The Study of Hydrogeochemical Environments and Microbial Communities along a Groundwater Salinity Gradient in the Pearl River Delta, China



Abstract
1. Introduction
2. Materials and Methods
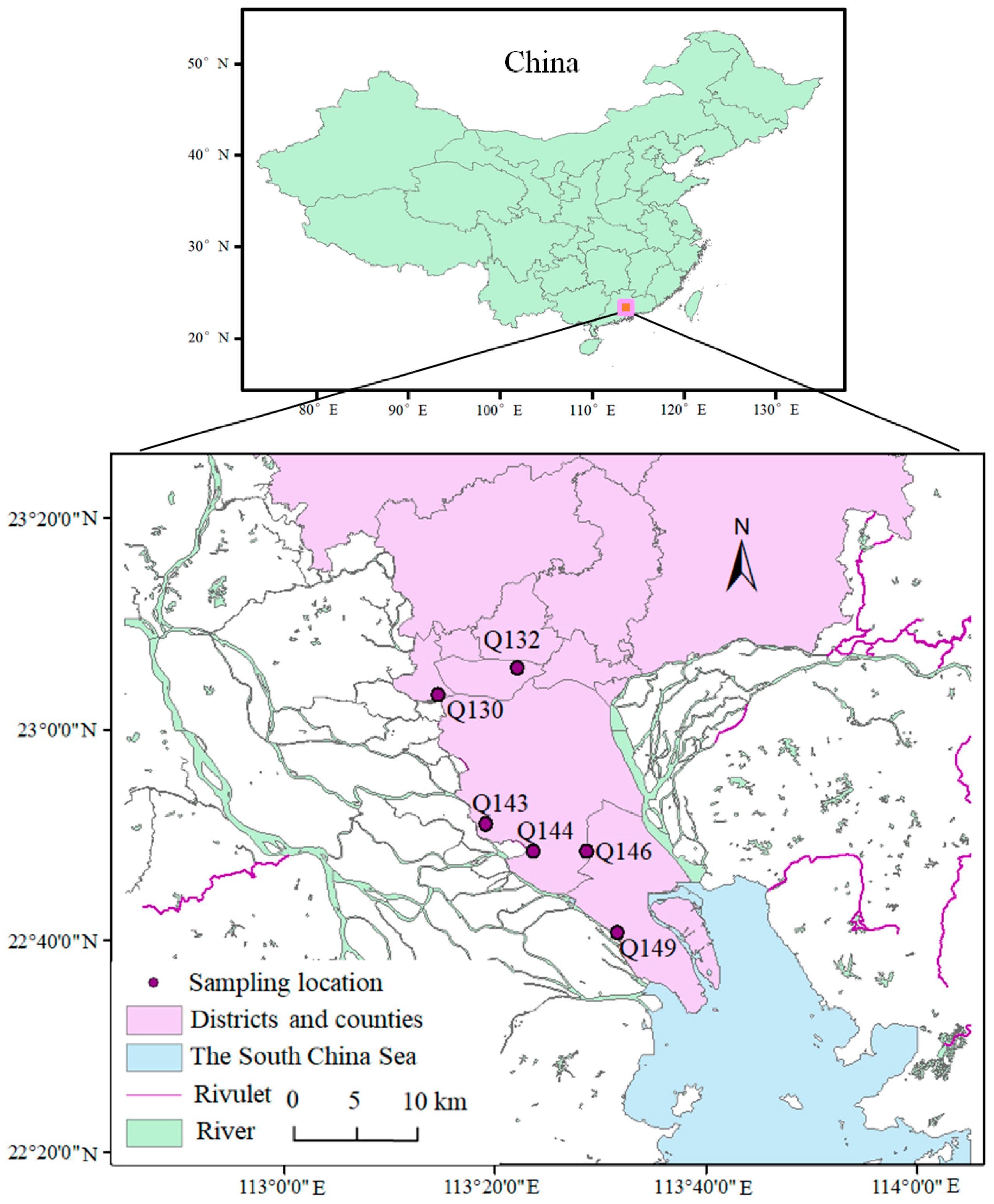
2.1. Site Description
2.2. Sample Collection and Physicochemical Analysis
2.3. Microbial Community Characterization
3. Results and Discussion
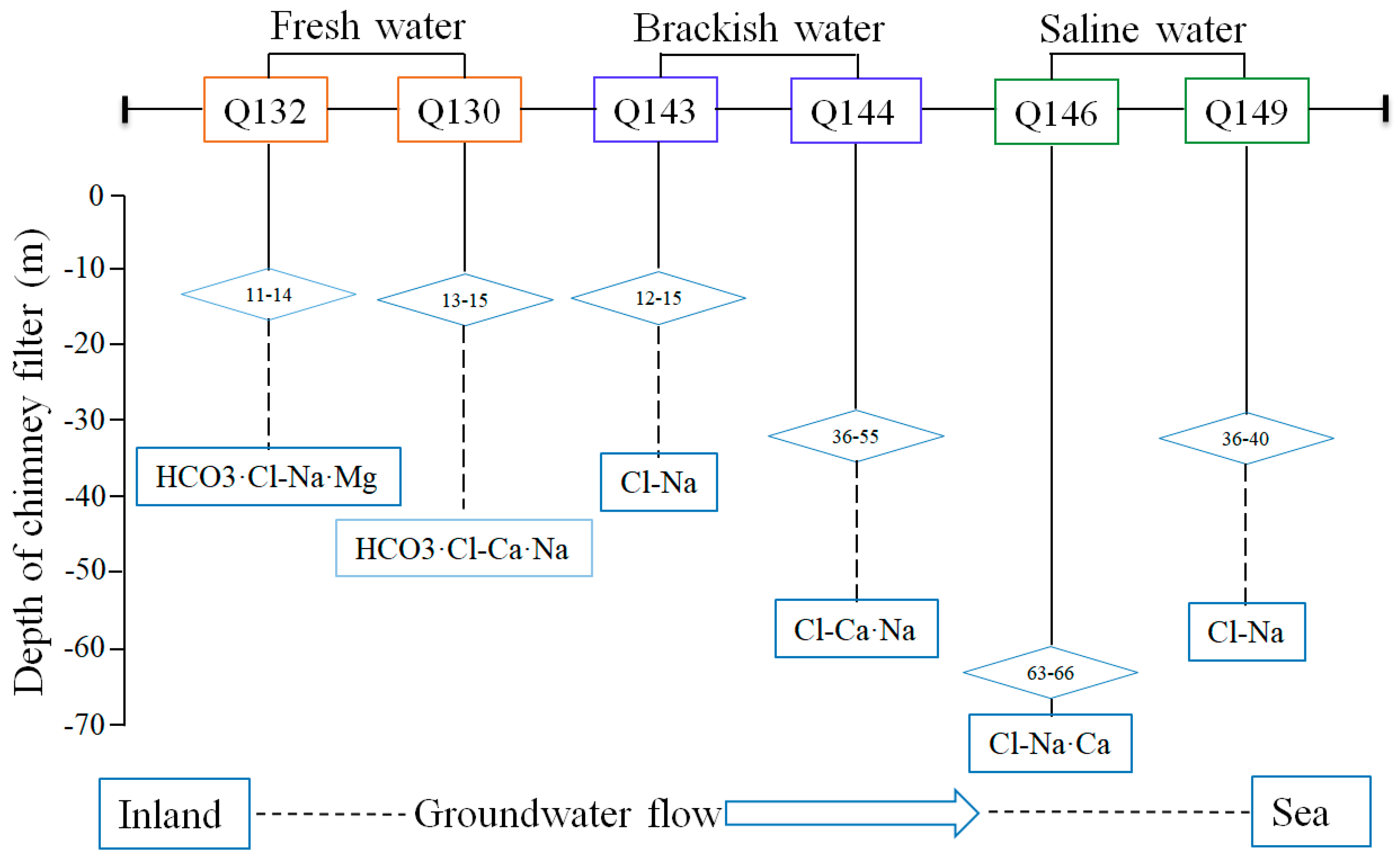
3.1. Hydrochemical Characterization
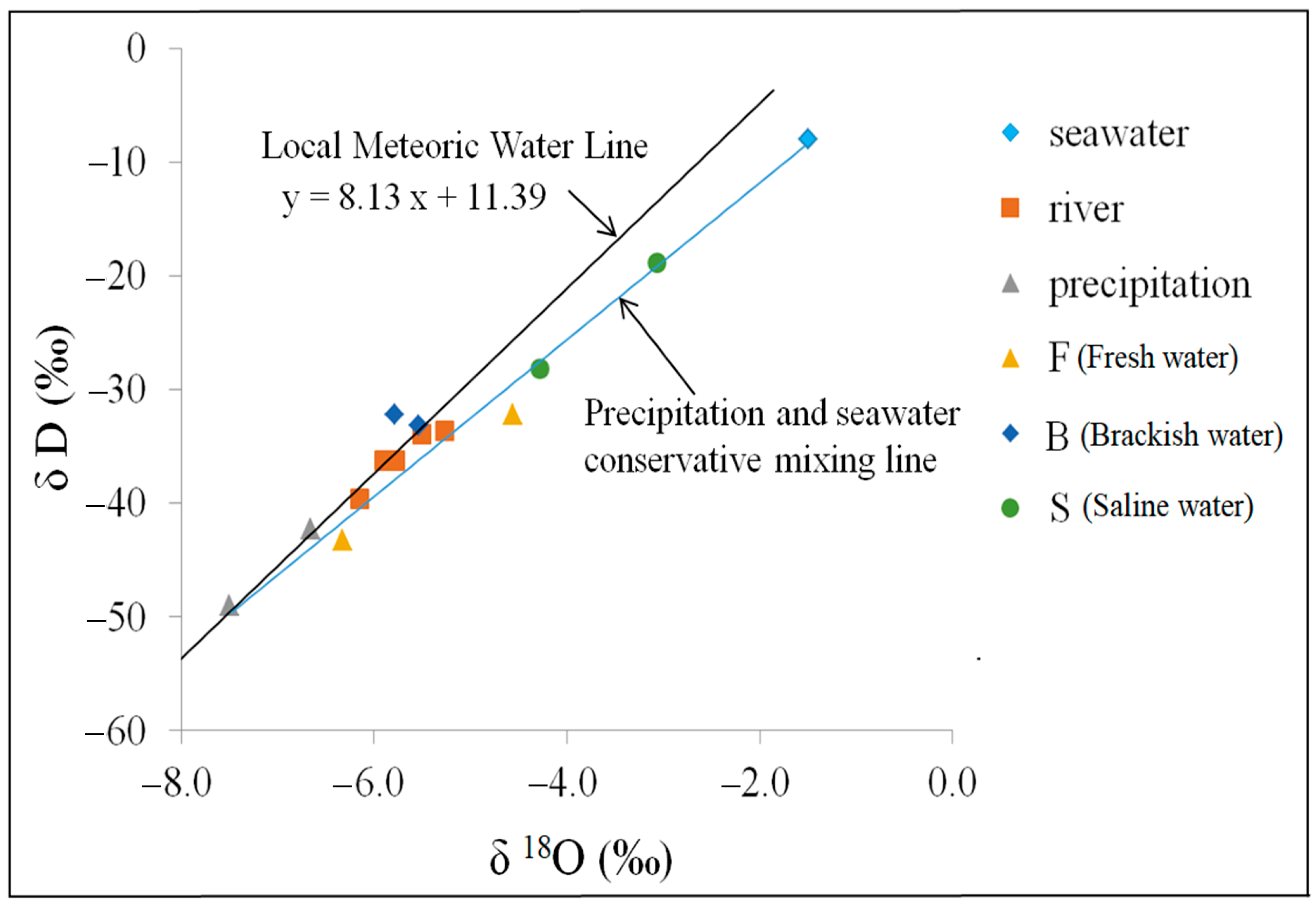
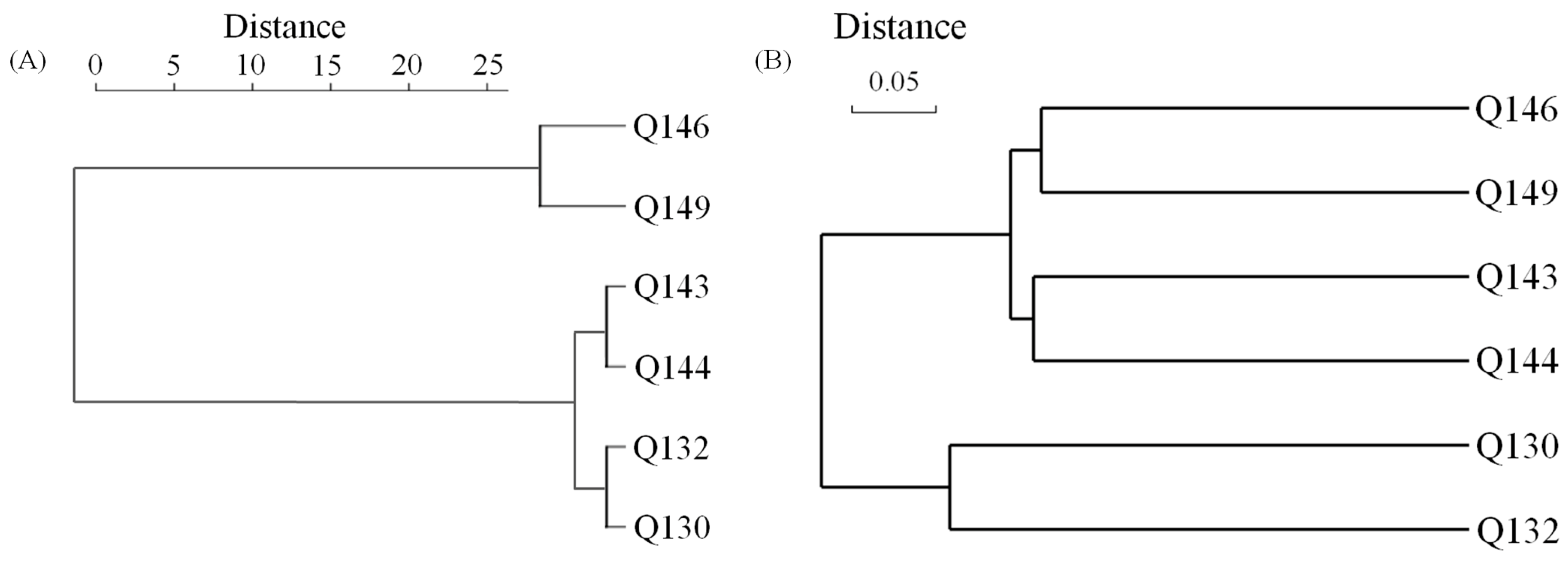
3.2. Isotopic Geochemical Characterization
3.3. Alpha Diversity of the Microbial Assemblages
3.4. Taxonomic Composition of Microbial Assemblages
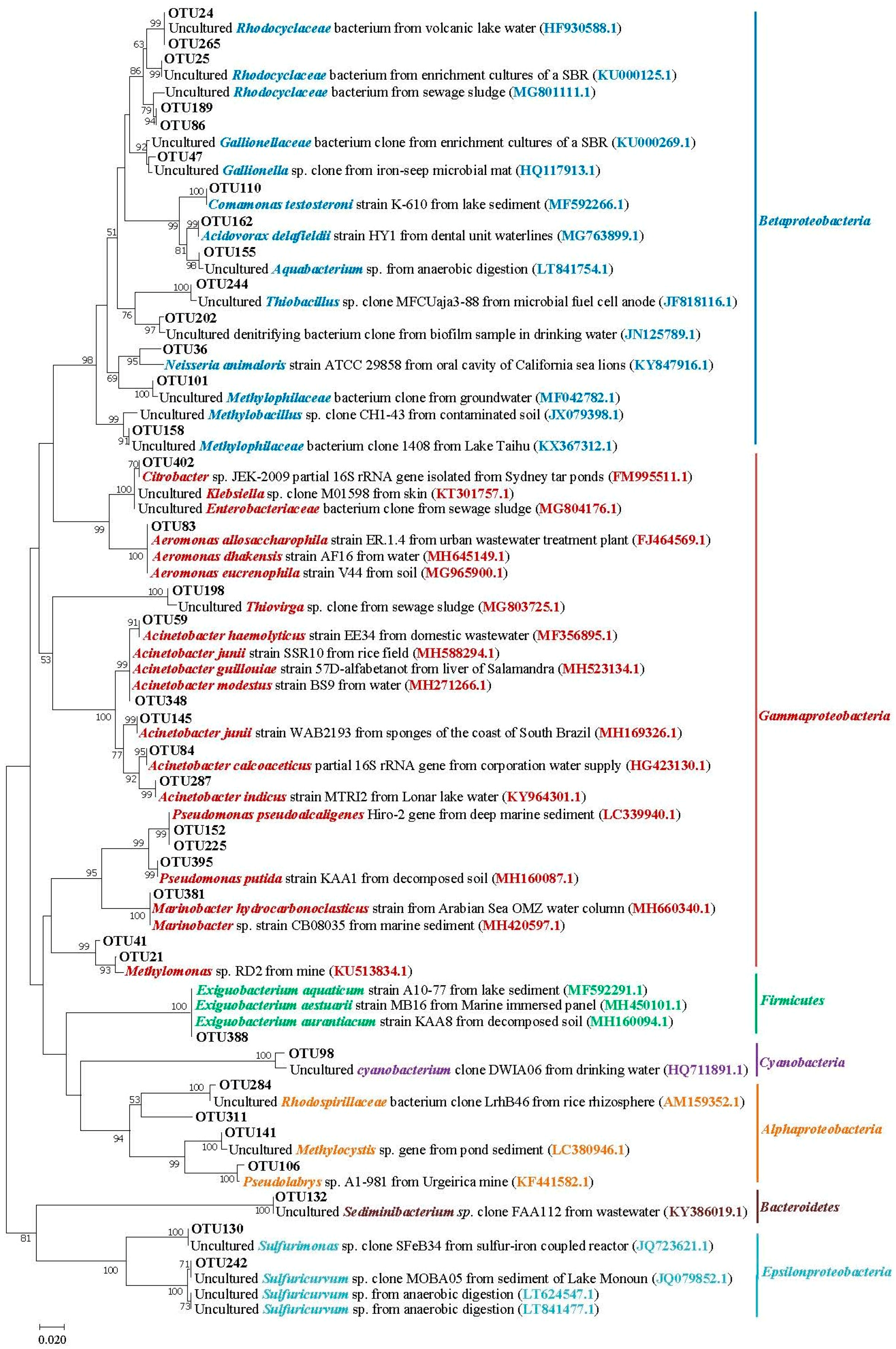
3.5. Phylogenetic Analysis of Main Phylum/Class Taxa within Different Salinity Groundwater
3.6. Linking the Distribution of Microbial Assemblages to the Hydrogeochemical Characteristics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ghabayen, S.M.; McKee, M.; Kemblowski, M. Ionic and isotopic ratios for identification of salinity sources and missing data in the Gaza aquifer. J. Hydrol. 2006, 318, 360–373. [Google Scholar] [CrossRef]
- Wang, Y.; Jiao, J.J. Origin of groundwater salinity and hydrogeochemical processes in the confined Quaternary aquifer of the Pearl River Delta, China. J. Hydrol. 2012, 438, 112–124. [Google Scholar] [CrossRef]
- Unno, T.; Kim, J.; Kim, Y.; Nguyen, S.G.; Guevarra, R.B.; Kim, G.P.; Lee, J.; Sadowsky, M.J. Influence of seawater intrusion on microbial communities in groundwater. Sci. Total Environ. 2015, 532, 337–343. [Google Scholar] [CrossRef]
- Hery, M.; Volant, A.; Garing, C.; Luquot, L.; Poulichet, F.E.; Gouze, P. Diversity and geochemical structuring of bacterial communities along a salinity gradient in a carbonate aquifer subject to seawater intrusion. FEMS Microbiol. Ecol. 2014, 90, 922–934. [Google Scholar] [CrossRef]
- Shi, L.; Jiao, J.J. Seawater intrusion and coastal aquifer management in China: A review. Environ. Earth Sci. 2014, 72, 2811–2819. [Google Scholar] [CrossRef]
- Teng, Y.; Su, J.; Wang, J.; Dai, N.; Li, J.; Song, L.; Zuo, R. Soil microbial community response to seawater intrusion into coastal aquifer of Donghai Island, South China. Environ. Earth Sci. 2014, 72, 3329–3338. [Google Scholar] [CrossRef]
- Pronk, M.; Goldscheider, N.; Zopfi, J. Microbial communities in karst groundwater and their potential use for biomonitoring. Hydrogeol. J. 2009, 17, 37–48. [Google Scholar] [CrossRef]
- Hahn, H.J.; Fuchs, A. Distribution patterns of groundwater communities across aquifer types in south-western Germany. Freshw. Biol. 2009, 54, 848–860. [Google Scholar] [CrossRef]
- Korbel, K.L.; Hancock, P.J.; Serov, P.; Lim, R.P.; Hose, G.C. Groundwater ecosystems vary with land use across a mixed agricultural landscape. J. Environ. Qual. 2013, 42, 380–390. [Google Scholar] [CrossRef]
- Qi, S.; Qiu, Q. Environmental hazard from saltwater intrusion in the Laizhou Gulf, Shandong Province of China. Nat. Hazards 2011, 56, 563–566. [Google Scholar] [CrossRef]
- van Dijk, G.; Smolders, A.J.; Loeb, R.; Bout, A.; Roelofs, J.G.; Lamers, L.P. Salinization of coastal freshwater wetlands; effects of constant versus fluctuating salinity on sediment biogeochemistry. Biogeochemistry 2015, 126, 71–84. [Google Scholar] [CrossRef]
- Edmonds, J.W.; Weston, N.B.; Joye, S.B.; Mou, X.; Moran, M.A. Microbial community response to seawater amendment in low-salinity tidal sediments. Microb. Ecol. 2009, 58, 558–568. [Google Scholar] [CrossRef]
- Zhi, F.; Yao, P.; Jiang, S.; Tu, S.; Kong, D.; Wang, L. Hydrochemical evidence of salt water formation in the Pearl River Delta. Hydrogeol. Eng. Geol. 2015, 42, 133–139. (In Chinese) [Google Scholar]
- Liu, K.; Jiao, J.J.; Gu, J. Investigation on bacterial community and diversity in the multilayer aquifer-aquitard system of the Pearl River Delta, China. Ecotoxicology 2014, 23, 2041–2052. [Google Scholar] [CrossRef]
- Lu, F.; Ni, H.; Liu, F.; Zeng, E.Y. Occurrence of nutrients in riverine runoff of the Pearl River Delta, South China. J. Hydrol. 2009, 376, 107–115. [Google Scholar] [CrossRef]
- Chen, S.; Fang, L.; Zhang, L.; Huang, W. Remote sensing of turbidity in seawater intrusion reaches of Pearl River Estuary–A case study in Modaomen water way, China. Estuar. Coast. Shelf Sci. 2009, 82, 119–127. [Google Scholar] [CrossRef]
- Jiao, J.J.; Wang, Y.; Cherry, J.A.; Wang, X.; Zhi, B.; Du, H.; Wen, D. Abnormally High Ammonium of Natural Origin in a Coastal Aquifer-Aquitard System in the Pearl River Delta, China. Environ. Sci. Technol. 2010, 44, 7470–7475. [Google Scholar] [CrossRef]
- Zong, Y.; Huang, G.; Switzer, A.D.; Yu, F.; Yim, W. An evolutionary model for the Holocene formation of the Pearl River delta, China. Holocene 2009, 19, 129–142. [Google Scholar] [CrossRef]
- Sheng, Y.; Wang, G.; Zhao, D.; Hao, C.; Liu, C.; Cui, L.; Zhang, G. Groundwater Microbial Communities Along a Generalized Flowpath in Nomhon Area, Qaidam Basin, China. Groundwater 2018, 56, 719–731. [Google Scholar] [CrossRef]
- Yamanaka, T.; Tsujimura, M.; Oyunbaatar, D.; Davaa, G. Isotopic variation of precipitation over eastern Mongolia and its implication for the atmospheric water cycle. J. Hydrol. 2007, 333, 21–34. [Google Scholar] [CrossRef]
- Wu, X.; Wang, X.; Wang, Y.; Hu, B.X. Origin of water in the Badain Jaran Desert, China: New insight from isotopes. Hydrol. Earth Syst. Sci. 2017, 21, 4419–4431. [Google Scholar] [CrossRef]
- Reckerth, A.; Stichler, W.; Schmidt, A.; Stumpp, C. Long-term data set analysis of stable isotopic composition in German rivers. J. Hydrol. 2017, 552, 718–731. [Google Scholar] [CrossRef]
- Wang, Y.; Jiao, J.J.; Cherry, J.A.; Lee, C.M. Contribution of the aquitard to the regional groundwater hydrochemistry of the underlying confined aquifer in the Pearl River Delta, China. Sci. Total Environ. 2013, 461, 663–671. [Google Scholar] [CrossRef]
- Lee, K.; Wang, Y.; Wang, Y.; Gu, J.; Jiao, J.J. Abundance and diversity of aerobic/anaerobic ammonia/ammonium-oxidizing microorganisms in an ammonium-rich aquitard in the Pearl River Delta of South China. Microb. Ecol. 2016, 76, 81–91. [Google Scholar] [CrossRef]
- Kim, H.; Kaown, D.; Mayer, B.; Lee, J.; Hyun, Y.; Lee, K. Identifying the sources of nitrate contamination of groundwater in an agricultural area (Haean basin, Korea) using isotope and microbial community analyses. Sci. Total Environ. 2015, 533, 566–575. [Google Scholar] [CrossRef]
- Bethencourt, M.; Fernández-Montblanc, T.; Izquierdo, A.; González-Duarte, M.M.; Muñoz-Mas, C. Study of the influence of physical, chemical and biological conditions that influence the deterioration and protection of Underwater Cultural Heritage. Sci. Total Environ. 2018, 613, 98–114. [Google Scholar] [CrossRef]
- Liu, Z.; Huang, S.; Sun, G.; Xu, Z.; Xu, M. Diversity and abundance of ammonia-oxidizing archaea in the Dongjiang River, China. Microbiol. Res. 2011, 166, 337–345. [Google Scholar] [CrossRef]
- Wang, Y.; Jiao, J.J.; Cherry, J.A. Occurrence and geochemical behavior of arsenic in a coastal aquifer–aquitard system of the Pearl River Delta, China. Sci. Total Environ. 2012, 427, 286–297. [Google Scholar] [CrossRef]
- Lu, Y.; Tang, C.; Chen, J.; Sakura, Y. Impact of septic tank systems on local groundwater quality and water supply in the Pearl River Delta, China: Case study. Hydrol. Process. Int. J. 2008, 22, 443–450. [Google Scholar] [CrossRef]
- Aitchison, J.C.; Ali, J.R.; Davis, A.M. When and where did India and Asia collide? J. Geophys. Res. Solid Earth 2007, 112, 1–2. [Google Scholar] [CrossRef]
- Freeze, R.A.; Cherry, J.A. Groundwater; Prentice-Hall, Inc.: Englewood Cliffs, NJ, USA, 1974; Volume 604, pp. 21–51. [Google Scholar]
- Renquan, Z.; Xing, L.; Menggui, J.; Li, W.; Qingchun, Y. Fundamentals of Hydrogeology; Geological Publishing House: Beijing, China, 2011. [Google Scholar]
- Christophersen, N.; Neal, C. Linking hydrological, geochemical, and soil chemical processes on the catchment scale: An interplay between modeling and field work. Water Resour. Res. 1990, 26, 3077–3086. [Google Scholar]
- Fritz, P.; Fontes, J. Handbook of Environmental Isotope Geochemistry; Elsevier: Amsterdam, The Netherlands, 1980; pp. 1–19. [Google Scholar]
- Wen, X.; Diao, M.; Wang, D.; Gao, M. Hydrochemical characteristics and salinization processes of groundwater in the shallow aquifer of Eastern Laizhou Bay, China. Hydrol. Process. 2012, 26, 2322–2332. [Google Scholar] [CrossRef]
- Carreira, P.M.; Marques, J.M.; Nunes, D. Source of groundwater salinity in coastline aquifers based on environmental isotopes (Portugal): Natural vs. human interference. A review and reinterpretation. Appl. Geochem. 2014, 41, 163–175. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, L.; Liu, J.; Liu, F.L.; Chen, Z.Y. D and 18O isotopes in atmospheric precipitation in Hongkong area. Ecol. Environ. Sci. 2009, 18, 572–577. [Google Scholar]
- Bethke, C.M.; Johnson, T.M. Groundwater age and groundwater age dating. Annu. Rev. Earth Planet. Sci. 2008, 36, 121–152. [Google Scholar] [CrossRef]
- Clark, I.D.; Fritz, P. Tracing the carbon cycle. In Environmental Isotopes in Hydrogeology; CRC Press: Boca Raton, FL, USA, 1997; pp. 111–134. [Google Scholar]
- Abed, R.M.; Kohls, K.; de Beer, D. Effect of salinity changes on the bacterial diversity, photosynthesis and oxygen consumption of cyanobacterial mats from an intertidal flat of the Arabian Gulf. Environ. Microbiol. 2007, 9, 1384–1392. [Google Scholar] [CrossRef]
- Campbell, B.J.; Kirchman, D.L. Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME J. 2013, 7, 210. [Google Scholar] [CrossRef]
- Griebler, C.; Lueders, T. Microbial biodiversity in groundwater ecosystems. Freshw. Biol. 2009, 54, 649–677. [Google Scholar] [CrossRef]
- Manes, C.D.O.; Barbe, C.; West, N.J.; Rapenne, S.; Lebaron, P. Impact of seawater-quality and water treatment procedures on the active bacterial assemblages at two desalination sites. Environ. Sci. Technol. 2011, 45, 5943–5951. [Google Scholar] [CrossRef]
- Fimlaid, K.A.; Shen, A. Diverse mechanisms regulate sporulation sigma factor activity in the Firmicutes. Curr. Opin. Microbiol. 2015, 24, 88–95. [Google Scholar] [CrossRef]
- Xu, Z.; Knight, R. Dietary effects on human gut microbiome diversity. Br. J. Nutr. 2015, 113, S1–S5. [Google Scholar] [CrossRef]
- Thomas, F.; Hehemann, J.; Rebuffet, E.; Czjzek, M.; Michel, G. Environmental and gut Bacteroidetes: The food connection. Front. Microbiol. 2011, 2, 93. [Google Scholar] [CrossRef]
- Redfield, E.; Barns, S.M.; Belnap, J.; Daane, L.L.; Kuske, C.R. Comparative diversity and composition of cyanobacteria in three predominant soil crusts of the Colorado Plateau. FEMS Microbiol. Ecol. 2002, 40, 55–63. [Google Scholar] [CrossRef]
- Issa, O.M.; Défarge, C.; le Bissonnais, Y.; Marin, B.; Duval, O.; Bruand, A.; Acqui, L.P.D.; Nordenberg, S.; Annerman, M. Effects of the inoculation of cyanobacteria on the microstructure and the structural stability of a tropical soil. Plant Soil 2007, 290, 209–219. [Google Scholar] [CrossRef]
- Leloup, J.; Fossing, H.; Kohls, K.; Holmkvist, L.; Borowski, C.; Jørgensen, B.B. Sulfate-reducing bacteria in marine sediment (Aarhus Bay, Denmark): Abundance and diversity related to geochemical zonation. Environ. Microbiol. 2009, 11, 1278–1291. [Google Scholar] [CrossRef]
- Amano, T.; Yoshinaga, I.; Okada, K.; Yamagishi, T.; Ueda, S.; Obuchi, A.; Sako, Y.; Suwa, Y. Detection of anammox activity and diversity of anammox bacteria-related 16S rRNA genes in coastal marine sediment in Japan. Microbes Environ. 2007, 22, 232–242. [Google Scholar] [CrossRef][Green Version]
- Lefort, T.; Gasol, J.M. Global-scale distributions of marine surface bacterioplankton groups along gradients of salinity, temperature, and chlorophyll: A meta-analysis of fluorescence in situ hybridization studies. Aquat. Microb. Ecol. 2013, 70, 111–130. [Google Scholar] [CrossRef]
- Logares, R.; Brate, J.; Bertilsson, S.; Clasen, J.L.; Shalchian-Tabrizi, K.; Rengefors, K. Infrequent marine-freshwater transitions in the microbial world. Trends Microbiol. 2009, 17, 414–422. [Google Scholar] [CrossRef]
- Takai, K.; Campbell, B.J.; Cary, S.C.; Suzuki, M.; Oida, H.; Nunoura, T.; Hirayama, H.; Nakagawa, S.; Suzuki, Y.; Inagaki, F.; et al. Enzymatic and genetic characterization of carbon and energy metabolisms by deep-sea hydrothermal chemolithoautotrophic isolates of Epsilonproteobacteria. Appl. Environ. Microb. 2005, 71, 7310–7320. [Google Scholar] [CrossRef]
- Xiong, J.; Liu, Y.; Lin, X.; Zhang, H.; Zeng, J.; Hou, J.; Yang, Y.; Yao, T.; Knight, R.; Chu, H. Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau. Environ. Microbiol. 2012, 14, 2457–2466. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, T.; Jiao, N.; Tian, L.; Hu, A.; Yu, W.; Li, S. Microbial diversity in the snow, a moraine lake and a stream in Himalayan glacier. Extremophiles 2011, 15, 411. [Google Scholar] [CrossRef]
- Rodrigues, D.F.; Goris, J.; Vishnivetskaya, T.; Gilichinsky, D.; Thomashow, M.F.; Tiedje, J.M. Characterization of Exiguobacterium isolates from the Siberian permafrost. Description of Exiguobacterium sibiricum sp. nov. Extremophiles 2006, 10, 285–294. [Google Scholar] [CrossRef]
- Knudston, K.E.; Haas, E.J.; Iwen, P.C.; Ramaley, W.C.; Ramaley, R.F. Characterization of a Gram-positive, non-spore-forming Exiguobacterium-like organism isolated from a Western Colorado (USA) hot spring. Abstr. Annu. Meet. Am. Soc. Microbiol. 2001, 1, 30. [Google Scholar]
- Selvakumar, G.; Joshi, P.; Nazim, S.; Mishra, P.K.; Kundu, S.; Gupta, H.S. Exiguobacterium acetylicum strain 1P (MTCC 8707) a novel bacterial antagonist from the North Western Indian Himalayas. World J. Microbiol. Biotechnol. 2009, 25, 131–137. [Google Scholar] [CrossRef]
- Mohanty, G.; Mukherji, S. Biodegradation rate of diesel range n-alkanes by bacterial cultures Exiguobacterium aurantiacum and Burkholderia cepacia. Int. Biodeter. Biodegr. 2008, 61, 240–250. [Google Scholar] [CrossRef]
- Tan, L.; Qu, Y.; Zhou, J.; Li, A.; Gou, M. Identification and characteristics of a novel salt-tolerant Exiguobacterium sp. for azo dyes decolorization. Appl. Biochem. Biotechnol. 2009, 159, 728. [Google Scholar] [CrossRef]
- Rocha, L.L.; Colares, G.B.; Angelim, A.L.; Grangeiro, T.B.; Melo, V.M. Culturable populations of Acinetobacter can promptly respond to contamination by alkanes in mangrove sediments. Mar. Pollut. Bull. 2013, 76, 214–219. [Google Scholar] [CrossRef]
- Labbé, D.; Margesin, R.; Schinner, F.; Whyte, L.G.; Greer, C.W. Comparative phylogenetic analysis of microbial communities in pristine and hydrocarbon-contaminated Alpine soils. FEMS Microbiol. Ecol. 2007, 59, 466–475. [Google Scholar] [CrossRef]
- Beyer, A.; Rzanny, M.; Weist, A.; Moeller, S.; Burow, K.; Gutmann, F.; Neumann, S.; Lindner, J.; Muesse, S.; Brangsch, H.; et al. Aquifer community structure in dependence of lithostratigraphy in groundwater reservoirs. Environ. Sci. Pollut. R. 2015, 22, 19342–19351. [Google Scholar] [CrossRef]
- Mosier, A.C.; Francis, C.A. Relative abundance and diversity of ammonia-oxidizing archaea and bacteria in the San Francisco Bay estuary. Environ. Microbiol. 2008, 10, 3002–3016. [Google Scholar] [CrossRef]
- Saito, T.; Ishii, S.; Otsuka, S.; Nishiyama, M.; Senoo, K. Identification of novel Betaproteobacteria in a succinate-assimilating population in denitrifying rice paddy soil by using stable isotope probing. Microbes Environ. 2008, 23, 192–200. [Google Scholar] [CrossRef][Green Version]
- Nishizawa, T.; Uei, Y.; Tago, K.; Isobe, K.; Otsuka, S.; Senoo, K. Taxonomic composition of denitrifying bacterial isolates is different among three rice paddy field soils in Japan. Soil Sci. Plant Nutr. 2013, 59, 305–310. [Google Scholar] [CrossRef]
- Ye, L.; Shao, M.; Zhang, T.; Tong, A.H.Y.; Lok, S. Analysis of the bacterial community in a laboratory-scale nitrification reactor and a wastewater treatment plant by 454-pyrosequencing. Water Res. 2011, 45, 4390–4398. [Google Scholar] [CrossRef]
- Barton, H.A.; Northup, D.E. Geomicrobiology in cave environments: Past, current and future perspectives. J. Cave Karst Stud. 2007, 69, 163–178. [Google Scholar]
- Rhee, J.Y.; Jung, D.S.; Peck, K.R. Clinical and therapeutic implications of Aeromonas bacteremia: 14 years nation-wide experiences in Korea. Infect. Chemother. 2016, 48, 274–284. [Google Scholar] [CrossRef]
- Sheng, Y.; Bibby, K.; Grettenberger, C.; Kaley, B.; Macalady, J.L.; Wang, G.; Burgos, W.D. Geochemical and temporal influences on the enrichment of acidophilic iron-oxidizing bacterial communities. Appl. Environ. Microb. 2016, 82, 3611–3621. [Google Scholar] [CrossRef]
- Tripathi, B.M.; Kim, M.; Kim, Y.; Byun, E.; Yang, J.; Ahn, J.; Lee, Y.K. Variations in bacterial and archaeal communities along depth profiles of Alaskan soil cores. Sci. Rep. 2018, 8, 504. [Google Scholar] [CrossRef]
- Hatosy, S.M.; Martiny, J.B.; Sachdeva, R.; Steele, J.; Fuhrman, J.A.; Martiny, A.C. Beta diversity of marine bacteria depends on temporal scale. Ecology 2013, 94, 1898–1904. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.; Konopka, A.E.; Fredrickson, J.K. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012, 6, 1653. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Geochemical Type | Parameter | Q132 | Q130 | Q143 | Q144 | Q146 | Q149 |
|---|---|---|---|---|---|---|---|
| Physiochemical characterization | Depth (m) | 11–14.1 | 13–15.2 | 12.2–15 | 36–55 | 63–66 | 36–40 |
| Longitude (°) | 113.37 | 113.24 | 113.32 | 113.39 | 113.48 | 113.53 | |
| Latitude (°) | 23.10 | 23.06 | 22.85 | 22.81 | 22.81 | 22.68 | |
| Temperature (°C) | 24.5 | 25.8 | 24.95 | 25.1 | 25.1 | 26 | |
| TOC (mg/L) | 3.53 | 0.3 | 0.53 | 2.01 | 2.85 | 3.08 | |
| TN (mg/L) | 32.42 | 47.59 | 1.37 | 3.78 | 1.27 | 87.27 | |
| TP (umol/L) | 2.58 | 0.56 | 0.68 | 0.56 | 1.04 | 0.8 | |
| NO2− (μg/L) | 27.69 | 28.43 | 6.74 | 6.44 | 7.25 | 2.33 | |
| Cl− (mg/L) | 92.518 | 228.19 | 2089.1 | 2484.6 | 9451.7 | 13441.8 | |
| DO (%) | 60.8 | 28.5 | 49 | 55.2 | 28.8 | 1.4 | |
| pH | 7.26 | 6.88 | 6.97 | 7.09 | 7.04 | 7.68 | |
| ORP(mV) | −30.6 | 71.2 | 29.1 | 32.5 | −100.7 | −220.1 | |
| TDS (g/L) | 0.41 | 0.74 | 3.52 | 3.61 | 10.79 | 15.3 | |
| NO3− (mg/L) | 0.01 | 0.66 | 0.06 | 0.07 | 0.03 | 0.01 | |
| SO42− (mg/L) | 2.458 | 1.19 | 337.5 | 1.4 | 137 | 57 | |
| HCO3− (mmol/L) | 6.4 | 9.39 | 7.98 | 2.27 | 0.18 | 0.26 | |
| K+ (mg/L) | 15.6 | 8.01 | 48.36 | 36.34 | 65.27 | 121.59 | |
| Na+ (mg/L) | 104.87 | 158.35 | 1252.29 | 538.25 | 2060.74 | 2456.13 | |
| Ca2+ (mg/L) | 47.68 | 158.94 | 286.51 | 831.53 | 1754.09 | 849.48 | |
| Mg2+ (mg/L) | 32.65 | 37.29 | 128.48 | 127.44 | 382.72 | 539.06 | |
| Isotopic geochemical characterization | δD(‰) | −43.25 | −32.18 | −33.21 | −32.25 | −28.13 | −18.84 |
| δ18O(‰) | −6.33 | −4.56 | −5.54 | −5.79 | −4.28 | −3.07 | |
| 3H(T.U) | <1.3 | <1.3 | <1.3 | <1.3 | <1.3 | <1.3 | |
| 14C(BP) | - | 1750 ± 15 | - | - | 4720 ± 80 | 8820 ± 40 | |
| water type | confined water | confined water | confined water | confined water | confined water | confined water |
| Samples | Observed OTUs | Shannon | Simpson | ACE | Chao | Heip | Coverage (%) |
|---|---|---|---|---|---|---|---|
| Q130 | 283 | 3.31 | 0.07 | 314.31 | 312.22 | 0.09 | 99.83 |
| Q132 | 288 | 3.54 | 0.06 | 310.27 | 307.50 | 0.12 | 99.86 |
| Q143 | 267 | 2.48 | 0.24 | 291.99 | 290.76 | 0.04 | 99.84 |
| Q144 | 308 | 2.60 | 0.18 | 341.51 | 343.36 | 0.04 | 99.80 |
| Q146 | 237 | 1.85 | 0.26 | 271.93 | 264.13 | 0.02 | 99.81 |
| Q149 | 243 | 2.15 | 0.21 | 279.99 | 278.78 | 0.03 | 99.80 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sang, S.; Dai, H.; Hu, B.X.; Hao, Y.; Zhou, T.; Zhang, J. The Study of Hydrogeochemical Environments and Microbial Communities along a Groundwater Salinity Gradient in the Pearl River Delta, China. Water 2019, 11, 804. https://doi.org/10.3390/w11040804
Sang S, Dai H, Hu BX, Hao Y, Zhou T, Zhang J. The Study of Hydrogeochemical Environments and Microbial Communities along a Groundwater Salinity Gradient in the Pearl River Delta, China. Water. 2019; 11(4):804. https://doi.org/10.3390/w11040804
Chicago/Turabian StyleSang, Shilei, Heng Dai, Bill X. Hu, Yanru Hao, Tong Zhou, and Jin Zhang. 2019. "The Study of Hydrogeochemical Environments and Microbial Communities along a Groundwater Salinity Gradient in the Pearl River Delta, China" Water 11, no. 4: 804. https://doi.org/10.3390/w11040804
APA StyleSang, S., Dai, H., Hu, B. X., Hao, Y., Zhou, T., & Zhang, J. (2019). The Study of Hydrogeochemical Environments and Microbial Communities along a Groundwater Salinity Gradient in the Pearl River Delta, China. Water, 11(4), 804. https://doi.org/10.3390/w11040804