1. Introduction
It is well known that different therapeutic classes of pharmaceuticals are used for the treatment of some diseases in the human body since they are biologically active compounds used for this purpose. However, the occurrence of some pharmaceuticals in surface water may be due to the bodily excretion of metabolized and un-metabolized pharmaceutical compounds into septic wastewater, which is then discharged to surface water. Although these concentrations are very low (ng/L), they are a big concern for their potential impact on the aquatic environment [
1]. In the environmental analysis of pharmaceuticals, many methods have been reported in literature using liquid chromatography (LC). The reason why LC was used in the analysis of these pharmaceuticals is related to the low volatility and high hydrophilicity of most of the pharmaceuticals. A gas chromatography instrument has also been used for the analysis of pharmaceuticals [
2,
3]; however, the derivatization of pharmaceuticals is needed to attain volatility and stability prior to the injection into the gas chromatography instrument [
4]. The quantification analysis of pharmaceuticals is challenging due to high interference with other organic pollutants and low concentrations present in real samples [
5]. Generally, a sample preparation method is required prior to instrumental analysis. A liquid-phase micro-extraction is a relatively newly developed extraction process consuming low solvent consumption [
6].
This procedure requires small volume, and it is more suitable for biological samples such as blood and urine. [
7]. So far, solid phase extraction is considered one of the most frequently employed extraction techniques in the analysis of pharmaceuticals in water, as it offers high selectivity, precision, and extraction efficiency [
8,
9,
10]. The most common solid phase extraction (SPE) materials that allow the retention of a wide variety of compounds are the copolymer poly (divinylbenzene-co-
N-vinylpyrrolidone) (Oasis HLB) that has both hydrophilic and lipophilic retention characteristics, and it can be used to retain both polar and non-polar compounds [
10,
11]. So far, very limited literature on the multi-residue analysis method for pharmaceuticals has been reported in Malaysia.
Therefore, the aim of this present study is to investigate the possibility of analyzing different therapeutic classes of pharmaceuticals in different bodies of water using a single solid phase extraction method by developing a very accurate and selective liquid chromatography-time of flight/mass spectrometry (LC-ToF/MS) method.
Hence, this study was conducted to develop a sensitive and accurate method for the determination of nine pharmaceuticals which are selected based on the national consumption report in Malaysia [
12].
The aim of this work is to develop and validate a comprehensive analytical LC-ToF/MS method that can simultaneously detect and quantify a wide spectrum of pharmaceuticals in water samples. One single extraction method is applied to investigate and quantify the studied pharmaceuticals in surface water, sewage treatment plant (STP) influent, STP effluent, and hospital effluent.
2. Materials and Methods
2.1. Consumption of the Pharmaceuticals
The general description of the studied pharmaceutical compounds was overviewed [
13]. Atenolol and metoprolol are called beta blockers, which are used for the treatment of high and low blood pressure and to prevent heart attack. A non-prescription compound also known as paracetamol, Acetaminophen is commonly used for its analgesic and antipyretic effects; its therapeutic effects are similar to salicylates. The non-prescription pharmaceutical stimulant xanthine compounds caffeine and theophylline are included in this present study. Theophylline is used to relax the muscles in the airway, making breathing easier, while caffeine is responsible for the stimulation of the central nervous system in the body.
Sulfamethoxazole is an antibacterial used to reduce the impact of bacterial synthesis of dihydrofolic acid. Prednisolone is one of the steroid compounds used to help reduce the symptoms of asthma, such as wheezing in children. Ketoprofen is nonsteroidal anti-inflammatory drug, which is used for the symptomatic treatment of acute and chronic rheumatoid arthritis. Glibenclamide is called glyburide, an antidiabetic drug, which is used to reduce the blood glucose in patients with non-insulin-dependent diabetes mellitus (Type II diabetes). In Malaysia, the Ministry of Health annually publishes a statistical report on drug consumption.
Table 1 presents the defined daily doses (DDD) of the studied pharmaceuticals per thousand inhabitants between 2011 and 2014 in Malaysia. The DDD values are based on the Anatomical Therapeutic Chemical (ATC) classification system by the World Health Organization (WHO) [
14]. The annual consumption of these pharmaceuticals can be calculated using the following formula:
where DDD is the defined daily dose and DDD/1000 inh is the number of daily doses consumed per 1000 inhabitants in one year.
It was observed from
Table 1 that acetaminophen has the highest consumption levels during the four years compared to the other pharmaceutical compounds. Furthermore, it could be considered an over-the-counter drug, and it is consumed in three different ways: orally, parenterally, and rectally.
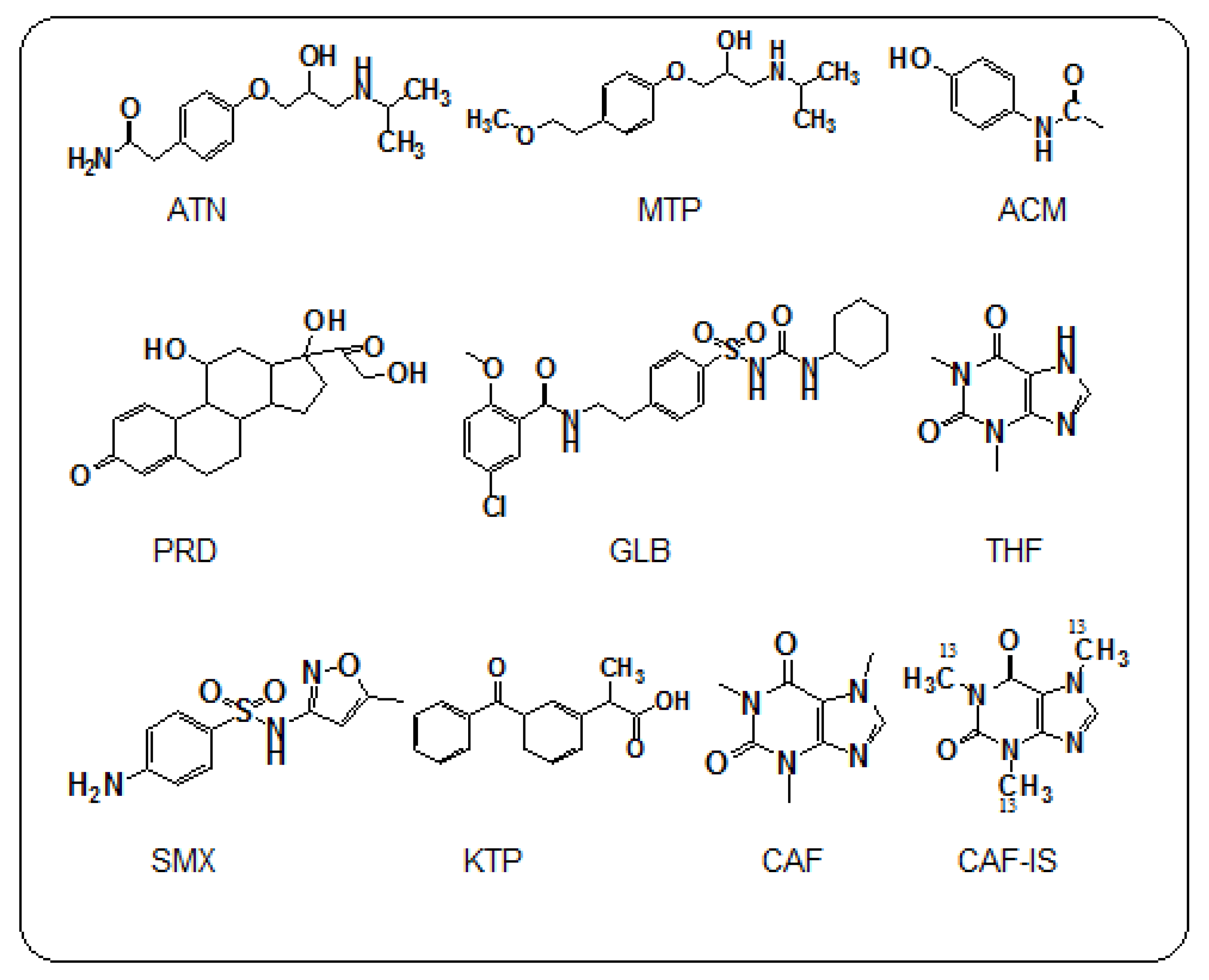
All of the studied pharmaceuticals have been presented in
Figure 1. Non-prescription pharmaceutical compounds, such caffeine and theophylline, were selected for their prevalence in very commonly consumed drinks, such as tea, coffee, milo, and Pepsi, and, furthermore, due to their frequent detection as reported in previous studies [
11,
15,
16,
17,
18,
19]. Acetaminophen was selected as a non-prescribed and/or prescribed pharmaceutical compound.
2.2. Reagents and Materials
All standards were pure with (≥98%) of atenolol, acetaminophen, theophylline, caffeine, caffeine-13C3 (internal standard, IS), metoprolol, sulfamethoxazole, prednisolone, ketoprofen, and glibenclamide, which were obtained from Sigma-Aldrich (St. Louis, MO, USA). Deionized water (DIW) was collected from the water analysis and research lab at Universiti Kebangsaan Malaysia. HPLC-grade methanol (MeOH), acetonitrile (ACN), methyl tertiary butyl ether (MTBE), acetone, and formic acid (FA) were supplied by Merck (Darmstadt, Germany). The cartridges used for SPE were Oasis HLB (3cc, Waters, Milford, MA, USA).
2.3. Sample Collection
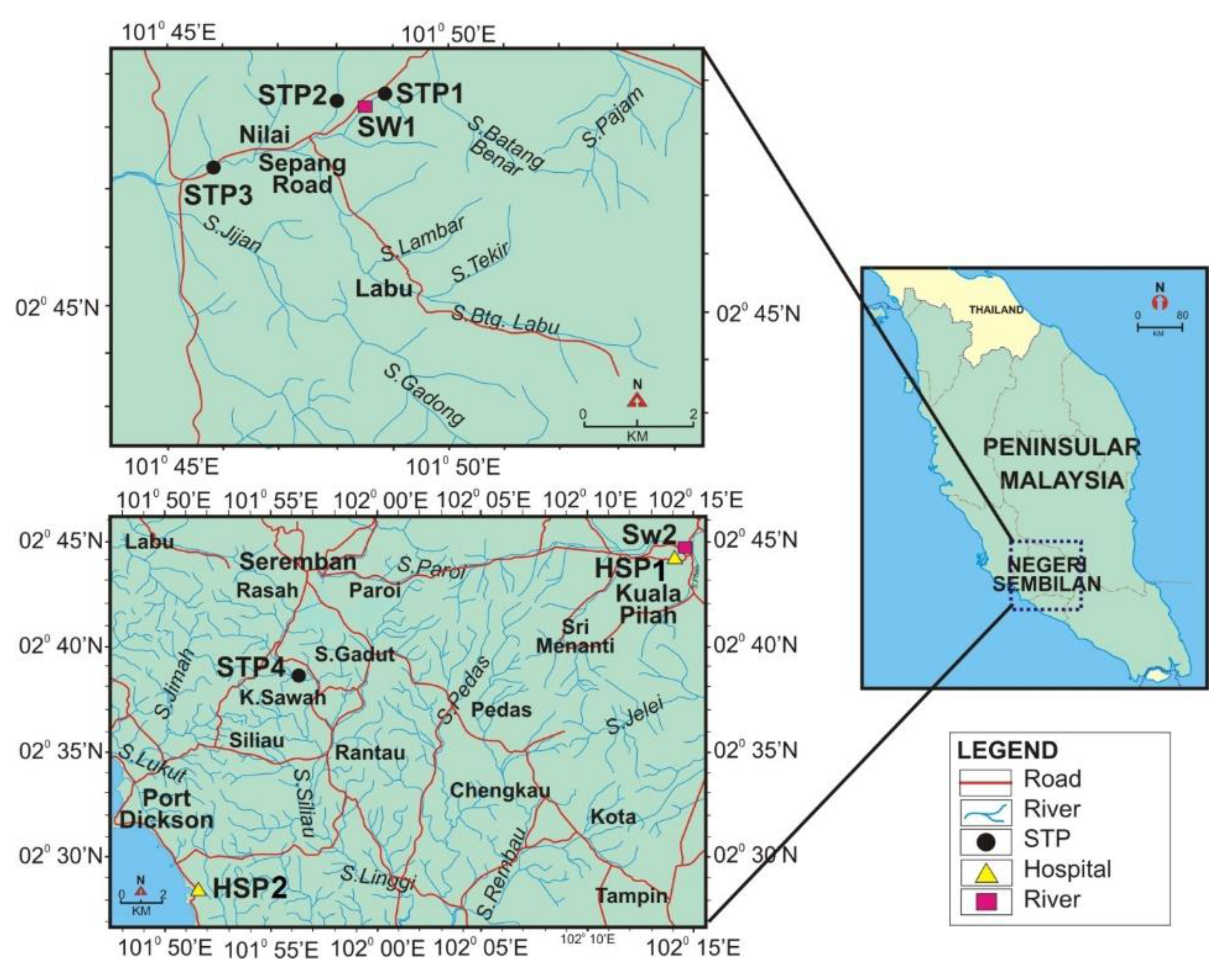
All samples were collected from Nilai and Seremban, Malaysia, and then shipped to the laboratory on the same day. Eight points, as shown in the map (see
Figure 2), were chosen to study the fate of nine pharmaceutical compounds. Samples were collected from four STPs (STP1, STP2, STP3, and STP4) and two hospitals (HSP1 and HSP2). Samples were also collected from the recipient rivers at two points (SW1 and SW2). The treatment process in all STPs was an oxidation ditch, while it was a rotating biological contractor in the hospitals. The frequency of sampling was for three months in 2014. Samples were collected on the same day, within three hours in the morning, at a fixed volume (1.0 L) for each point; the sampling interval was every month. One liter amber glass bottles were rinsed in the field twice before sample collection. A polyethylene plastic bucket was used to collect wastewater samples and fill the glass bottles. All safety was taken into account during sampling. The sampler used disposable gloves to prevent any contamination by the personal care products from the sample. A plastic bucket was used to collect the samples. All samples were filtered by 0.7 µm GF/F filter (Whatman, Little Chalfont, UK) to remove any solid matter suspended in the samples. All filtered samples were kept at 4 °C until the solid phase extraction experiments.
2.4. Instrumental and Extraction Method
Separation of pharmaceuticals was performed on the liquid chromatography (LC) instrument (Dionex, Sunnyvale, CA, USA). 30 µL of sample was injected at 0.3 mL/min. All selected pharmaceuticals were analyzed in the positive mode (PI). Two mobile phases were used; (A) 0.1% FA in DIW and (B) ACN-MeOH (3:1, v/v) at 0.3 mL/min. The gradient elution is as follow:
5% B (0 min) → 60% B (linear increased in 3 min) → 97% B (linear increased in 3 min) → 97% B (hold 5 min) → 5% B (linear decreased in 0.1 min) → 5% B (hold 5 min).
All analytes were acquired using an independent reference spray via the LockSpray interference to ensure accuracy and reproducibility (mass spectrometry (MS) capillary voltages, 4000 (PI); drying gas flow rate, 8.0 L/min; drying gas temperature, 190 °C; and nebulizer pressure, 4.0 bar). A mixture of sodium hydroxide and FA was used as the lock mass m/z 90.9766 to 974.8132. Accurate masses were calculated using the software Daltons Data Analysis incorporated in the instrument. Samples of 500, 250, 100 mL from surface water, sewage treatment plant and hospital effluent, and sewage treatment plant influent, respectively, were filtered by 0.7 µm GF/F filter (Whatman, UK) to remove any solid matter suspended in the samples. All filtered samples were kept at 4 °C until solid phase extraction. The sample extraction method was provided using Oasis hydrophilic-lypophilic balanced (HLB) (3 cc, 60 mg) cartridges (Waters, Milford, MA, USA). To achieve all SPE experiments, a vacuum manifold was used for this extraction procedure. SPE cartridges were pre-conditioned with 2 mL of methanol and 2 mL of deionized water (DIW) before sample loading. Water samples were loaded at a flow rate of 3 mL/min under vacuum. To exclude water residue from the cartridge, it was dried under vacuum for 15 min. After that, analytes were eluted by passing 5 mL of MTBE and 5 mL of (MeOH-ACN, 50:50, v/v). Then, eluents were dried by flowing a stream of nitrogen gas. A 0.5 mL of solvent was added to reconstitute the extracted analytes, which were filtered by 0.45 µm (Nylon syringe) before injection. Each experiment was repeated three times to find the precision of the injection using LC-ToF/MS. Individual stock standard solutions and caffeine 13C3 as an internal standard solution (1000 µg/mL) were prepared in MeOH by dissolving 0.01 g of compound in 10 mL of methanol. Stock solutions were kept at −20 °C until further experiments. Working solutions were prepared by serial dilutions of stock standard solution with MeOH-DIW (1:9, v/v) solvent.
2.5. Method Validation
Selectivity is the ability of an analytical method to differentiate and quantify the targeted compounds in the presence of other sample components [
20]. The method selectivity was investigated by analyzing a blank solvent sample MeOH:DIW (10:90,
v/
v), an effluent STP sample spiked with nine pharmaceuticals and one IS, and an effluent STP un-spiked sample. It was observed that the LC-ToF/MS method has a good ability to select the target compounds from different samples. To assess intra-day precision, three concentrations (8, 40 and 200 ng/mL) of mixture compounds were injected three times using liquid chromatography–time of flight/mass spectrometry. To assess inter-day precision, samples were analyzed with the same above concentrations on three separate days. Five replicates (
n = 5) were performed on the same day (intra-day precision) and at different days (inter-day precision). Recovery was investigated in different samples: surface water, the influent of a sewage treatment plant, and the effluent of a sewage treatment plant. Standard solution mixtures of 1 and 10 ng/mL (
n = 5) were spiked in the samples and extracted using solid phase extraction. The recoveries were evaluated by comparing the peak area of the extracted samples to the peak area of the standard solutions. The recoveries (R%) were calculated based on the following formula:
where A
SP is the peak area of a compound in an extract, A
UN is the peak area of a compound in a sample, and A
s is the peak area of a compound in the standard solution.
Linearity was investigated by generating the calibration curve for each analyte. Four to five points of calibration curves were generated by injecting mixture solutions prepared from the standard stock solution. Concentrations used to create the calibration curves ranged from each analyte’s instrumental quantification limit (IQL) up to 400 ng/mL. Calibration curves were generated for each compound by plotting the peak area against the concentration of each compound using the linear regression model. The determination coefficient,
R2 ≥ 0.993, was obtained for all analytes. The instrumental quantification limit (IQL) was the lowest concentration corresponding to the signal-to-noise ratio (S/N) ratio ≥ 10. The limit of quantification (LOQ) for the whole method in the different matrices was estimated using the following formula [
21]:
where IQL is the instrumental quantification limit (ng/L), R (%) is the recovery of the compound, and CF is the concentration factor which corresponds to 2000, 1000, 500, and 200 for drinking water, surface water, STP and HSP effluent, and STP influent, respectively. The identification and quantification of pharmaceutical compounds was based on retention times (Rt) and mass value (
m/z) for each analyte. A 0.02 Da narrow window was applied for all analytes to be extracted and quantified in real samples.
2.6. Matrix Effects
The matrix effect (ME%) was evaluated based on the signal intensity of the analytes in a sample. However, it was calculated according to this procedure: Sample extracts of STP influent and effluent were spiked at a level of 10 ng/mL of pharmaceuticals (
n = 3), and then it could be injected to LC–ToF/MS. The following formula was used to estimate the matrix effect.
where A
S is the peak area of the compound in the standard solution, A
SP is the peak area of the compound in the extract of the sample, and A
UN is the peak area of the compound in the un-spiked extract.
3. Results and Discussion
Atenolol, metoprolol, sulfamethoxazole, prednisolone, ketoprofen, and glibenclamide were selected as the top prescribed pharmaceuticals in Malaysia [
12]. The most commonly used non-prescription drugs were acetaminophen, theophylline, and caffeine. An example of chromatographical separation (
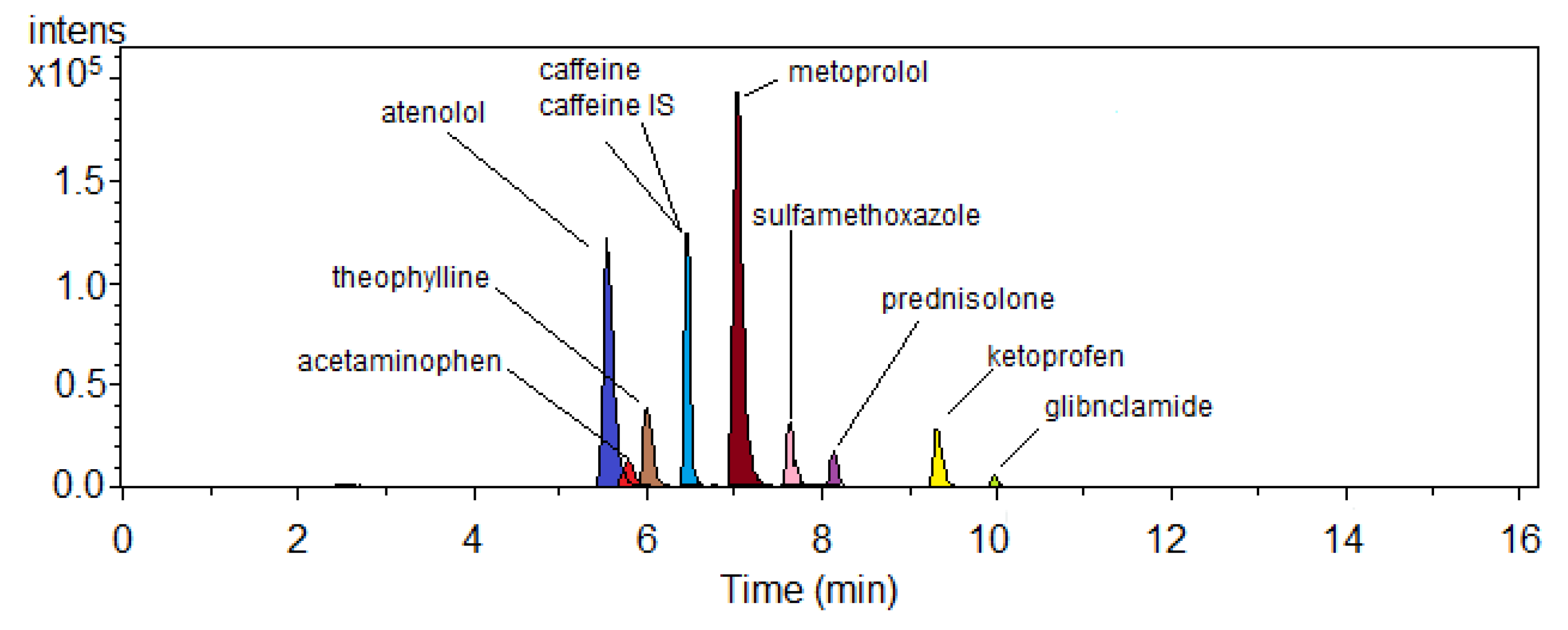
Figure 3) was provided by using a gradient elution program as described in the previous section. The intensity of the pharmaceutical compounds varied strongly. This variation may be due to the diversity of physico-chemical properties among the selected pharmaceuticals under electrospray ionization conditions. However, the LC-chromatogram was more sufficient for analysis of the studied pharmaceuticals at the expected ambient environmental concentrations.
3.1. Elution Solvent Effect
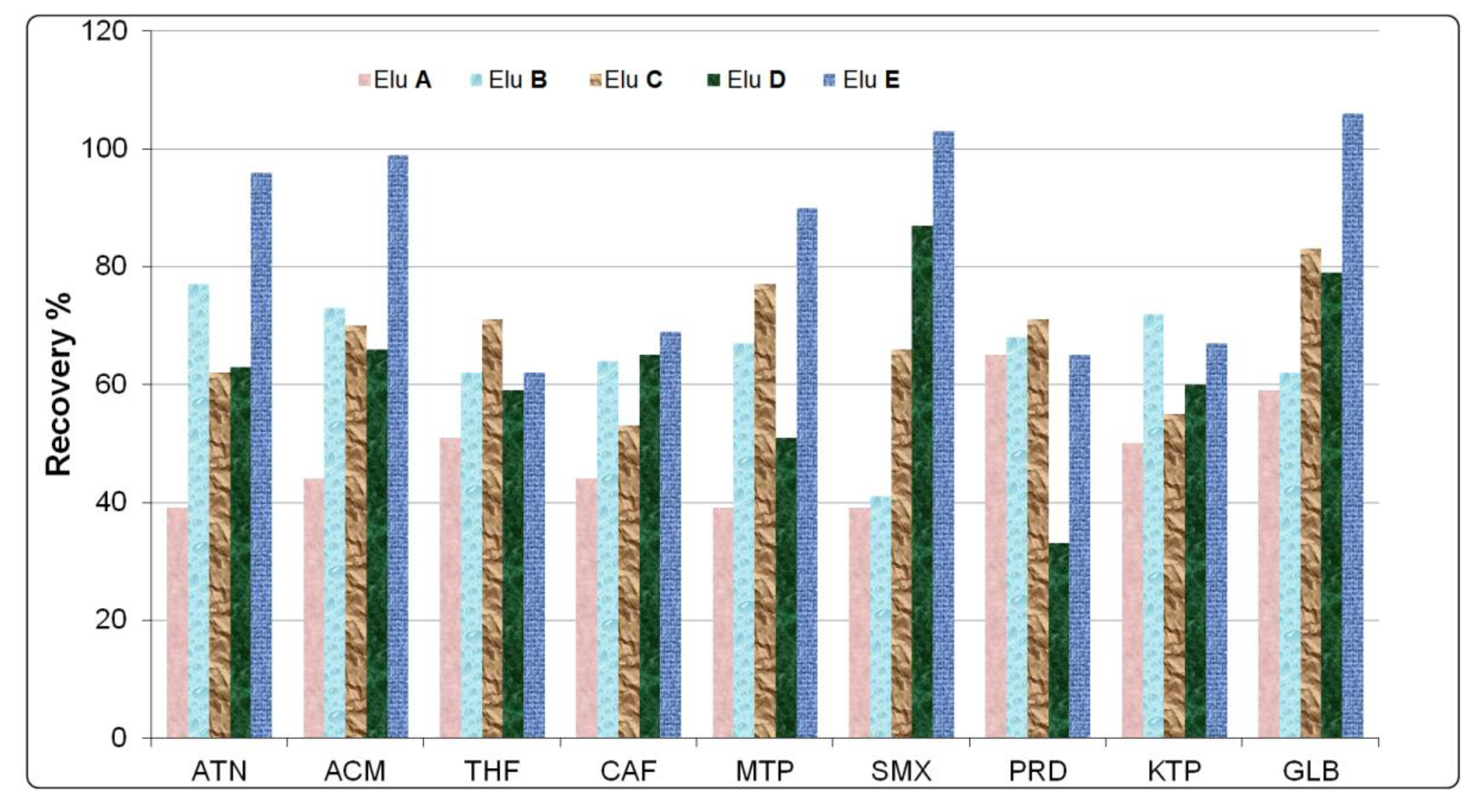
The variety of physico-chemical properties of pharmaceutical compounds provided varying results among the elution solvents tested in the preliminary experiments. The best overall solvent elution recoveries were achieved using 5 mL of MTBE + 5 mL of methanol:acetone (50:50,
v/
v) without a pH adjustment of the sample. Recoveries for the compounds tested are shown in
Figure 4. It is well known that extraction of pharmaceuticals from the Hydrophilic-Lipophilic Balance sorbent (HLB-oasis cartridge) could be impacted by the polarity and non-polarity of the solvent and depends on the type of analytes. Various elution solvents combining methanol, acetone, and methyl tertiary butyl ether have been tested in this study. These elution solvents include the following: Elu
A: 10 mL of MeOH, Elu
B: 10 mL of acetone, Elu
C: 10 mL of methanol:acetone solution (50:50,
v/
v), Elu
D: 10 mL of MTBE, and Elu
E: 5 mL of MTBE + 5 mL of methanol:acetone (50:50,
v/
v). On average, the analytes were recovered by 84% with eluent
E, 63% with eluent
D, 67.5% with eluent
C, 65.1% with eluent
B, and 48% with eluent
A.
Slightly-polar compounds, such as atenolol, acetaminophen, metoprolol, sulfamethoxazole, and glibenclamide, were recovered at less than 60% with methanol as the elution solvent (A). By reducing the polarity of the solvent, it was observed that most of the compounds were well recovered. A polar–nonpolar elution solvent, such as eluent E, was the best choice in this present study. Low recovery for slightly-polar pharmaceutical compounds may be attributed to the poor elution from the HLB sorbent or poor retention on the sorbent, whilst most of compounds were very well recovered (≥90%) using eluent E (5 mL of MTBE + 5 mL of methanol: acetone (50:50, v/v)). Recoveries of less than 50% were observed for the prednisolone and ketoprofen in the presence of eluent E. Theophylline and caffeine, in the same way, are believed to be poorly retained in the polymeric sorbent without pH adjustment. However, this low recovery is not an obstacle to quantify theophylline and caffeine in real samples, as they have a reliably low limit of quantification.
3.2. ToF Screening and Confirmation
Future strategies in LC-ToF/MS method development would include the use of electrospray ionization modes to enhance detection methodology. In addition, the development of good chromatographical separation using different mobile phases (data not shown) provides a highly sensitive and selective method to separate and quantify the compounds in real samples.
Two types of ionization, either positive or negative modes, were optimized. However, positive ionization (PI) was selected to monitor and quantify the analytes in the samples. It was found that, at the PI mode, the S/N ratio for all selected pharmaceuticals was the highest (data not shown). Two compounds, ketoprofen and glibenclamide, were also identified in the negative ionization (NI) mode, but their intensities were very low. Thus, all pharmaceutical compounds were analyzed in positive ionization mode.
The low detection limit and the possibility of interference with other organic pollutants in a real sample that has mass-to-charge value close to that of studied pharmaceuticals are one of the most challenging fields in quantitative analysis. In order to reduce this challenge and to increase the selectivity of ToF/MS measurements, a narrow, accurate mass interval was used to reconstruct the chromatographic traces levels. Extracted ion chromatograms (EIC) were typically extracted using a 0.02 Da for all studied pharmaceuticals. However, reducing the mass window resulted in an enhancement of the detection limit in influent and effluent sewage treatment plants and a complete loss of interferences from contaminants.
Petrovic et al. observed that reducing the mass window from 100 to 20 mDa resulted in an almost 15-fold increase of the signal-to-noise ratio and in an almost complete loss of the interferences from the isobaric contaminant ions for carbamazepine in urban wastewater [
22].
In all cases, the accurate mass of the protonated [M + H]
+ molecular ions were applied for confirmation and quantification purposes. Accurate mass data for the molecular ions was processed through the software Brucker Daltons Data Analysis, which provided the elemental formula and mass errors.
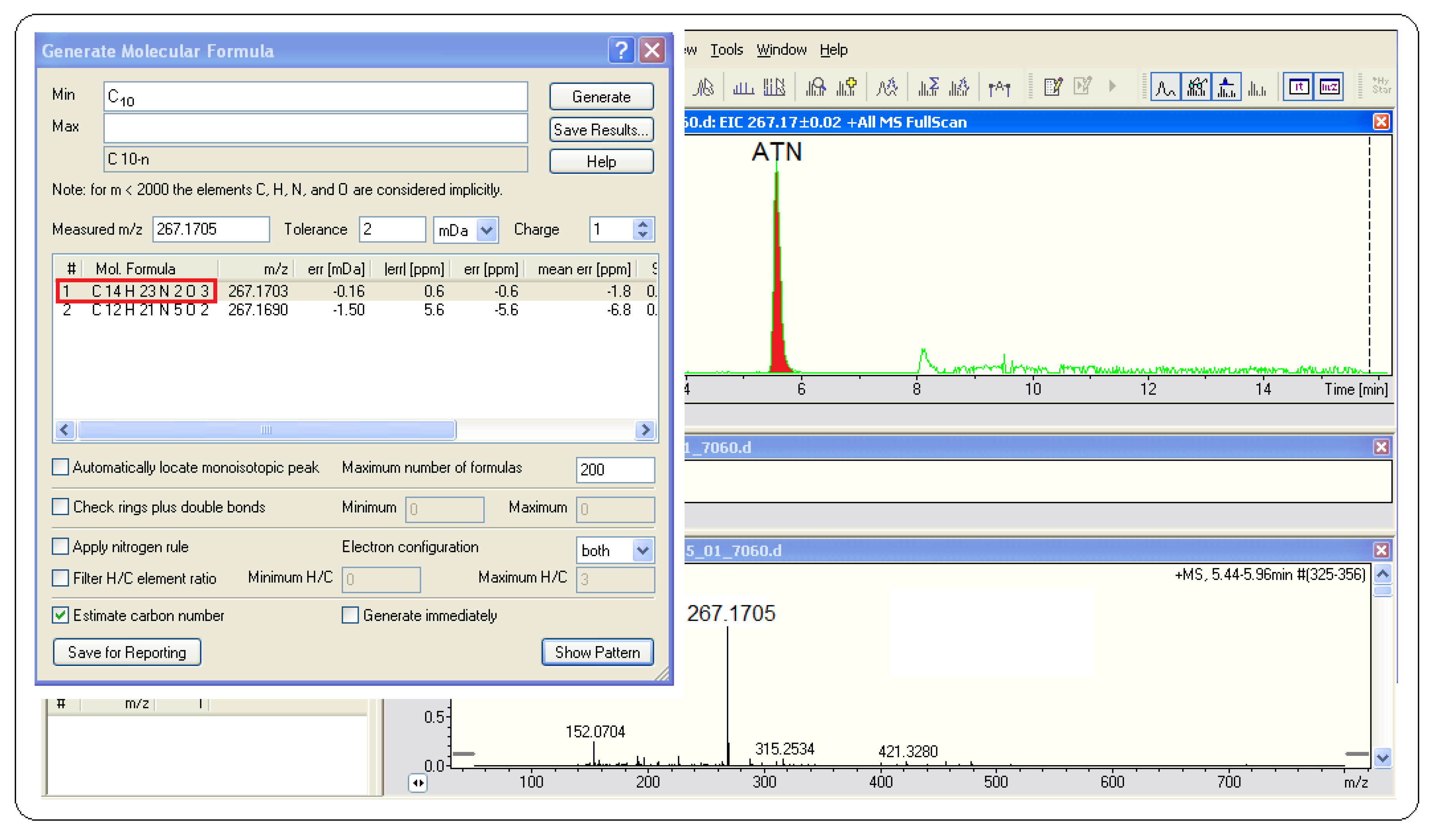
Figure 5 shows an example of atenolol analysis in the influent of sewage treatment plants using Brucker software. It could be observed that the elemental formula (C
14H
23N
2O
3) has −1.8 ppm, which is an accurate value to confirm that this formula belongs to atenolol. The other exact mass measurements, retention times, elemental composition, and mass errors were presented in
Table 2.
3.3. Method Validation
The linearity of the external calibration curve method ranged from IQL to 400 ng/mL for all compounds. It was observed from
Table 3 and
Figure S1 that (4–5) points were generated to achieve correlation coefficients (
R2) ≥ 0.993 using linear regression. The IQL for each analyte was determined using pure standards that were analyzed using the LC–ToF/MS method. The IQL was determined to be the concentration with an
S/N ratio ≥10. A wide range of IQLs were obtained, because they depend on the sensitivity of the instrument and the ionization efficiency of the analyte in an electrospray ionization (ESI) source. The IQLs for all pharmaceuticals ranged between 0.3 and 8 ng/mL except for prednisolone, which was 40 ng/mL. This high value is not an obstacle for developing the method since most pharmaceuticals have very good IQLs compared to prednisolone. The LOQs over the entire method were calculated using Equation (1), in which the concentration factors and matrix effects of different environmental samples were considered. In drinking water (DW), the LOQ ranged between 0.3 and 8.2 ng/L. In effluent of STP, the LOQ ranged between 6.5 and 50.3 ng/L, whereas the LOQ ranged between 11.1 and 83 ng/L in influent of STP. The findings in this present study were in agreement with our previous studies [
11,
19].
The precision of the method was evaluated based on the results of the analysis of three concentrations (8, 40, and 200 ng/mL) with three replications for each one on the same day and the results from inter-day precision from the other three different days. The values were compared with the standards; thus, all values demonstrated good results with RSD% ≤ 6.7% for intra-day precision and 11.7% for inter-day precision (see
Table 4). Recoveries of the solid phase extraction method were compared to the recoveries from drinking water, surface water, and STP influent and effluent samples. Five samples of surface water, STP influent, and STP effluent were spiked at 1 and 10 ng/mL, and then extracted using HLB sorbent and eluent
E. These set spikes are used to evaluate method performance over different matrices. CAF-
13C
3, as an internal standard, was spiked at the same concentrations to evaluate the relative recovery for caffeine as one of the selected compounds in this study. The absolute and relative recoveries are presented in
Table 5. Recoveries for SPE trials, extracted and analyzed in triplicate, ranged from 30.7 to 79.6% in STP influent, 37.4 to 82.4% in STP effluent, and 41.2 to 86.4% in surface water at 1 ng/mL spiking level with a mean of 54, 61, and 69%, respectively. For 10 ng/mL spiking level, recoveries were better and ranged from 50.1 to 100.6% in STP influent, 61.2 to 106.9% in STP effluent, and 66.8 to 109.6% in surface water.
The means of the recoveries were 77, 83, and 88% for STP influent, STP effluent, and surface water, respectively. Lower recoveries for the prednisolone and ketoprofen ranged from 30.7 to 41.2% and 40.6 to 48.8%, respectively, at 1 ng/mL in all samples; these are likely attributable to the unsuitability elution with eluent E as a non-polar to polar solvent. Although prednisolone and ketoprofen exhibited low recoveries, other pharmaceutical compounds were recovered well in this extraction method. In comparison to other previous studies, the recovery results were not considered surprising compared to those pharmaceutical compounds that recovered between 10 and 15% in wastewater samples, as reported by Ferrer et al. [
23]. In the same way, Shaaban et al. reported that few compounds were recovered between 12.7 and 32.2% at a 100 µg/L spiking level [
24]. Thus, this method could be acceptable for extracting all nine pharmaceuticals using a single solid phase extraction cartridge. All pharmaceuticals were eluted within 16.1 min, with atenolol as the first elute and glibenclamide as the last. A perfect chromatogram of nine pharmaceuticals and one internal standard spike in STP effluent is presented in
Figure 6.
3.4. Matrix Effect
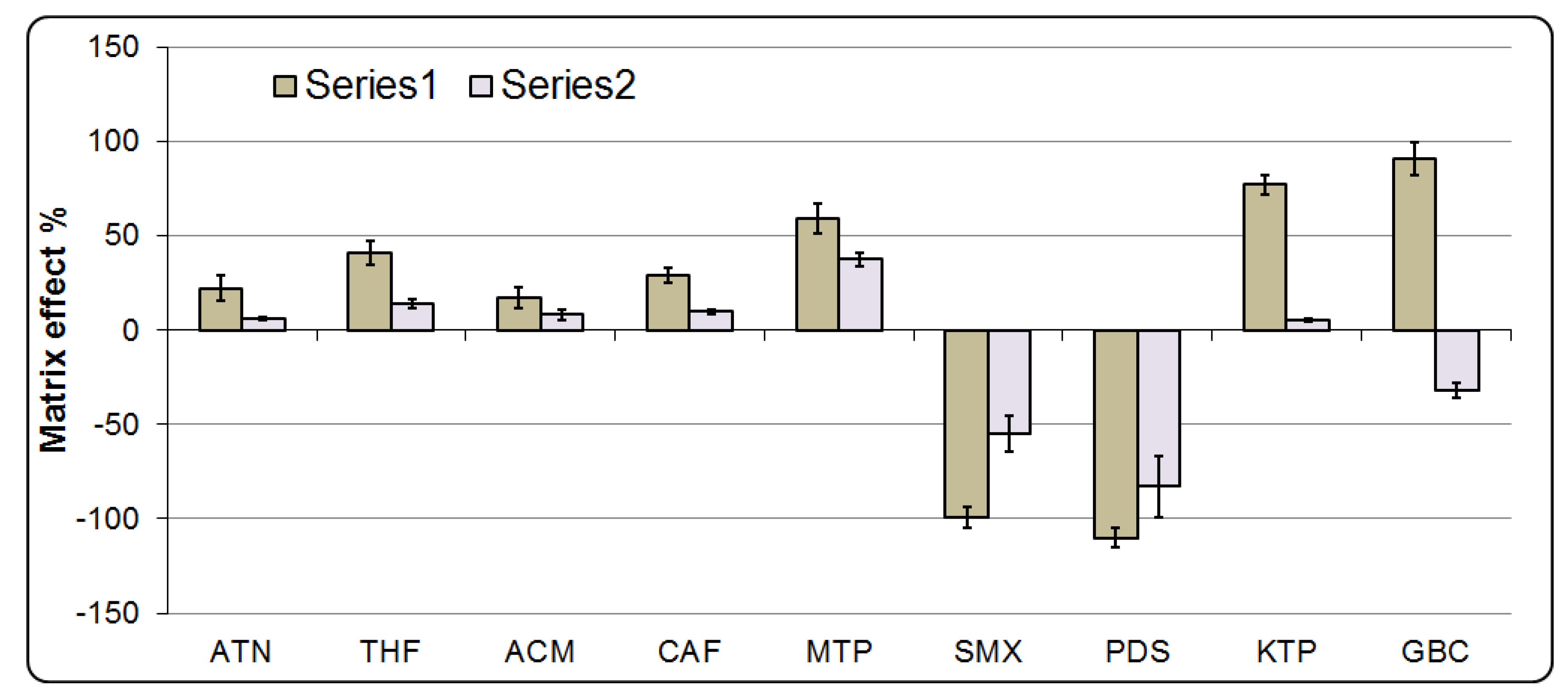
As the developed sample preparation procedure involves an extraction process, ion suppression or enhancement is assessed through spiking the extracted influent and effluent of STP before injection to LC-ToF/MS at 10 ng/mL (see
Figure 7). The matrix effect was evaluated as an enhancement or suppression according to Equation (3). Some pharmaceuticals at higher portions of acetonitrile (Rt > 7 min) were affected, in which signal suppression was 77 and 91% for ketoprofen and glibenclamide in influent of STP, respectively. Signal enhancement was also observed to be found at −99 and −110% for sulfamethxazole and prednisolone, respectively. These results indicate that the organic pollutants present in a matrix that elute at higher proportions of acetonitrile could suppress and/or enhance the ionization of the pharmaceutical compounds eluting at retention times longer than 7 min. The reason is related to the fact that at this time the polarity of elution mobile phase increases according to the elution program. The same phenomenon has been previously reported by Hernando et al. [
25]. The matrix effect is highly dependent on the chromatographic gradient elution and the composition of the mobile phase; however, it was reported that some pharmaceuticals eluting at the beginning of the LC gradient were more heavily affected by the matrix effect as well [
25].
3.5. Occurrence of Pharmaceuticals in Water Samples
The LC-ToF/MS method described herein has been applied to samples collected from surface water, STP influent and effluent, and HSP effluent in Malaysia. The pharmaceuticals detected frequently by this method were six compounds (see
Table 6), and some of them were presented in
Figure S2. The most frequently detected compounds were non-prescription compounds, such as acetaminophen (75%), theophylline (100%), and caffeine (83.3%). Mean concentrations measured were 74 ng/L for acetaminophen, 38 ng/L for theophylline, and 540 ng/L for caffeine; the highest concentrations for these compounds were 110, 60, and 821 ng/L, respectively, in surface water. For the influent of sewage treatment, the mean concentrations measured were 3305, 1805, and 3900 ng/L for acetaminophen, theophylline, and caffeine, respectively; the highest concentrations for these compounds were 4919, 2722, and 8700 ng/L, respectively. All non-prescription pharmaceutical compounds were detected in all samples (100%).
For the effluent of sewage treatment plants, only theophylline was completely frequently detected (100%). Acetaminophen and caffeine were detected at 75% and 50%, respectively. The highest mean concentration measured, 360 ng/L, was for caffeine. The maximum concentrations for acetaminophen, theophylline, and caffeine were 122, 108, and 1190 ng/L, respectively. The frequency of detection was 100% for theophylline and caffeine and 50% for acetaminophen in hospital effluent, with the highest maximum concentrations of 3314, 628, and 2860 ng/L, respectively. Actually, the frequent detection of caffeine and theophylline in water samples is not surprising, as it is widely available in many drinks such as tea, coffee, cocoa, sport drinks, and soft drinks. Al-Qaim et al. reported that caffeine was detected in different beverages and tea drinks in Malaysia [
26]. Pedrouzo et al. and Huggett et al. reported that caffeine was detected at a maximum concentration, reaching up to 9945 and 1056 ng/L, in wastewater and surface water, respectively [
27,
28]. In the same way, acetaminophen (prescribed and/or non-prescribed drug) was detected at a high concentration as well in all water samples. This finding may be due to its high levels of consumption by people as a therapeutic drug. However, the mean consumption of acetaminophen from 2011 to 2014 was 261,577 kg/y.
Six prescribed pharmaceutical compounds were less frequently present in water samples. Two prescribed pharmaceuticals, prednisolone and glibenclamide, were not detected in all samples, while they are considered the top consumed pharmaceuticals in Malaysia. The reason may be attributed to their low consumption of 613 and 475 kg/y within four years (2011–2014). Ketoprofen and sulfamethoxazole were not listed as the top consumed pharmaceutical compounds from 2011 to 2014. Ketoprofen was also not detected in all samples. Although sulfamethoxazole was not listed as a top consumed compound in Malaysia, it was detected with 75% in STP influent and effluent. The mean concentration for sulfamethoxazole ranged between 52 and 650 ng/L in STP influent and effluent. Atenolol and metoprolol were the most prescribed pharmaceuticals present in water samples, and both compounds are the top consumed pharmaceutical in Malaysia. Atenolol was consumed by the human body, averaging 8485 kg/y within four years; it was frequently detected in STP influent and effluent, hospital effluent, and surface water. The highest concentration detected for atenolol was 1009 ng/L in STP influent. The mean concentration ranged from 35 to 561 ng/L.
The concentration of atenolol was 273 ng/L in the River Taff sample and 2702 ng/L in the wastewater effluent sample in the UK [
29].
Metoprolol was frequently detected at 100%, 75%, 100%, and 100% in STP influent, STP effluent, HSP effluent, and surface water, respectively. The highest concentration detected for metoprolol was 606 ng/L in HSP effluent followed by 190 ng/L in surface water, 153 ng/L in STP influent, and 36 ng/L in STP effluent; however, these results were in line with the previous study [
30].
4. Conclusions
The analysis and determination of pharmaceutical compounds within sewage treatment plant influent and effluent, hospital effluent, and surface water using SPE and LC-ToF/MS has been observed to be practical and effective. The prescription and non-prescription pharmaceuticals most likely found in Malaysian wastewater and surface waters were investigated and determined. The method performance presented indicates that the SPE and LC-ToF/MS techniques applied to routine analysis of sewage treatment plant influent and effluent, hospital effluent, and surface water for pharmaceuticals is sensitive and accurate for the majority of compounds tested, with detection limits averaging 29, 16, 7, and 2 ng/L in STP influent and effluent, surface water, and drinking water, respectively.
Re-constituted concentration and sample preparation were achieved by a solid phase extraction method after optimization of elution solvent. All studied pharmaceuticals were analyzed in the positive ionization mode, and they were separated in 16.1 min. Caffeine-13C3 was applied as an internal standard to investigate the method extraction efficiency; however, recovery was quite acceptable, wherein the means of the most analyzed pharmaceuticals ranged 50.1 to 100%, 61.2 to 106.9%, and 70.2 to 109.9% in STP influent, STP effluent, and surface water, respectively. The matrix effect was high for most of the compounds, especially those eluted after 7 min.
The results showed that six out of nine pharmaceuticals, namely atenolol, acetaminophen, theophylline, caffeine, metoprolol, and sulfamethoxazole, were detected in STP influent, STP effluent, and surface water. However, the mean of concentration was 561, 3305, 1805, 3900, 78, and 308 ng/L for atenolol, acetaminophen, theophylline, caffeine, metoprolol, and sulfamethoxazole, respectively, in STP influent.
The highest prescribed compounds detected in water samples were atenolol and metoprolol, with levels of 1009 and 606 ng/L, respectively. Non-prescription pharmaceuticals, caffeine, acetaminophen, and theophylline, were detected frequently and at high concentrations of 8700, 4919, and 2722 ng/L, respectively, in STP influent.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}