The Origin of Major Ions of Groundwater in a Loess Aquifer



Abstract
:1. Introduction
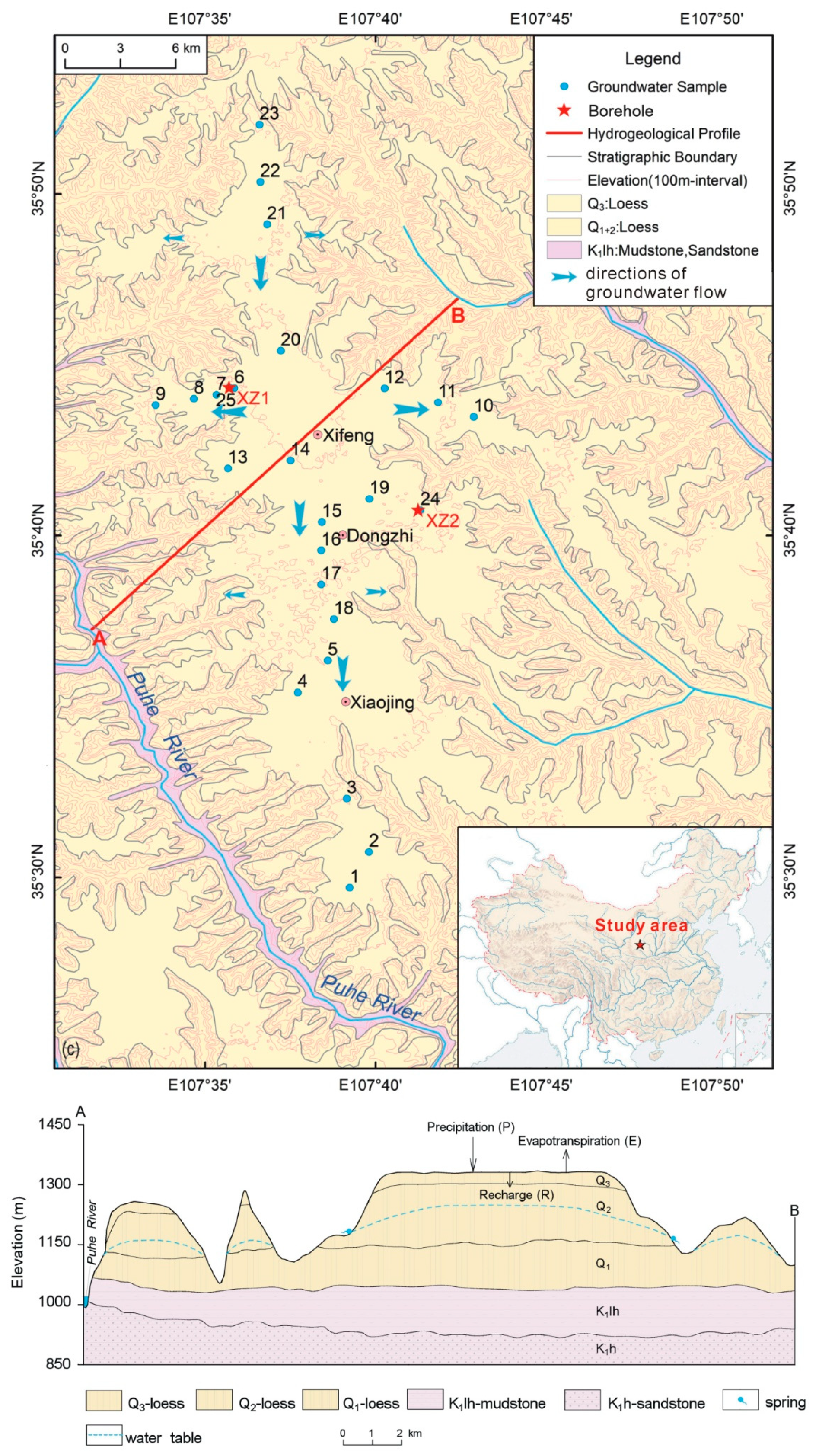
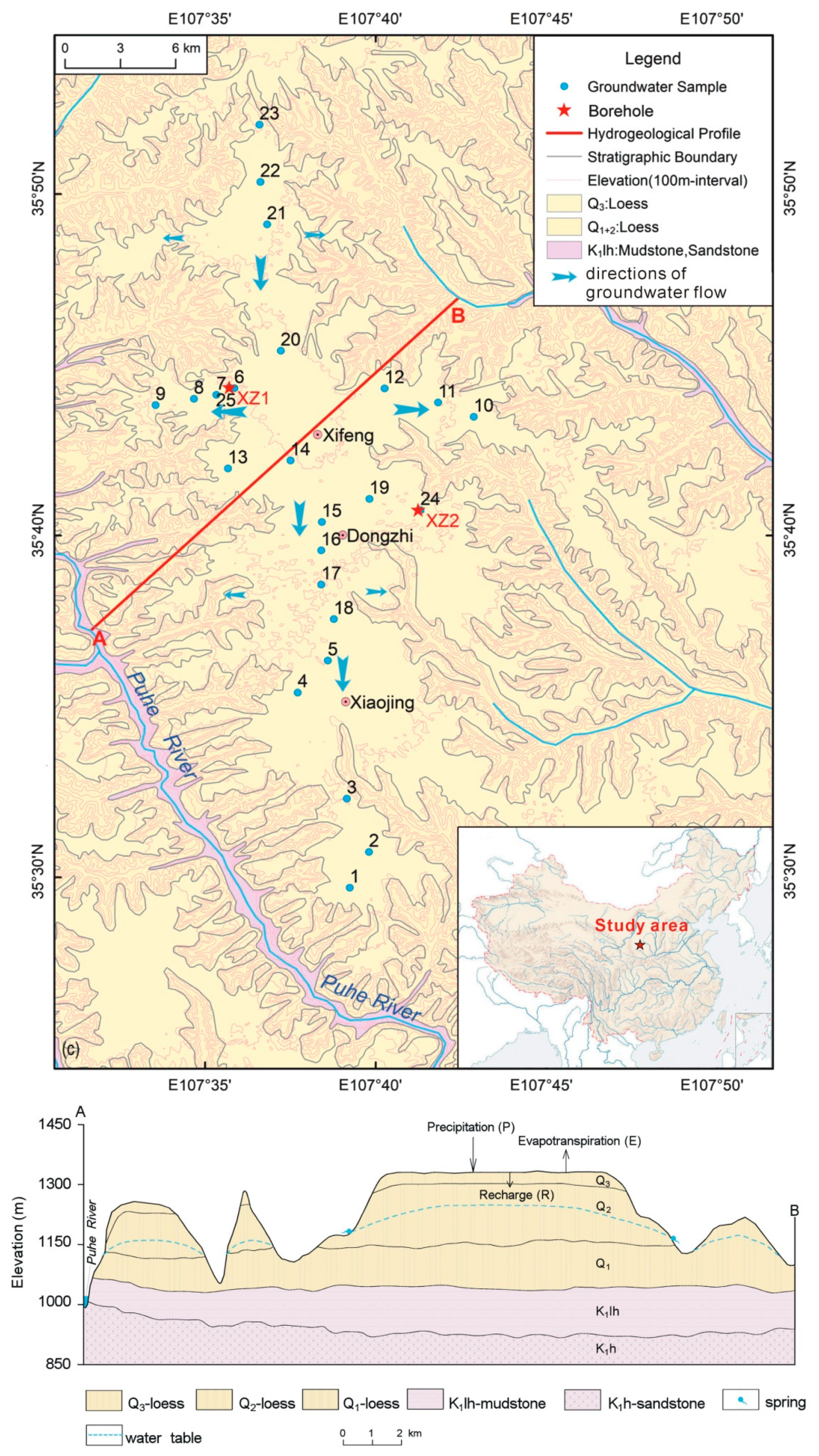
2. Study Area and Sampling
3. Experiment and Analyses
4. Results
4.1. Geochemical Characteristics of Loess
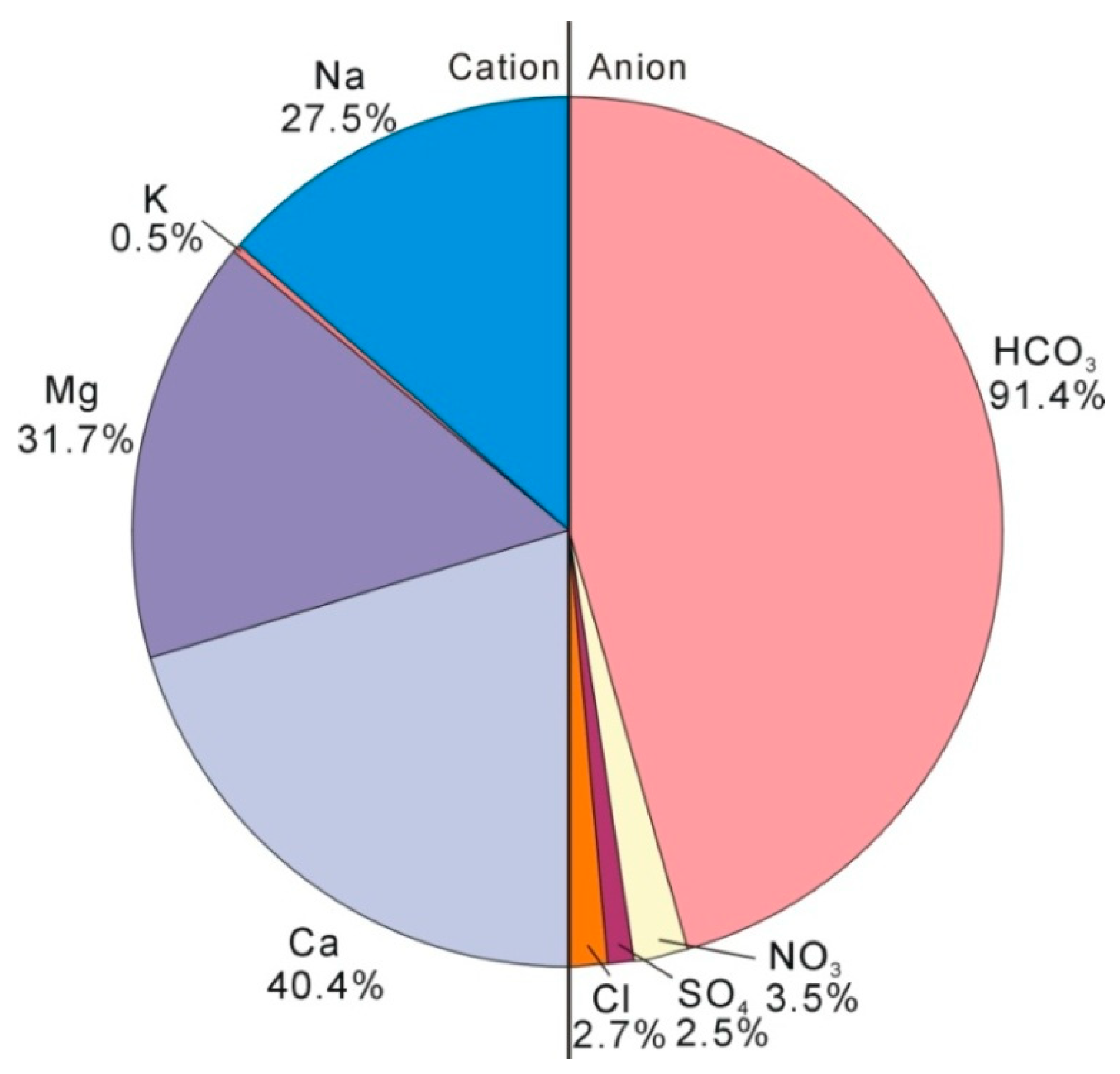
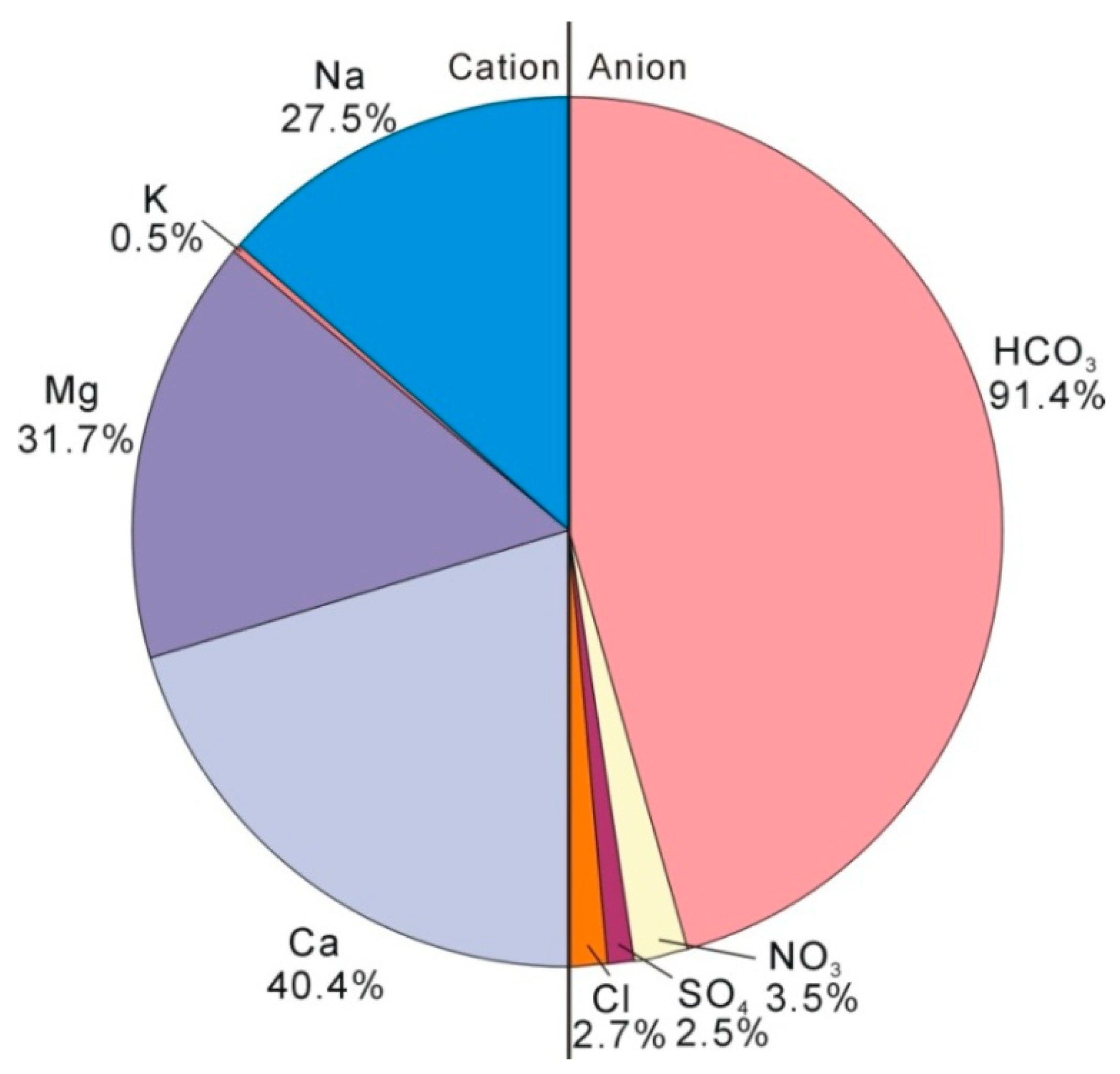
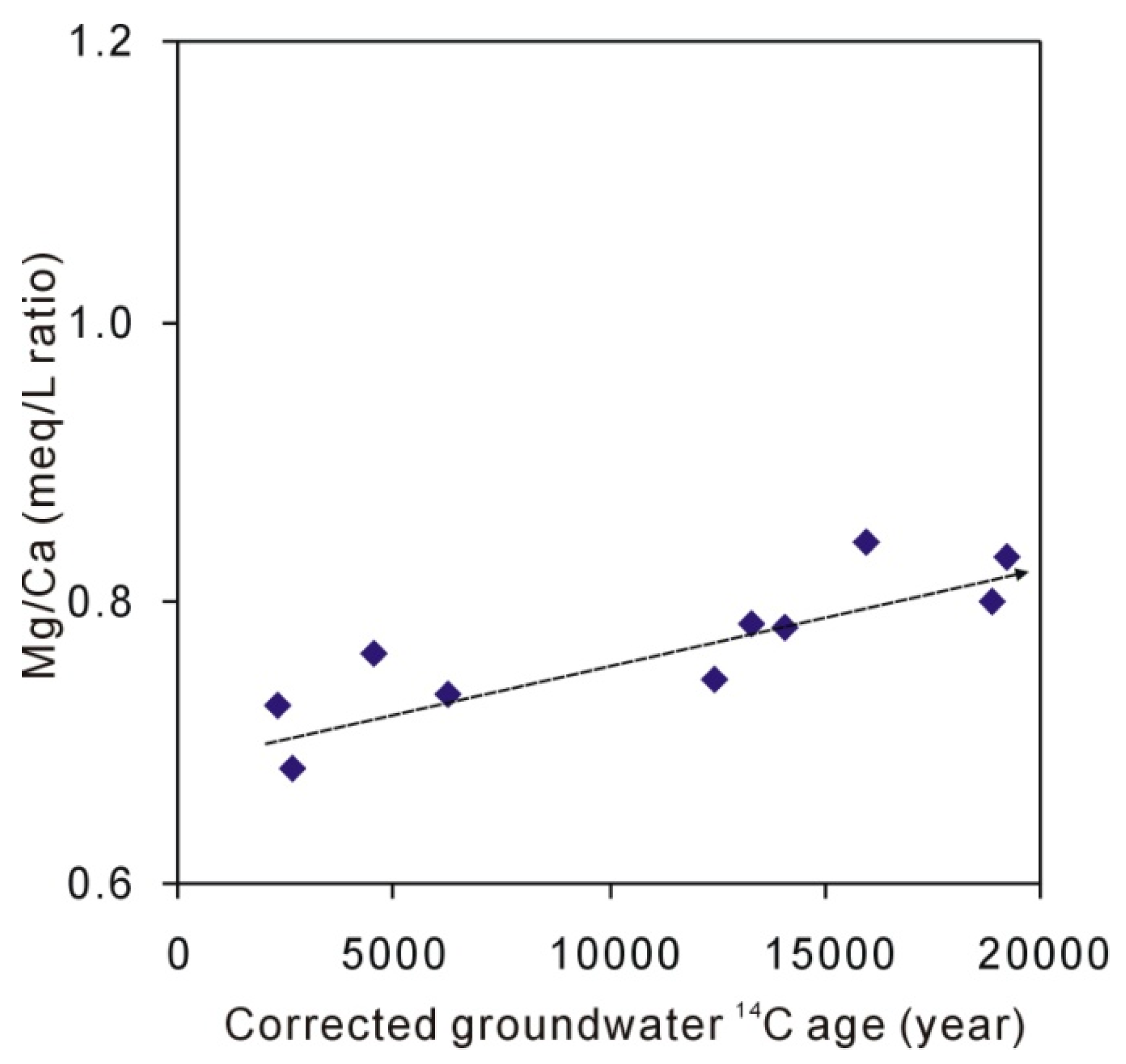
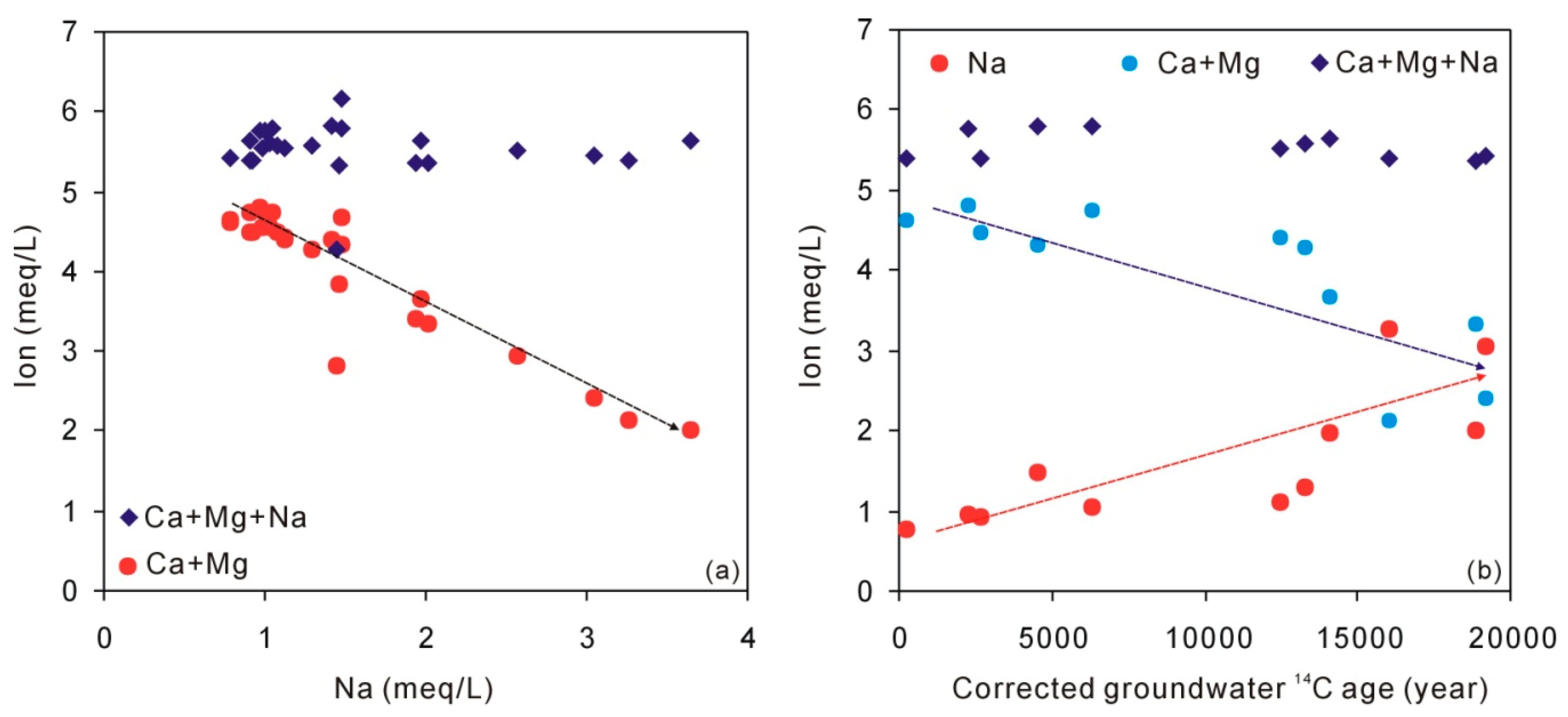
4.2. Hydrogeochemical Characteristics of Groundwater
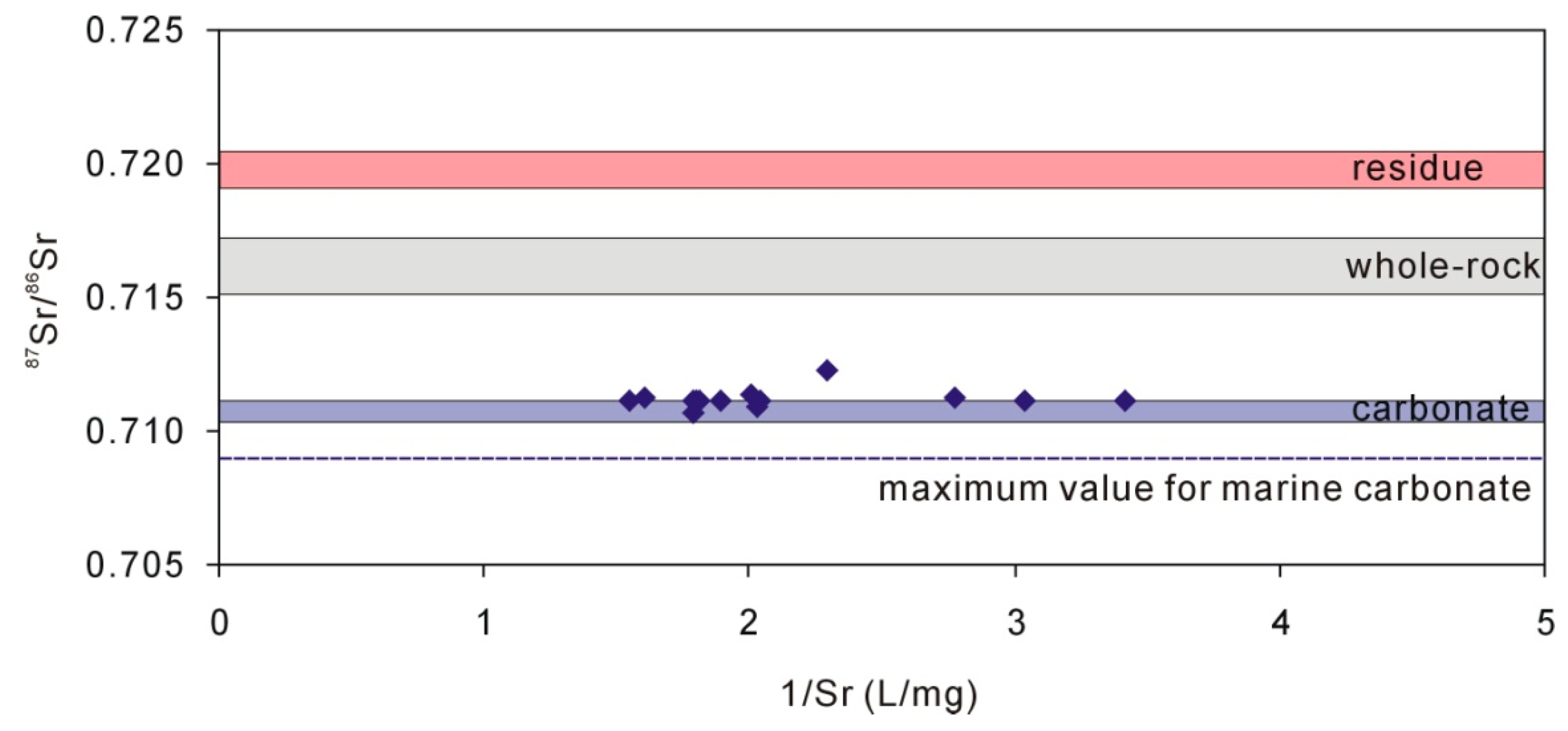
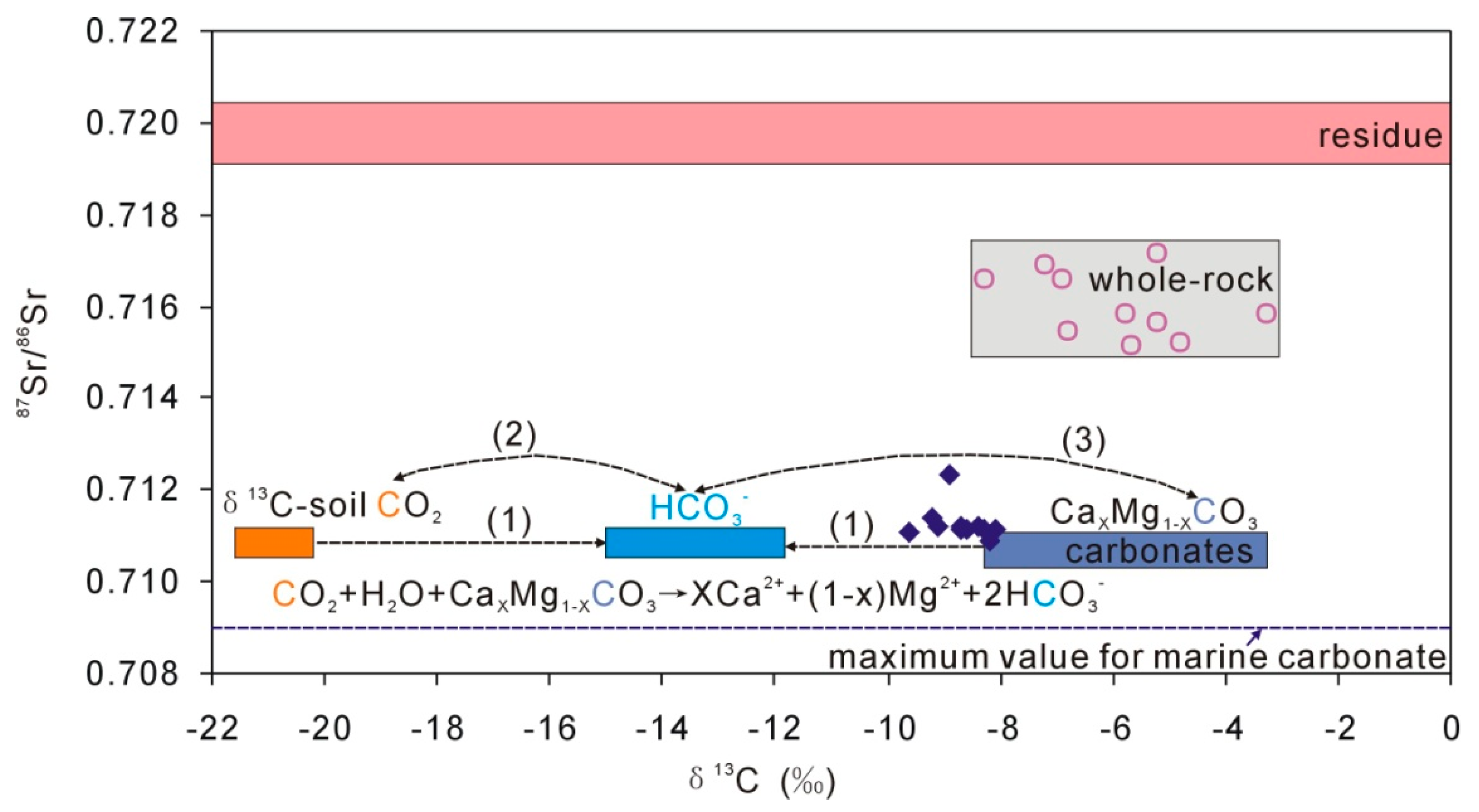
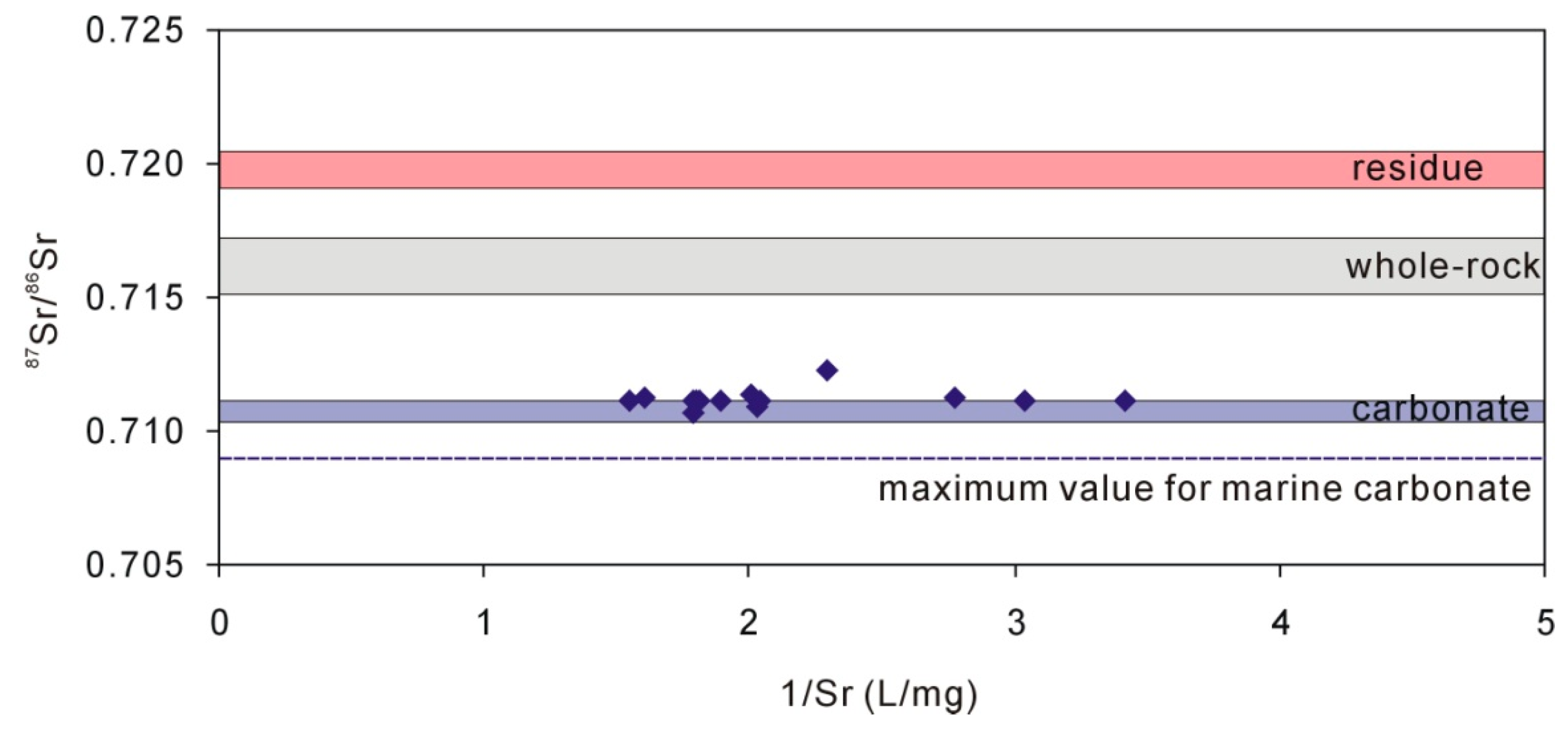
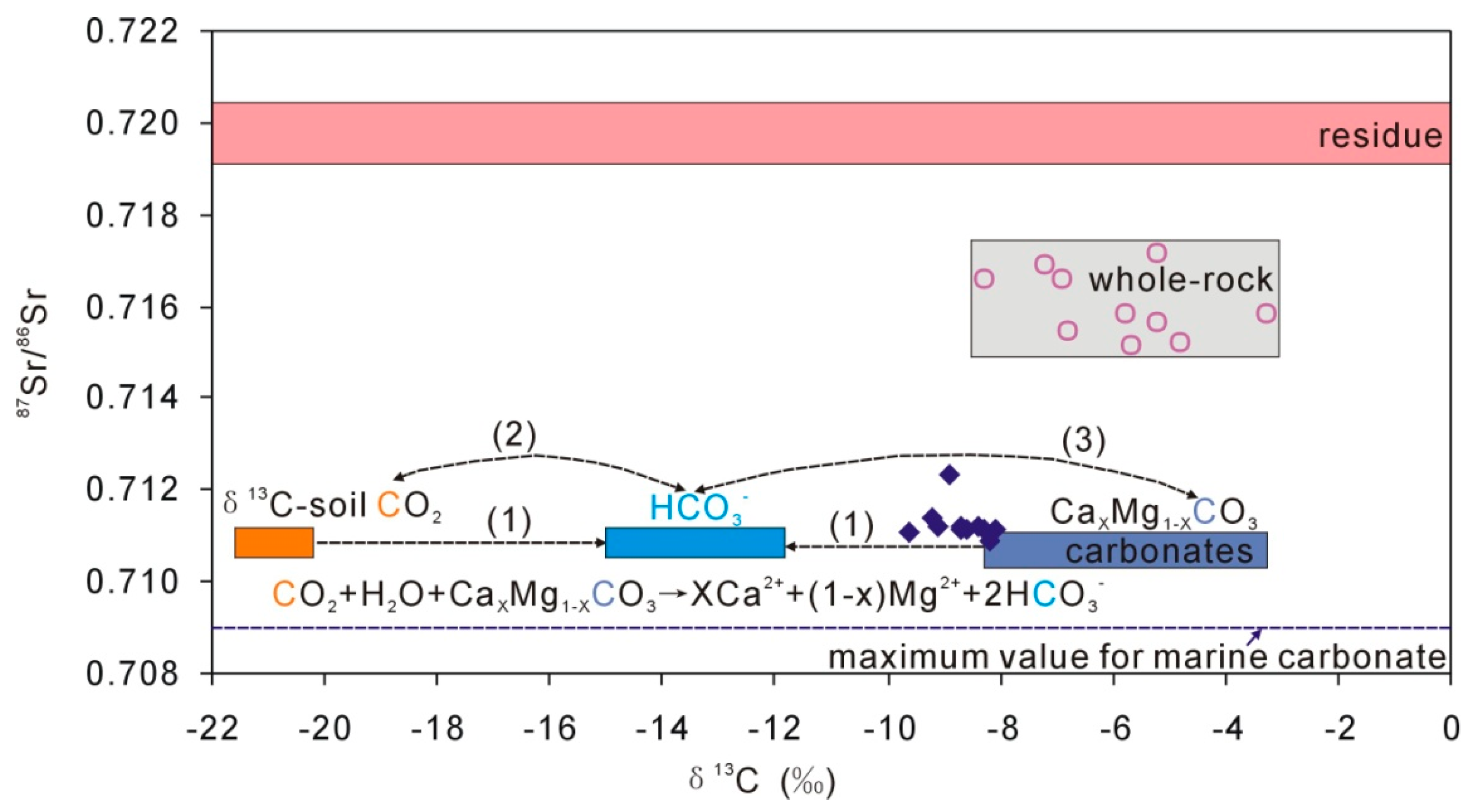
4.3. Characteristics of Carbonate in Loess
5. Discussion
5.1. Atmospheric Deposition
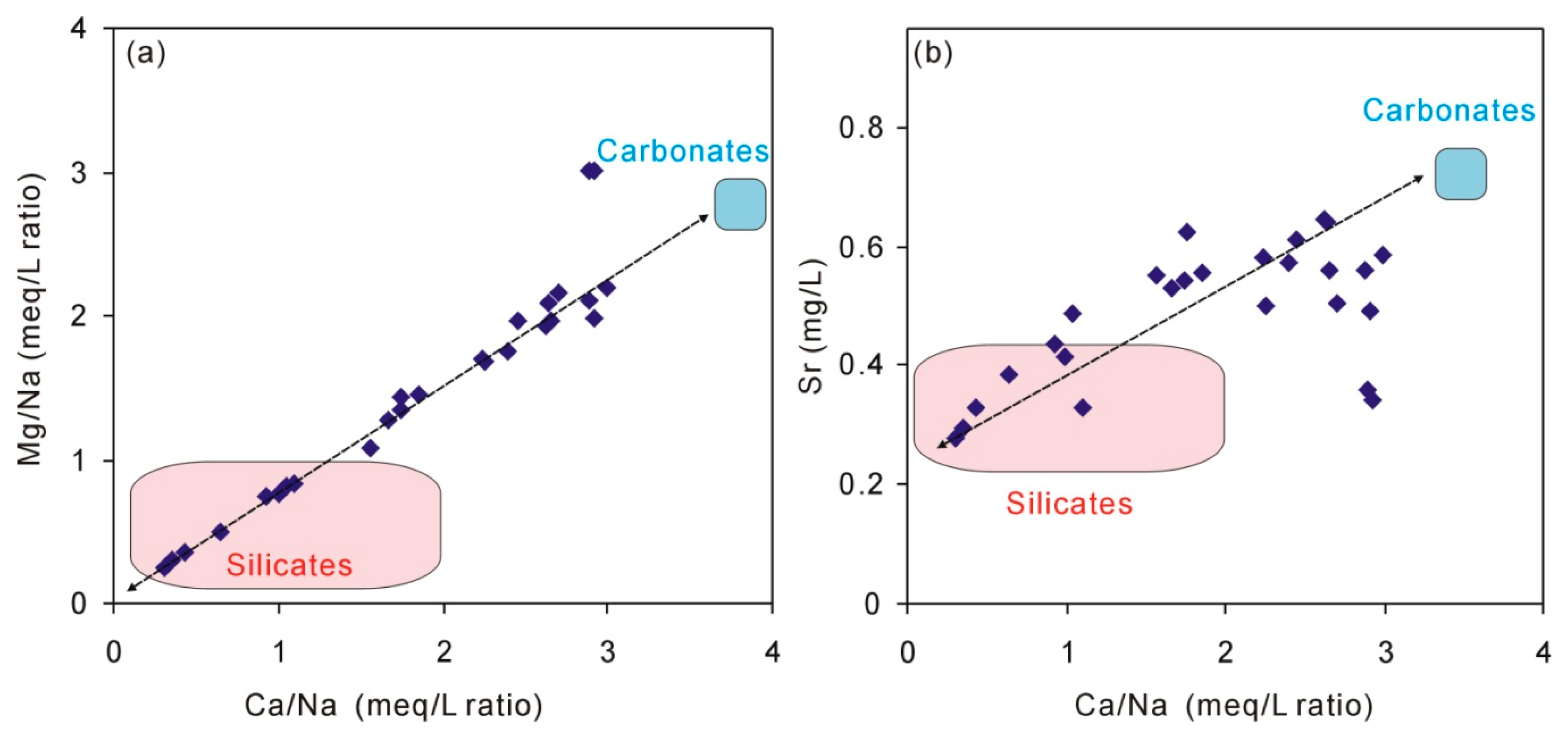
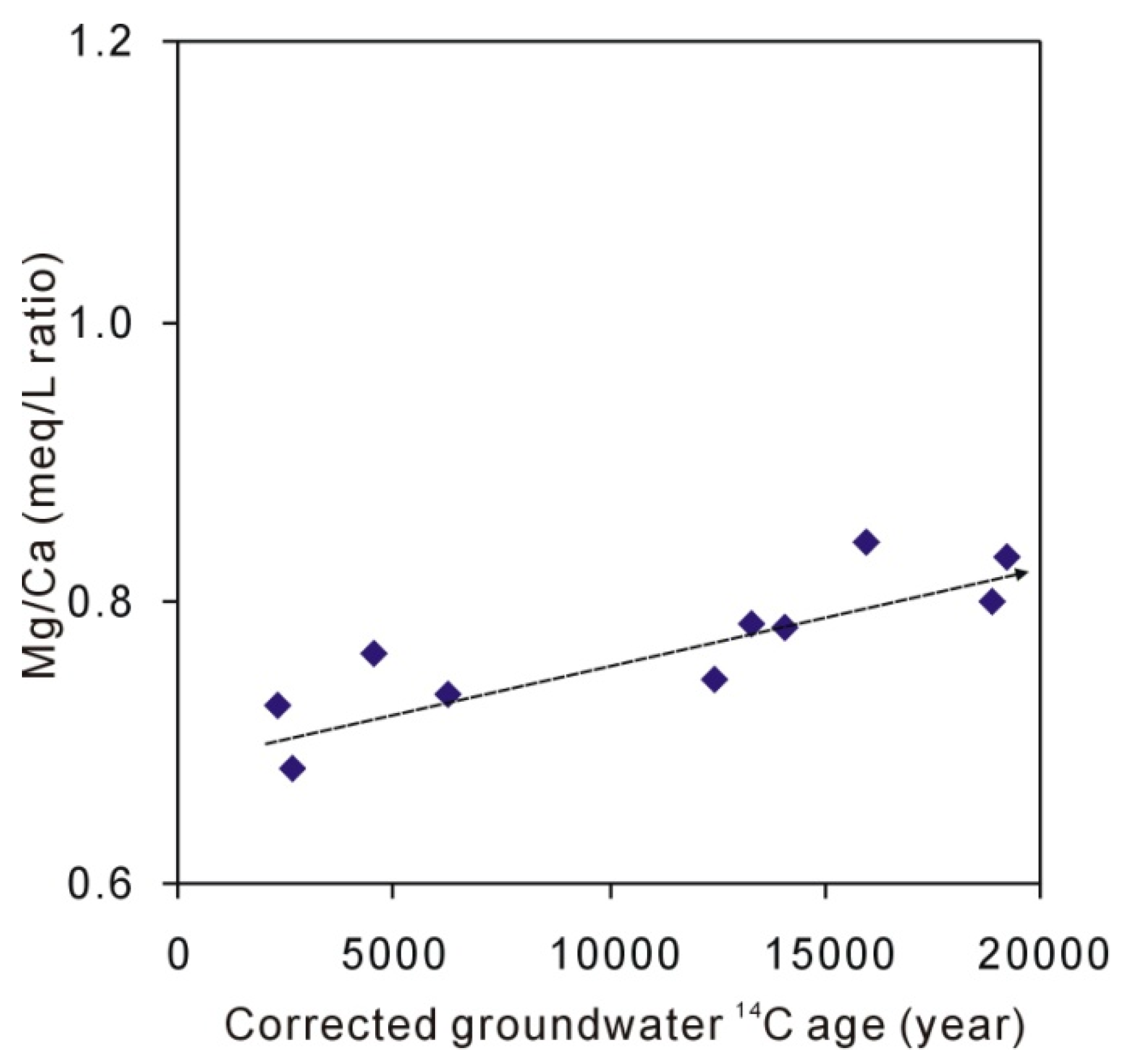
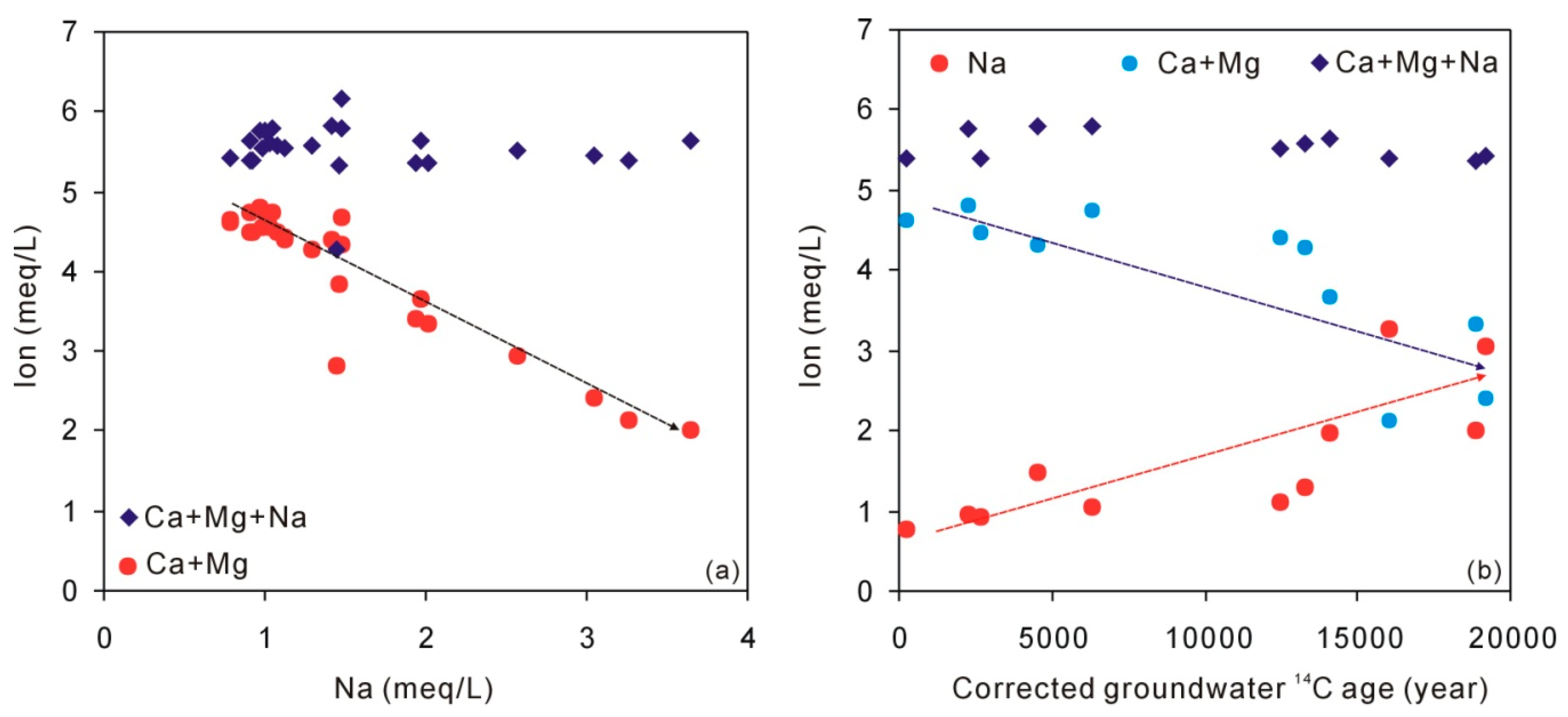
5.2. The Origin of Na, Ca, and Mg
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shiklomanov, I.A.; Rodda, J.C. World Water Resources at the Beginning of the 21st Century; Cambridge University Press: Cambridge, UK, 2003; pp. 1–18. [Google Scholar]
- De Vries, J.J.; Simmers, I. Groundwater recharge: An overview of processes and challenges. Hydrogeol. J. 2002, 10, 5–17. [Google Scholar] [CrossRef]
- Kemper, K.E. Groundwater from development to management. Hydrogeol. J. 2004, 12, 3–5. [Google Scholar] [CrossRef]
- Appelo, C.A.J.; Postma, D. Geochemistry, Groundwater and Pollution, 2nd ed.; A. A. Balkema Publishers: Amsterdam, The Netherlands, 2005; pp. 1–634. [Google Scholar]
- Edmunds, W.M. Limits to the availability of groundwater in Africa. Environ. Res. Lett. 2012, 7, 021003. [Google Scholar] [CrossRef]
- Edmunds, W.M. Geochemistry’s vital contribution to solving water resource problems. Appl. Geochem. 2009, 24, 1058–1073. [Google Scholar] [CrossRef]
- Huang, T.; Pang, Z.; Li, J.; Xiang, Y.; Zhao, Z. Mapping groundwater renewability using age data in the Baiyang alluvial fan, NW China. Hydrogeol. J. 2017, 25, 743–755. [Google Scholar] [CrossRef]
- Shen, Z.; Zhu, Y.; Zhong, Y. Hydrogeochemistry; Geological Publishing House: Beijing, China, 1993; pp. 1–189. [Google Scholar]
- Marcus, Y.; Kertes, A.S. Ion Exchange and Solvent Extraction of Metal Complexes; Wiley Interscience: New York, NY, USA, 1969; pp. 1–1037. [Google Scholar]
- Salifu, M.; Aiglsperger, T.; Hällström, L.; Martinsson, O.; Billström, K.; Ingri, J.; Dold, B.; Alakangas, L. Strontium (87Sr/86Sr) isotopes: A tracer for geochemical processes in mineralogically-complex mine wastes. Appl. Geochem. 2018, 99, 42–54. [Google Scholar] [CrossRef]
- Tipper, E.T.; Bickle, M.J.; Galy, A.; West, A.J.; Pomiès, C.; Chapman, H.J. The short term climatic sensitivity of carbonate and silicate weathering fluxes: Insight from seasonal variations in river chemistry. Geochim. Cosmochim. Acta 2006, 70, 2737–2754. [Google Scholar] [CrossRef]
- McNutt, R.H. Strontium Isotopes. In Environmental Tracers in Subsurface Hydrology; Cook, P.G., Herczeg, A.L., Eds.; Kluwer Academic Publishers: New York, NY, USA, 2000; pp. 233–260. [Google Scholar]
- Palmer, M.R.; Edmond, J.M. Controls over the strontium isotope composition of river water. Geochim. Cosmochim. Acta 1992, 56, 2099–2111. [Google Scholar] [CrossRef]
- Han, G.; Liu, C.Q. Water geochemistry controlled by carbonate dissolution: A study of the river waters draining karst-dominated terrain, Guizhou Province, China. Chem. Geol. 2004, 204, 1–21. [Google Scholar] [CrossRef]
- Shand, P.; Darbyshire, D.P.F.; Love, A.J.; Edmunds, W.M. Sr isotopes in natural waters: Applications to source characterisation and water–rock interaction in contrasting landscapes. Appl. Geochem. 2009, 24, 574–586. [Google Scholar] [CrossRef]
- Bickle, M.J.; Harris, N.B.W.; Bunbury, J.M.; Chapman, H.J.; Fairchild, I.J.; Ahmad, T. Controls on the 87Sr/86Sr Ratio of Carbonates in the Garhwal Himalaya, Headwaters of the Ganges. J. Geol. 2001, 109, 737–753. [Google Scholar] [CrossRef]
- Yang, J.; Chen, J.; An, Z.; Shield, G.; Tao, X.; Zhu, H.; Ji, J.; Chen, Y. Variations in 87Sr/86Sr ratios of calcites in Chinese loess: A proxy for chemical weathering associated with the East Asian summer monsoon. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2000, 157, 151–159. [Google Scholar] [CrossRef]
- Chen, J.; Li, G.; Yang, J.; Rao, W.; Lu, H.; Balsam, W.; Sun, Y.; Ji, J. Nd and Sr isotopic characteristics of Chinese deserts: Implications for the provenances of Asian dust. Geochim. Cosmochim. Acta 2007, 71, 3904–3914. [Google Scholar] [CrossRef]
- Yang, S.; Ding, Z.; Li, Y.; Wang, X.; Jiang, W.; Huang, X. Warming-induced northwestward migration of the East Asian monsoon rain belt from the last glacial maximum to the mid-Holocene. Proc. Natl. Acad. Sci. USA 2015, 112, 13178–13183. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chen, J. Geochemistry of Stable Isotopes; Science Press: Beijing, China, 2000; pp. 193–217. [Google Scholar]
- Huang, T.; Ma, B.; Pang, Z.; Li, Z.; Li, Z.; Long, Y. How does precipitation recharge groundwater in loess aquifers? Evidence from multiple environmental tracers. J. Hydrol. 2019. in revision. [Google Scholar]
- Liu, T.S. Loess and the Environment; Science Press: Beijing, China, 1985; pp. 1–481. [Google Scholar]
- Geng, A.; Wen, Q. Some geochemical characteristics of carbonates in Luochuan loess, Shaanxi Province. Geochimica 1988, 17, 267–275. [Google Scholar]
- Ning, Y.; Liu, W.; An, Z. Variation of soil Δδ13C values in Xifeng loess–paleosol sequence and its paleoenvironmental implication. Chin. Sci. Bull. 2006, 51, 1350–1354. [Google Scholar] [CrossRef]
- Qu, H. Assessment of Groundwater Resources in the Arid and Semiarid Land of China; Science Press: Beijing, China, 1991; pp. 1–457. [Google Scholar]
- Sheng, X.; Yang, J.; Li, C.; Chen, J.; Tao, X. A method for separation of calcite and dolomite in loess and sedimentary rocks. Rock Miner. Anal. 2000, 19, 264–267. [Google Scholar]
- Yokoo, Y.; Nakano, T.; Nishikawa, M.; Quan, H. Mineralogical variation of Sr–Nd isotopic and elemental compositions in loess and desert sand from the central Loess Plateau in China as a provenance tracer of wet and dry deposition in the northwestern Pacific. Chem. Geol. 2004, 204, 45–62. [Google Scholar] [CrossRef]
- Barta, G. Secondary carbonates in loess-paleosoil sequences: A general review. Cent. Eur. J. Geosci. 2011, 3, 129–146. [Google Scholar] [CrossRef]
- Keith, M.L.; Weber, J.N. Carbon and oxygen isotopic composition of selected limestones and fossils. Geochim. Cosmochim. Acta 1964, 18, 1787–1816. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. User’s Guide to PHREEQC (Version 2)—A Compute Program for Speciation, Batch–Reaction, One–Dimensional Transport, and Inverse Geochemical Calculations; USGS: Reston, VA, USA, 1999; pp. 1–312.
- Herczeg, A.L.; Edmunds, W.M. Inorganic ions as tracers. In Environmental Tracers in Subsurface Hydrology; Cook, P.G., Herczeg, A.L., Eds.; Kluwer Academic Publishers: New York, NY, USA, 2000; pp. 31–78. [Google Scholar]
- Hem, J.D. Study and Interpretation of the Chemical Characteristics of Natural Water; Water Supply Paper, no. 2254; US Geological Survey: Reston, VA, USA, 1985; pp. 1–264.
- Allison, G.B.; Hughes, M.W. The use of environmental chloride and tritium to estimate total recharge to an unconfined aquifer. Aust. J. Soil Res. 1978, 16, 181–195. [Google Scholar] [CrossRef]
- Edmunds, W.M.; Walton, N.R.G. A geochemical and isotopic approach to recharge evaluation in semi-arid zones—Past and present. In Arid–Zone Hydrology: Investigations with Isotope Techniques, Proceeding of Advisory Group Meeting; Fontes, J.C., Ed.; IAEA: Vienna, Austria, 1980; pp. 47–68. [Google Scholar]
- Eriksson, E.; Khunakasem, V. Chloride concentration in groundwater, recharge rate and rate of deposition of chloride in the Israel Coastal Plain. J. Hydrol. 1969, 7, 178–197. [Google Scholar] [CrossRef]
- Wood, W.W.; Sanford, W.E. Chemical and isotopic methods for quantifying groundwater recharge in a regional, semiarid environment. Ground Water 1995, 33, 458–468. [Google Scholar] [CrossRef]
- Mazor, E. Chemical and Isotopic Groundwater Hydrology, 3rd ed.; Dekker: New York, NY, USA, 2004; pp. 1–453. [Google Scholar]
- Edmunds, W.M.; Ma, J.; Aeschbach–Hertig, W.; Kipfer, R.; Darbyshire, D.P.F. Groundwater recharge history and hydrogeochemical evolution in the Minqin Basin, North West China. Appl. Geochem. 2006, 21, 2148–2170. [Google Scholar] [CrossRef]
- Huang, T.; Pang, Z.; Liu, J.; Ma, J.; Gates, J. Groundwater recharge mechanism in an integrated tableland of the Loess Plateau, northern China: Insights from environmental tracers. Hydrogeol. J. 2017, 25, 2049–2065. [Google Scholar] [CrossRef]
- Huang, T.; Pang, Z. Estimating groundwater recharge following land-use change using chloride mass balance of soil profiles: A case study at Guyuan and Xifeng in the Loess Plateau of China. Hydrogeol. J. 2011, 19, 177–186. [Google Scholar] [CrossRef]
- Feth, J.H. Chloride in Natural Continental Water: A Review; Water Supply Paper, no. 2176; US Geological Survey: Reston, VA, USA, 1981; pp. 1–30.
- The Acid Deposition Monitoring Network in East Asia (EANET). Available online: http://www.eanet.asia (accessed on 12 October 2018).
- Wang, T.; Wang, P.; Theys, N.; Tong, D.; Hendrick, F.; Zhang, Q.; van Roozendael, M. Spatial and temporal changes in SO2 regimes over China in the recent decade and the driving mechanism. Atmos. Chem. Phys. 2018, 18, 18063–18078. [Google Scholar] [CrossRef]
- Huang, T.; Fan, Y.; Long, Y.; Pang, Z. Quantitative calculation for the contribution of acid rain to carbonate weathering. J. Hydrol. 2019, 568, 360–371. [Google Scholar] [CrossRef]
- Gaillardet, J.; Dupré, B.; Louvat, P.; Allègre, C.J. Global silicate weathering and CO2 consumption rates deduced from the chemistry of large rivers. Chem. Geol. 1999, 159, 3–30. [Google Scholar] [CrossRef]
- Petelet-Giraud, E.; Luck, J.M.; Othman, D.B.; Joseph, C.; Négrel, P. Chemical and isotopic fingerprinting of small ungauged watershed: How far the hydrological functioning can be understood? Comptes Rendus Geosci. 2016, 348, 379–386. [Google Scholar] [CrossRef]
- Bishop, P.K.; Smalley, P.C.; Emery, D.; Dickson, J.A.D. Strontium isotopes as indicators of the dissolving phase in a carbonate aquifer: Implications for 14C dating of groundwaters. J. Hydrol. 1994, 154, 301–321. [Google Scholar] [CrossRef]
- Giletti, B.J.; Casserly, J.E.D. Strontium diffusion kinetics in plagioclase feldspars. Geochim. Cosmochim. Acta 1994, 58, 3785–3793. [Google Scholar] [CrossRef]
- Welch, W.S.A.; Ullman, J. Feldspar dissolution in acidic and organic solutions: Compositional and pH dependence of dissolution rate. Geochim. Cosmochim. Acta 1996, 60, 2939–2948. [Google Scholar] [CrossRef]
- Rao, Z.; Zhang, X.; Xue, S.; Xu, Y.; Liu, X. Primary organic carbon isotopic study result of Xifeng loess/red clay profile. Quat. Sci. 2012, 32, 825–827. [Google Scholar]
- Breeuwsma, A.; Wösten, J.H.M.; Vleeshouwer, J.J.; van Slobbe, A.M.; Bouma, J. Derivation of land qualities to assess environmental problems from soil surveys. Soil Sci. Soc. Am. J. 1986, 50, 186–190. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Quartz | Potassium Feldspar | Plagioclase | Calcite | Dolomite | Clay |
|---|---|---|---|---|---|---|
| S1 | 43.0 | 3.4 | 16.5 | 9.1 | 0.0 | 28.0 |
| S2 | 44.7 | 2.9 | 11.8 | 13.6 | 0.0 | 27.0 |
| S3 | 44.5 | 4.3 | 15.3 | 16.4 | 3.4 | 16.1 |
| S4 | 33.1 | 2.8 | 9.8 | 22.1 | 0.0 | 32.2 |
| Sample | Smectite | Illite-Smectite | Illite | Kaolinite | Chlorite |
|---|---|---|---|---|---|
| S1 | 0 | 32 | 49 | 5 | 14 |
| S2 | 0 | 20 | 60 | 7 | 13 |
| S3 | 0 | 21 | 58 | 9 | 12 |
| S4 | 0 | 13 | 68 | 7 | 12 |
| Sample | SiO2 | Al2O3 | Fe2O3 | MgO | CaO | Na2O | K2O | MnO | TiO2 | P2O5 | LOI |
|---|---|---|---|---|---|---|---|---|---|---|---|
| S1 | 59.14 | 12.02 | 4.51 | 2.11 | 7.05 | 1.78 | 2.34 | 0.087 | 0.649 | 0.141 | 9.77 |
| S2 | 58.98 | 12.56 | 4.89 | 2.05 | 6.36 | 1.58 | 2.38 | 0.087 | 0.677 | 0.139 | 9.72 |
| S3 | 59.51 | 11.83 | 4.35 | 2.15 | 7.28 | 1.72 | 2.28 | 0.083 | 0.637 | 0.167 | 9.79 |
| S4 | 54.73 | 11.89 | 4.57 | 1.95 | 9.42 | 1.39 | 2.26 | 0.085 | 0.636 | 0.204 | 12.37 |
| Location | Depth Range (m) | δ13C (‰) | 87Sr/86Sr |
|---|---|---|---|
| XZ1 | 9.75–10 (S1) | −6.8 | 0.715481 |
| XZ1 | 19.75–20 | −4.8 | 0.715244 |
| XZ1 | 29.5–30 (S2) | −6.9 | 0.716614 |
| XZ1 | 39.5–40 | −5.2 | 0.715639 |
| XZ1 | 52–52.5 | −5.8 | 0.715851 |
| XZ1 | 54.5–55 (S3) | −3.3 | 0.715873 |
| XZ2 | 9.75–10 | −8.3 | 0.716586 |
| XZ2 | 19.75–20 | −5.7 | 0.715136 |
| XZ2 | 29.5–30 | −5.2 | 0.717155 |
| XZ2 | 44.5–45 (S4) | −7.2 | 0.716909 |
| Item | Unit | Min. | Max. | Average | Median |
|---|---|---|---|---|---|
| TDS * | mg/L | 225 | 316 | 288 | 289 |
| pH * | - | 7.6 | 8.2 | 7.9 | 7.8 |
| Cl * | mg/L | 4.0 | 10.9 | 5.5 | 5.3 |
| SO4 * | mg/L | 3.2 | 13.1 | 6.8 | 6.9 |
| NO3 * | mg/L | 6.5 | 19.2 | 12.4 | 11.9 |
| HCO3 * | mg/L | 246 | 357 | 322 | 324 |
| F * | mg/L | 0.3 | 0.6 | 0.4 | 0.4 |
| Br * | mg/L | 0.019 | 0.029 | 0.025 | 0.027 |
| Na * | mg/L | 18 | 84 | 35 | 28 |
| K * | mg/L | 0.5 | 1.8 | 1.0 | 1.0 |
| Mg * | mg/L | 11 | 28 | 21 | 23 |
| Ca * | mg/L | 22 | 56 | 45 | 49 |
| δ13C-DIC * | ‰ | −9.6 | −8.1 | −8.7 | −8.7 |
| 3H * | TU | <0.4 | <0.4 | <0.4 | <0.4 |
| 14C age * | years | 220 | 19220 | 9990 | 12424 |
| saturation indices (SI) | calcite | 0.20 | 0.83 | 0.44 | 0.43 |
| dolomite | 0.36 | 1.67 | 0.83 | 0.78 | |
| fluorite | −2.14 | −1.65 | −1.90 | −1.91 | |
| gypsum | −3.47 | −2.59 | −2.97 | −2.90 | |
| magnesite | −0.41 | 0.26 | −0.17 | −0.21 | |
| strontianite | −1.44 | −0.62 | −1.04 | −1.03 | |
| Sr | mg/L | 0.278 | 0.646 | 0.489 | 0.516 |
| 87Sr/86Sr | - | 0.710677 | 0.712319 | 0.711207 | 0.711157 |
| Sample | Ca mg/L | Mg mg/L | Na mg/L | K mg/L | Sr μg/L | 87Sr/86Sr | CaCO3 (%) | MgCO3 (%) | CaMg(CO3)2 (%) |
|---|---|---|---|---|---|---|---|---|---|
| S1-1 | 5870 | 161 | 11.7 | 3.7 | 8304 | 0.710750 | 14.68 | 0.56 | 0.76 |
| S1-2 | 485 | 117 | 8.7 | 4.4 | 785 | 0.710818 | |||
| S2-1 | 5260 | 147 | 12.4 | 4.0 | 6394 | 0.711038 | 13.15 | 0.51 | 0.48 |
| S2-2 | 406 | 72 | 8.2 | 4.2 | 683 | 0.710969 | |||
| S3-1 | 5770 | 127 | 11.9 | 5.1 | 8081 | 0.710455 | 14.43 | 0.44 | 1.43 |
| S3-2 | 691 | 219 | 8.2 | 4.3 | 896 | 0.710329 | |||
| S4-1 | 7445 | 128 | 12.7 | 5.5 | 5111 | 0.711085 | 18.61 | 0.45 | 0.47 |
| S4-2 | 1034 | 76 | 8.4 | 6.0 | 996 | 0.710958 |
| Sample | Quartz | Potassium Feldspar | Plagioclase | Calcite | Dolomite | Clay | 87Sr/86Sr |
|---|---|---|---|---|---|---|---|
| S1-res | 51.2 | 4.0 | 19.2 | 0 | 0 | 25.6 | 0.719091 |
| S2-res | 54.9 | 2.6 | 13.4 | 0 | 0 | 29.1 | 0.720415 |
| S3-res | 53.5 | 1.9 | 16.2 | 0 | 0 | 28.4 | 0.719876 |
| S4-res | 54.0 | 2.2 | 11.8 | 0 | 0 | 32.0 | 0.720438 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, T.; Ma, B. The Origin of Major Ions of Groundwater in a Loess Aquifer. Water 2019, 11, 2464. https://doi.org/10.3390/w11122464
Huang T, Ma B. The Origin of Major Ions of Groundwater in a Loess Aquifer. Water. 2019; 11(12):2464. https://doi.org/10.3390/w11122464
Chicago/Turabian StyleHuang, Tianming, and Baoqiang Ma. 2019. "The Origin of Major Ions of Groundwater in a Loess Aquifer" Water 11, no. 12: 2464. https://doi.org/10.3390/w11122464
APA StyleHuang, T., & Ma, B. (2019). The Origin of Major Ions of Groundwater in a Loess Aquifer. Water, 11(12), 2464. https://doi.org/10.3390/w11122464