–219. [Google Scholar] [CrossRef]
- Sanzani, S.M.; Schena, L.; Nigro, F.; de Girolamo, A.; Ippolito, A. Effect of quercetin and umbelliferone on the transcript level of Penicillium expansum genes involved in patulin biosynthesis. Eur. J. Plant Pathol. 2009, 125, 223–233. [Google Scholar] [CrossRef]
- Ben Taheur, F.; Mansour, C.; Kouidhi, B.; Chaieb, K. Use of lactic acid bacteria for the inhibition of Aspergillus flavus and Aspergillus carbonarius growth and mycotoxin production. Toxicon 2019, 166, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Dogi, C.A.; Fochesato, A.; Armando, R.; Pribull, B.; de Souza, M.M.S.; da Silva Coelho, I.; Araújo de Melo, D.; Dalcero, A.; Cavaglieri, L. Selection of lactic acid bacteria to promote an efficient silage fermentation capable of inhibiting the activity of Aspergillus parasiticus and Fusarium gramineraum and mycotoxin production. J. Appl. Microbiol. 2013, 114, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Hatab, S.; Yue, T.; Mohamad, O. Reduction of Patulin in Aqueous Solution by Lactic Acid Bacteria. J. Food Sci. 2012, 77, M238–M241. [Google Scholar] [CrossRef]
- Hatab, S.; Yue, T.; Mohamad, O. Removal of patulin from apple juice using inactivated lactic acid bacteria. J. Appl. Microbiol. 2012, 112, 892–899. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, X.; Hatab, S.; Wang, Z.; Wang, Y.; Luo, Y.; Yue, T. Patulin reduction in apple juice by inactivated Alicyclobacillus spp. Lett. Appl. Microbiol. 2014, 59, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Roze, L.V.; Chanda, A.; Linz, J.E. Compartmentalization and molecular traffic in secondary metabolism: A new understanding of established cellular processes. Fungal Genet. Biol. 2011, 48, 35–48. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Set | F Primer Sequence (5′ to 3′) | R Primer Sequence (5′ to 3′) | Product Length (bp) | qPCR Efficiency (%) |
|---|---|---|---|---|---|
| idh | EK-RT-2F/EK-RT-2R | GCAGTTTCGCGATCGATGT | GTAGGGAGTAGCCGCCTTGA | 59 | 92.1 |
| β-tubulin | BTub-2F/BTub-2R | GGTCCCTTCGGCAAGCTT | TGTTACCAGCACCGGACTGA | 64 | 93.0 |
| Conditions | Patulin Relative Fold Change to Control |
|---|---|
| P 72 h − P + M 72 h | 0.322 ± 0.048 (p < 0.0001) |
| P 96 h − P + M 96 h | 3.349 ± 1.627 (p = 0.067) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afonso, T.B.; Simões, L.C.; Lima, N. Methylobacterium oryzae Influences Isoepoxydon Dehydrogenase Gene Expression and Patulin Production by Penicillium expansum. Water 2021, 13, 1427. https://doi.org/10.3390/w13101427
Afonso TB, Simões LC, Lima N. Methylobacterium oryzae Influences Isoepoxydon Dehydrogenase Gene Expression and Patulin Production by Penicillium expansum. Water. 2021; 13(10):1427. https://doi.org/10.3390/w13101427
Chicago/Turabian StyleAfonso, Tiago Barros, Lúcia Chaves Simões, and Nelson Lima. 2021. "Methylobacterium oryzae Influences Isoepoxydon Dehydrogenase Gene Expression and Patulin Production by Penicillium expansum" Water 13, no. 10: 1427. https://doi.org/10.3390/w13101427
APA StyleAfonso, T. B., Simões, L. C., & Lima, N. (2021). Methylobacterium oryzae Influences Isoepoxydon Dehydrogenase Gene Expression and Patulin Production by Penicillium expansum. Water, 13(10), 1427. https://doi.org/10.3390/w13101427