Recent Advances in Atmospheric Chemistry of Mercury



Abstract
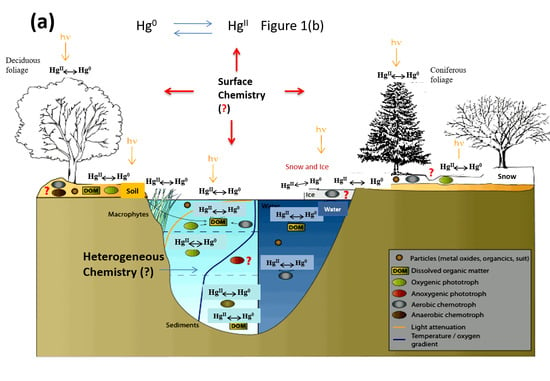
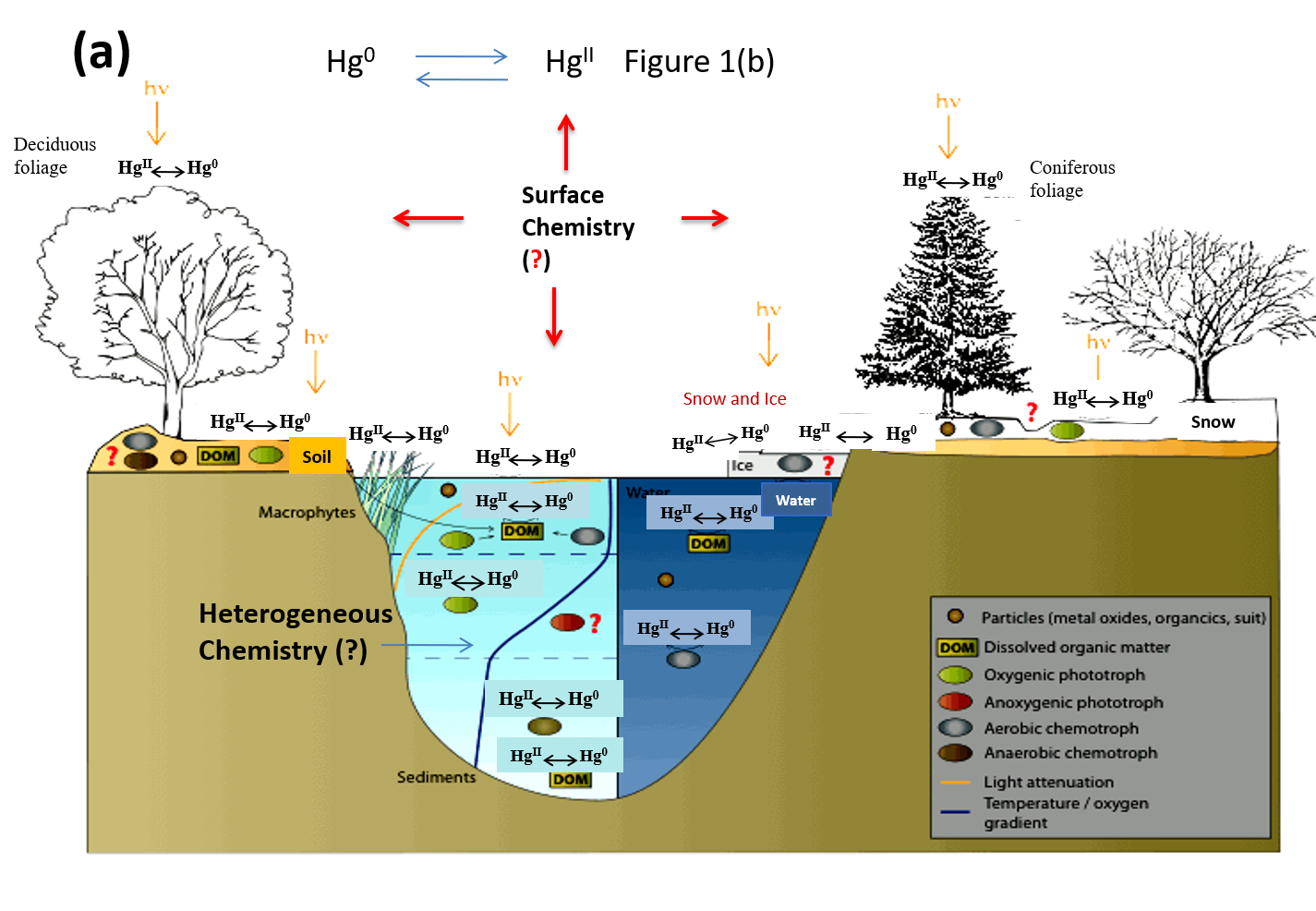
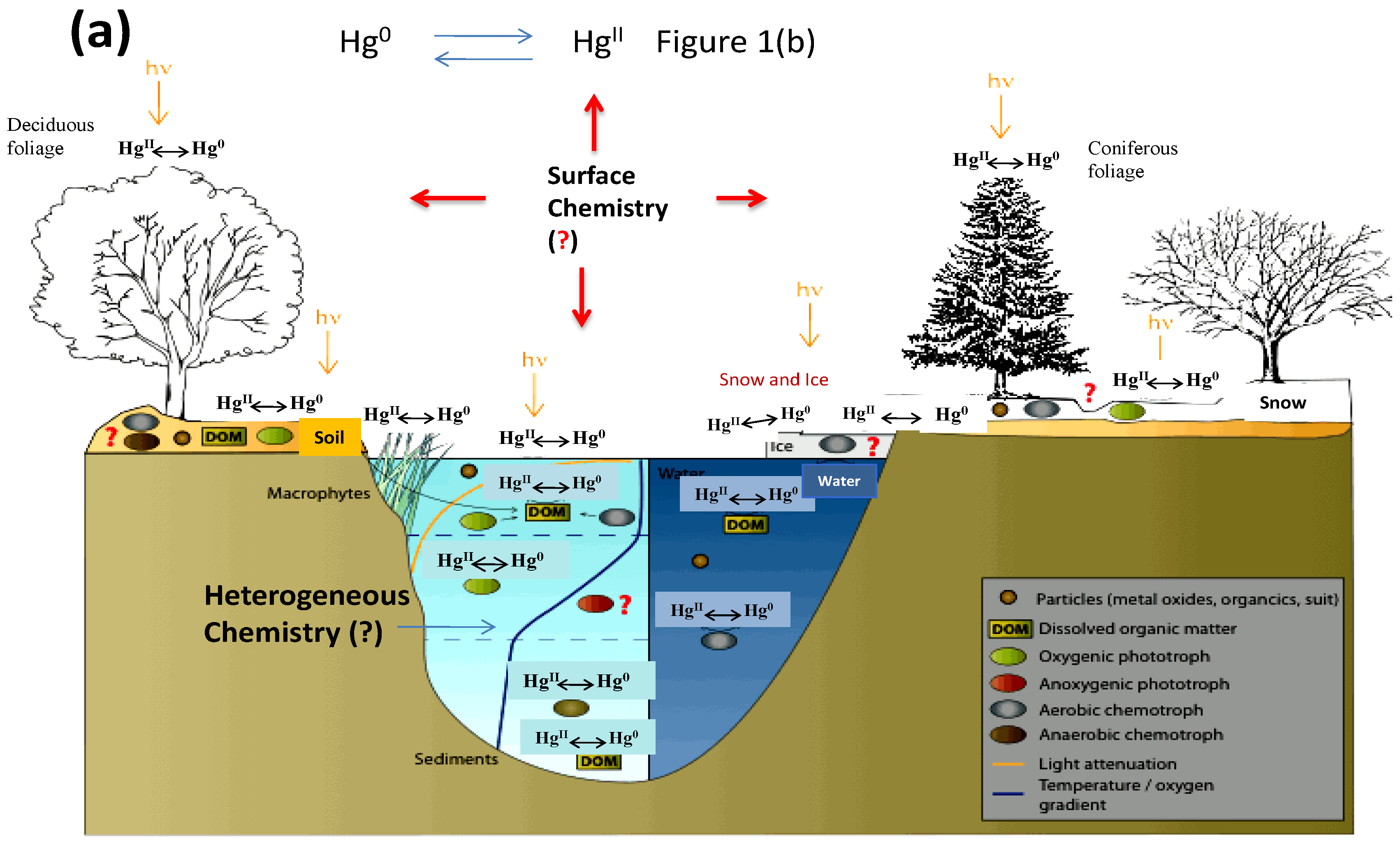
1. Introduction
2. Chemical Redox Pathways in the Gas Phase
2.1. Br-Initiated Oxidation of Hg0
2.2. Cl-Initiated Oxidation of Hg0
2.3. Oxidation of Hg0 by NO3
2.4. Dominant Gaseous Oxidant for Hg0: O3/OH, Br or Others?
3. Chemical Redox Reactions of Hg in the Aqueous Phase
3.1. Field Evidence for the Reduction of HgII
3.2. Photoreduction of HgII-Organic Complexes
3.3. Direct Reduction of HgII by Sulfite
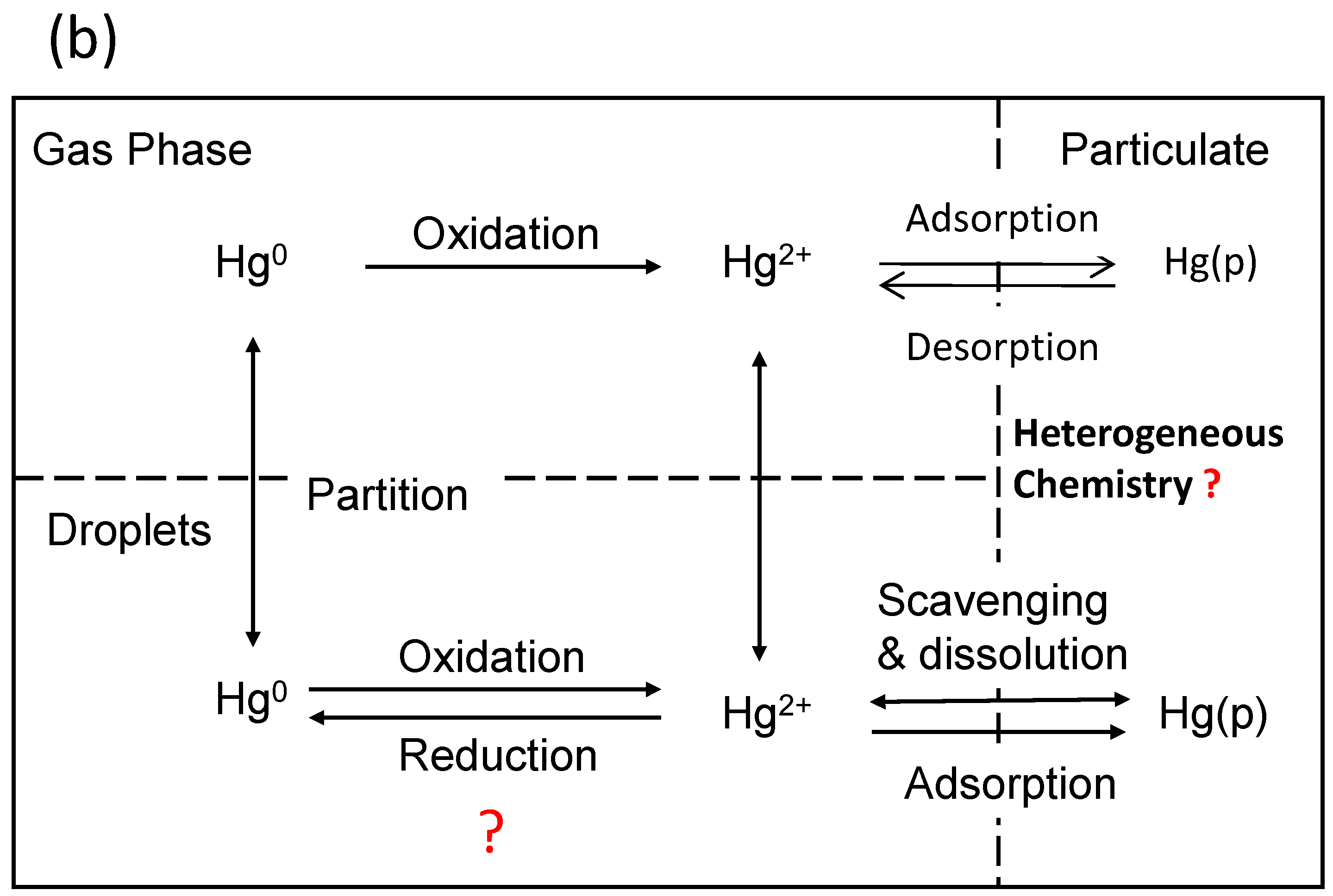
4. Heterogeneous Redox Reactions of Hg
5. Future Research Directions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Miretzky, P.; Cirelli, A.F. Hg(II) removal from water by chitosan and chitosan derivatives: A review. J. Hazard. Mater. 2009, 167, 10–23. [Google Scholar] [CrossRef] [PubMed]
- UNEP. Global Mercury Assessment 2013: Sources, Emissions, Releases, and Environmental Transport; UNEP Chemicals Branch: Geneva, Switzerland, 2013. [Google Scholar]
- Khalizov, A.F.; Viswanathan, B.; Larregaray, P.; Ariya, P.A. Theoretical Study on the Reactions of Hg with Halogens: Atmospheric Implications. J. Phys. Chem. A 2003, 107, 6360–6365. [Google Scholar] [CrossRef]
- Ariya, P.A.; Amyot, M.; Dastoor, A.; Deeds, D.; Feinberg, A.; Kos, G.; Poulain, A.; Ryjkov, A.; Semeniuk, K.; Subir, M.; et al. Mercury Physicochemical and Biogeochemical Transformation in the Atmosphere and at Atmospheric Interfaces: A Review and Future Directions. Chem. Rev. 2015, 115, 3760–3802. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.; Jaeglé, L.; Gratz, L.E.; Ambrose, J.L.; Jaffe, D.A.; Selin, N.E.; Song, S.; Campos, T.L.; Flocke, F.M.; Reeves, M.; et al. Origin of oxidized mercury in the summertime free troposphere over the southeastern US. Atmos. Chem. Phys. 2016, 16, 1511–1530. [Google Scholar] [CrossRef]
- Driscoll, C.T.; Mason, R.P.; Chan, H.M.; Jacob, D.J.; Pirrone, N. Mercury as a Global Pollutant: Sources, Pathways, and Effects. Environ. Sci. Technol. 2013, 47, 4967–4983. [Google Scholar] [CrossRef] [PubMed]
- Bergan, T.; Gallardo, L.; Rodhe, H. Mercury in the global troposphere: A three dimensional model study. Atmos. Environ. 1999, 33, 1575–1585. [Google Scholar] [CrossRef]
- Zhang, L.; Lyman, S.; Mao, H.; Lin, C.-J.; Gay, D.A.; Wang, S.; Gustin, M.S.; Feng, X.; Wania, F. A synthesis of research needs for improving the understanding of atmospheric mercury cycling. Atmos. Chem. Phys. 2017, 17, 9133–9144. [Google Scholar] [CrossRef]
- Pacyna, J.M.; Travnikov, O.; Simone, F.D.; Hedgecock, I.M.; Sundseth, K.; Pacyna, E.G.; Steenhuisen, F.; Pirrone, N.; Munthe, J.; Kindbom, K. Current and future levels of mercury atmospheric pollution on a global scale. Atmos. Chem. Phys. 2016, 16, 12495–12511. [Google Scholar] [CrossRef]
- Schroeder, W.; Munthe, J. Atmospheric mercury—An overview. Atmos. Environ. 1998, 5, 809–822. [Google Scholar] [CrossRef]
- Ryaboshapko, A.; Bullock, R.; Ebinghaus, R.; Ilyin, I.; Lohman, K.; Munthe, J.; Peterson, G.; Seigneur, C.; Wangberg, I. Comparison of mercury chemistry models. Atmos. Environ. 2002, 36, 3881–3898. [Google Scholar] [CrossRef]
- Fitzgerald, W.F.; Mason, R.P. Biogeochemical cycling of mercury in the marine environment. Metal Ions Biol. Syst. 1997, 34, 53–111. [Google Scholar]
- Ariya, P.A.; Skov, H.; Grage, M.M.L.; Goodsite, E.M. Gaseous elemental mercury in the ambient atmosphere: Review of the application of theoretical calculations and experimental studies for determination of reaction coefficients and mechanisms with halogens and other reactants. Adv. Quantum Chem. 2008, 55, 43–55. [Google Scholar]
- Lin, C.-J.; Singhasuk, P.; Pehkonen, S.O. Atmospheric Chemistry of Mercury. In Environmental Chemistry and Toxicology of Mercury; Liu, G., Cai, Y., O’Driscoll, N., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 113–153. [Google Scholar]
- Jaffe, D.; Strode, S. Sources, fate and transport of atmospheric mercury from Asia. Environ. Chem. 2008, 5, 121–126. [Google Scholar] [CrossRef]
- Lin, C.-J.; Pehkonen, S.O. The chemistry of atmospheric mercury: A review. Atmos. Environ. 1999, 33, 2067–2079. [Google Scholar] [CrossRef]
- Ariya, P.A.; Peterson, K.; Snider, G.; Amyot, M. Mercury Chemical Transformations in the Gas, Aqueous and Heterogeneous Phases: State-of-the-Art Science and Uncertainties. In Mercury Fate and Transport in the Global Atmosphere; Springer: Dordrecht, The Netherlands, 2009; pp. 459–501. [Google Scholar]
- Subir, M.; Ariya, P.A.; Dastoor, A.P. A review of uncertainties in atmospheric modeling of mercury chemistry I. Uncertainties in existing kinetic parameters—Fundamental limitations and the importance of heterogeneous chemistry. Atmos. Environ. 2011, 45, 5664–5676. [Google Scholar] [CrossRef]
- Ariya, P.A.; Khalizov, A.F.; Gidas, A. Reaction of Gaseous Mercury with Atomic and Molecular Halogens: Kinetics, Product Studies, and Atmospheric Implications. J. Phys. Chem. A 2002, 106, 7310–7320. [Google Scholar] [CrossRef]
- Spicer, C.W.; Satola, J.; Abbgy, A.A.; Plastridge, R.A.; Cowen, K.A. Kinetics of Gas-Phase Elemental Mercury Reaction with Halogen Species, Ozone, and Nitrate Radical Under Atmospheric Conditions. In Final Report to Florida Department of Environmental Protection; Battelle: Columbus, OH, USA, 2002. [Google Scholar]
- Goodsite, M.E.; Plane, J.M.C.; Skov, H. A theoretical study of the oxidation of Hg0 to HgBr2 in the troposphere. Environ. Sci. Technol. 2004, 38, 1772–1776. [Google Scholar] [CrossRef] [PubMed]
- Donohoue, D.L.; Bauer, D.; Cossairt, B.; Hynes, A.J. Temperature and pressure dependent rate coefficients for the reaction of Hg with Br and the reaction of Br with Br: A pulsed laser photolysis-pulsed laser induced fluorescence study. J. Phys. Chem. A 2006, 110, 6623–6632. [Google Scholar] [CrossRef] [PubMed]
- Shepler, B.C.; Balabanov, N.B.; Peterson, K.A. Hg+Br–>HgBr recombination and collision induced dissociation dynamics. J. Chem. Phys. 2007, 127, 164–304. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Sommar, J.; Feng, X.; Lin, C.-J.; Ge, M.; Wang, W.; Yin, R.; Fu, X.; Shang, L. Mass-dependent and -independent fractionation of mercury isotope during gas-phase oxidation of elemental mercury vapor by atomic Cl and Br. Environ. Sci. Technol. 2016, 50, 9232–9241. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Dibble, T.S. First kinetic study of the atmospherically important reactions BrHg + NO2 and BrHg + HOO. Phys. Chem. Chem. Phys. 2017, 19, 1826–1838. [Google Scholar] [CrossRef] [PubMed]
- Greig, G.; Gunning, H.E.; Strausz, O.P. Reactions of metal atoms. II. The combination of mercury and bromine atoms and the dimerization of HgBr. J. Chem. Phys. 1970, 52, 3684–3690. [Google Scholar] [CrossRef]
- Horne, D.G.; Gosavi, R.; Strausz, O.P. Reactions of metal atoms: Combination of mercury and chlorine atoms and the dimerization of HgCl. J. Chem. Phys. 1968, 48, 4758–4764. [Google Scholar] [CrossRef]
- Donohoue, D.L.; Bauer, D.; Hynes, A.J. Temperature and pressure dependent rate coefficients for the reaction of Hg with Cl and the reaction of Cl with Cl: A pulsed laser photolysispulsed laser induced fluorescence study. J. Phys. Chem. A 2005, 109, 7732–7741. [Google Scholar] [CrossRef] [PubMed]
- Byun, Y.; Cho, M.; Namkung, W.; Lee, K.; Koh, D.J.; Shin, D.N. Insight into the unique oxidation chemistry of elemental mercury by chlorine-containing species: Experiment and simulation. Environ. Sci. Technol. 2010, 44, 1624–1629. [Google Scholar] [CrossRef] [PubMed]
- Slemr, F.; Schuster, G.; Seiler, W. Distribution, speciation, and budget of atmospheric mercury. J. Atmos. Chem. 1985, 3, 407–434. [Google Scholar] [CrossRef]
- P’yankov, V.A. O kinetike Reaktsii Parov Rtuti s Ozonom (Kinetics of the Reaction of Mercury Vapour with Ozone). Zhurmal Obscej. Chem. Akatemijaneuk SSSR 1949, 19, 224–229. [Google Scholar]
- Schroeder, W.; Yarwood, G.; Niki, H. Transformation processes involving mercury species in the atmosphere—Results from a literature survey. Water Air Soil Pollut. 1991, 56, 653–666. [Google Scholar] [CrossRef]
- Iverfeldt, A.A.; Lindqvist, O. Atmospheric oxidation of elemental mercury by ozone in the aqueous phase. Atmos. Environ. 1986, 20, 1567–1573. [Google Scholar] [CrossRef]
- Hall, B. The gas-phase oxidation of elemental mercury by ozone. Water Air Soil Pollut. 1995, 80, 301–315. [Google Scholar] [CrossRef]
- Pal, B.; Ariya, P.A. Studies of ozone initiated reactions of gaseous mercury: Kinetics, product studies, and atmospheric implications. Phys. Chem. Chem. Phys. 2004, 6, 572–579. [Google Scholar] [CrossRef]
- Sumner, A.; Spicer, C.; Satola, J.; Mangaraj, R.; Cowen, K.; Landis, M.; Stevens, R.; Atkeson, T. Environmental chamber studies of mercury reactions in the atmosphere. In Dynamics of Mercury Pollution on Regional and Global Scales; Springer: New York, NY, USA, 2005; pp. 193–212. [Google Scholar]
- Snider, G.; Raofie, F.; Ariya, P.A. Effects of relative humidity and CO(g) on the O3-initiated oxidation reaction of Hg0(g): Kinetic & product studies. Phys. Chem. Chem. Phys. 2008, 10, 5616–5623. [Google Scholar] [PubMed]
- Rutter, A.P.; Shakya, K.M.; Lehr, R.; Schauer, J.J.; Griffin, R.J. Oxidation of gaseous elemental mercury in the presence of secondary organic aerosols. Atmos. Environ. 2012, 59, 86–92. [Google Scholar] [CrossRef]
- Sommar, J.; Hallquist, M.; Ljungstrom, E.; Lindqvist, O. On the gas phase reactions between volatile biogenic mercury species and the nitrate radical. J. Atmos. Chem. 1997, 27, 233–247. [Google Scholar] [CrossRef]
- Miller, G.C.; Quashnick, J.; Hebert, V. Reaction rate of metallic mercury with hydroxyl radical in the gas phase. Abstr. Paper Am. Chem. Soc. 2001, 221, 16-AGRO. [Google Scholar]
- Bauer, D.; D’Ottone, L.; Campuzano-Jost, P.; Hynes, A.J. Gas phase elemental mercury: Acomparison of LIF detection techniques and study of the kinetics of reaction with thehydroxyl radical. J. Photochem. Photobiol. A 2003, 157, 247–256. [Google Scholar] [CrossRef]
- Pal, B.; Ariya, P.A. Gas-phase HO center dot-initiated reactions of elemental mercury: Kinetics, product studies, and atmospheric implications. Environ. Sci. Technol. 2004, 38, 5555–5566. [Google Scholar] [CrossRef] [PubMed]
- Raofie, F.; Snider, G.; Ariya, P.A. Reaction of gaseous mercury with molecular iodine, atomic iodine, and iodine oxide radicals—Kinetics, product studies, and atmospheric implications. Can. J. Chem. 2008, 86, 811–820. [Google Scholar] [CrossRef]
- Raofie, F.; Ariya, P.A. Product Study of the Gas-Phase BrO-Initiated Oxidation of Hg0: Evidence for Stable Hg1+ Compounds. Environ. Sci. Technol. 2004, 38, 4319–4326. [Google Scholar] [CrossRef] [PubMed]
- Seigneur, C.; Wrobel, J.; Constantinou, E. A chemical kinetic mechanism for atmospheric inorganic mercury. Environ. Sci. Technol. 1994, 28, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Tokos, J.J.S.; Hall, B.; Calhoun, J.A.; Prestbo, E.M. Homogeneous gas-phase reaction of Hg◦ with H2O2, O3, CH3I, and (CH3)2S: Implications for atmospheric Hg cycling. Atmos. Environ. 1998, 32, 823–827. [Google Scholar] [CrossRef]
- Dibble, T.S.; Zelie, M.J.; Mao, H. Thermodynamics of reactions of ClHg and BrHg radicals with atmospherically abundant free radicals. Atmos. Chem. Phys. 2012, 12, 10271–10279. [Google Scholar] [CrossRef]
- Subir, M.; Ariya, P.A.; Dastoor, A.P. A review of the sources of uncertainties in atmospheric mercury modeling II. Mercury surface and heterogeneous chemistry—A missing link. Atmos. Environ. 2012, 46, 1–10. [Google Scholar] [CrossRef]
- Hynes, A.J.; Donohoue, D.L.; Goodsite, M.E.; Hedgecock, I.M. Our current understanding of major chemical and physical processes affecting mercury dynamics in the atmosphere and at the air-water/terrestrial interfaces. In Mercury Fate and Transport in the Global Atmosphere; Springer: New York, NY, USA, 2009; pp. 427–457. [Google Scholar]
- Peleg, M.; Tas, E.; Obrist, D.; Matveev, V.; Moore, C.; Gabay, M.; Luria, M. Observational Evidence for Involvement of Nitrate Radicals in Nighttime Oxidation of Mercury. Environ. Sci. Technol. 2015, 49, 14008–14018. [Google Scholar] [CrossRef] [PubMed]
- Bergan, T.; Rodhe, H. Oxidation of elemental mercury in the atmosphere; constraints imposed by global scale modelling. J. Atmos. Chem. 2001, 40, 191–212. [Google Scholar] [CrossRef]
- Dastoor, A.P.; Larcoque, Y. Global circulation of atmospheric mercury: A modelling study. Atmos. Environ. 2004, 38, 147–161. [Google Scholar] [CrossRef]
- Selin, N.E.; Javob, D.J.; Park, R.J.; Yantosca, R.M.; Strode, S.; Jaeglé, L.; Jaffe, D. Chemical cycling and deposition of atmospheric mercury: Global constraints from observations. J. Geophys. Res. Atmos. 2007, 112, D02308. [Google Scholar] [CrossRef]
- Travnikov, O.; Ilyin, I. The EMEP/MSC-E Mercury Modeling System. In Mercury Fate and Transport in the Global Atmosphere: Emissions, Measurements, and Models; Pirrone, N., Mason, R.P., Eds.; Springer: New York, NY, USA, 2009; pp. 571–587. [Google Scholar]
- De Simone, F.; Gencarelli, C.N.; Hedgecock, I.M.; Pirrone, N. Global atmospheric cycle of mercury: A model study on the impact of oxidation mechanisms. Environ. Sci. Pollut. Res. 2014, 21, 4110–4123. [Google Scholar] [CrossRef] [PubMed]
- Gencarelli, C.N.; de Simone, F.; Hedgecock, I.M.; Sprovieri, F.; Pirrone, N. Development and Application of a Regional-Scale Atmospheric Mercury Model Based on WRF/Chem: A Mediterranean Area Investigation. Environ. Sci. Pollut. Res. 2014, 21, 4095–4109. [Google Scholar] [CrossRef] [PubMed]
- Travnikov, O.; Angot, H.; Artaxo, P.; Bencardino, M.; Bieser, J.; D’Amore, F.; Dastoor, A.; Simone, F.D.; Diéguez, M.d.C.; Dommergue, A.; et al. Multi-model study of mercury dispersion in the atmosphere: Atmospheric processes and model evaluation. Atmos. Chem. Phys. 2017, 17, 5271–5295. [Google Scholar] [CrossRef]
- Mason, R.P.; Sheu, G.R. Role of the ocean in the global mercury cycle. Glob. Biogeochem. Cycle 2002, 16, 1093. [Google Scholar] [CrossRef]
- Hedgecock, I.M.; Pirrone, N. Chasing quicksilver: Modeling the atmospheric lifetime of Hg-(g)(0) in the marine boundary layer at various latitudes. Environ. Sci. Technol. 2004, 38, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Dastoor, A.P.; Davignon, D.; Theys, N.; van Roozendael, M.; Steffen, A.; Ariya, P.A. Modeling dynamic exchange of gaseous elemental mercury at polar sunrise. Environ. Sci. Technol. 2008, 42, 5183–5188. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.D.; Jacob, D.J.; Yang, X. Global lifetime of elemental mercury against oxidation by atomic bromine in the free troposphere. Geophys. Res. Lett. 2006, 33, L20808. [Google Scholar] [CrossRef]
- Theys, N.; van Roozendael, M.; Hendrick, F.; Yang, X.; de Smedt, I.; Richter, A.; Begoin, M.; Errera, Q.; Johnston, P.V.; Kreher, K.; et al. Global observations of tropospheric BrO columns using GOME-2 satellite data. Atmos. Chem. Phys. 2011, 11, 1791–1811. [Google Scholar] [CrossRef]
- Wang, S.Y.; Schmidt, J.A.; Baidar, S.; Coburn, S.; Dix, B.; Koenig, T.K.; Apel, E.; Bowdalo, D.; Campos, T.L.; Eloranta, E.; et al. Active and widespread halogen chemistry in the tropical and subtropical free troposphere. Proc. Natl. Acad. Sci. USA 2015, 112, 9281–9286. [Google Scholar] [CrossRef] [PubMed]
- Gratz, L.E.; Ambrose, J.L.; Jaffe, D.A.; Shah, V.; Jaegle, L.; Stutz, J.; Festa, J.; Spolaor, M.; Tsai, C.; Selin, N.E.; et al. Oxidation of mercury by bromine in the subtropical Pacific free troposphere. Geophys. Res. Lett. 2015, 42, 10494–10502. [Google Scholar] [CrossRef]
- Finlayson-Pitts, B.J.; Pitts, J.N.J. Chemistry of the Upper and Lower Atmosphere, Theory, Experiments, and Applications; Academic Press: San Diego, CA, USA, 2000. [Google Scholar]
- Gustin, M.S.; Amos, H.M.; Huang, J.; Miller, M.B.; Heidecorn, K. Measuring and modeling mercury in the atmosphere: A critical review. Atmos. Chem. Phys. 2015, 15, 5697–5713. [Google Scholar] [CrossRef]
- Ye, Z.; Mao, H.; Lin, C.-J.; Kim, S.Y. Investigation of processes controlling summertime gaseous elemental mercury oxidation at midlatitudinal marine, coastal, and inland sites. Atmos. Chem. Phys. 2016, 16, 8461–8478. [Google Scholar] [CrossRef]
- Gencarelli, C.N.; Bieser, J.; Carbone, F.; Simone, F.D.; Hedgecock, I.M.; Matthias, V.; Travnikov, O.; Yang, X.; Pirrone, N. Sensitivity model study of regional mercury dispersion in the atmosphere. Atmos. Chem. Phys. 2017, 17, 627–643. [Google Scholar] [CrossRef]
- Bieser, J.; Slemr, F.; Ambrose, J.; Brenninkmeijer, C.; Brooks, S.; Dastoor, A.; Simone, F.D.; Ebinghaus, R.; Gencarelli, C.N.; Geyer, B.; et al. Multi-model study of mercury dispersion in the atmosphere: Vertical and interhemispheric distribution of mercury species. Atmos. Chem. Phys. 2017, 17, 6925–6955. [Google Scholar] [CrossRef]
- Munthe, J.; Xiao, Z.F.; Lindqvist, O. The aqueous reduction of divalent mercury by sulfite. Water Air Soil Pollut. 1991, 56, 621–630. [Google Scholar] [CrossRef]
- Van Loon, L.; Mader, E.; Scott, S.L. Reduction of the aqueous mercuric ion by sulfite: UV Spectrum of HgSO3 and Its Intramolecular Redox Reaction. J.Phys.Chem. A 2000, 104, 1621–1626. [Google Scholar] [CrossRef]
- Feinberg, A.I.; Kurien, U.; Ariya, P.A. The Kinetics of Aqueous Mercury(II) Reduction by Sulfite Over an Array of Environmental Conditions. Water Air Soil Pollut. 2015, 226, 1–12. [Google Scholar] [CrossRef]
- Xiao, Z.F.; Munthe, J.; Stromberg, D.; Lindqvist, O. Photochemical behavior of inorganic mercury compounds in aqueous solution. In Mercury as a Global Pollutant-Integration and Synthesis; Watras, C.J., Huckabee, J.W., Eds.; Lewis Publishers: Boca Raton, USA, 1994; pp. 581–592. [Google Scholar]
- Pehkonen, S.O.; Lin, C.-J. Aqueous photochemistry of divalent mercury with organic acids. J. Air Waste Manag. Assoc. 1998, 48, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Gardfeldt, K.; Jonsson, M. Is bimolecular reduction of Hg(II) complexes possible in aqueous systems of environmental importance. J. Phys. Chem. A 2003, 107, 4478–4482. [Google Scholar] [CrossRef]
- Si, L.; Ariya, P.A. Reduction of oxidized mercury species by dicarboxylic acids (C2-C4): Kinetic and product studies. Environ. Sci. Technol. 2008, 42, 5150–5155. [Google Scholar] [CrossRef] [PubMed]
- Munthe, J. Aqueous oxidation of elemental Hg by O3. Atmos. Environ. 1992, 26, 1461–1468. [Google Scholar] [CrossRef]
- Gardfeldt, K.; Sommar, J.; Stromberg, D.; Feng, X. Oxidation of atomic mercury by hydroxyl radicals and photoinduced decomposition of methylmercury in the aqueous phase. Atmos. Environ. 2001, 35, 3039–3047. [Google Scholar] [CrossRef]
- Hines, N.A.; Brezonik, P.L. Mercury dynamics in a small Northern Minnesota lake: Water to air exchange and photoreactions of mercury. Mar. Chem. 2004, 90, 137–149. [Google Scholar] [CrossRef]
- Wang, Z.; Pehkonen, S.O. Oxidation of elemental mercury by aqueous bromine: Atmospheric implications. Atmos. Environ. 2004, 38, 3675–3688. [Google Scholar] [CrossRef]
- Lin, C.-J.; Pehkonen, S.O. Oxidation of elemental mercury by aqueous chlorine (HOCl−/OCl−): Implication for tropospheric mercury chemistry. J. Geophys. Res. 1998, 103, 28093–28102. [Google Scholar] [CrossRef]
- Foy, B.D.; Tong, Y.; Yin, X.; Zhang, W.; Kang, S.; Zhang, Q.; Zhang, G.; Wang, X.; Schauer, J.J. First field-based atmospheric observation of the reduction of reactive mercury driven by sunlight. Atmos. Environ. 2016, 134, 27–39. [Google Scholar] [CrossRef]
- Haitzer, M.; Aiken, G.R.; Ryan, J.N. Binding of mercury(II) to dissolved organic matter: The role of the mercuryto-DOM concentration ratio. Environ. Sci. Technol. 2002, 36, 3564–3570. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, M. Interactions between mercury and dissolved organic matter—A review. Chemosphere 2004, 55, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Hintelmann, H. Mercury isotope fractionation during photoreduction in natural water is controlled by its Hg-DOC ratio. Geochim. Cosmochim. Acta 2009, 73, 6704–6715. [Google Scholar] [CrossRef]
- Amyot, M.; Mierle, G.; Lean, D.R.S.; McQueen, D.J. Sunlight-induced formation of dissovled gaseous mercury in lake waters. Environ. Sci. Technol. 1994, 28, 2366–2371. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.F.; Stromberg, D.; Lindqvist, O. Influence of humic substances on photolysis of divalent mercury in aqueous solution. Water Air Soil Pollut. 1995, 80, 789–798. [Google Scholar] [CrossRef]
- O’Driscoll, N.J.; Siciliano, S.D.; Lean, D.R.S.; Amyot, M. Gross photoreduction kinetics of mercury in temperate freshwater lakes and rivers: Application to a general model of DGM dynamics. Environ. Sci. Technol. 2006, 40, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Whalin, L.; Mason, R. A new method for the investigation of mercury redox chemistry in natural waters utilizing deflatable Teflon (R) bags and additions of isotopically labeled mercury. Anal. Chim. Acta 2006, 558, 211–221. [Google Scholar] [CrossRef]
- Si, L.; Ariya, P.A. Aqueous photoreduction of oxidized mercury species in presence of selected alkanethiols. Chemosphere 2011, 84, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, H.M.; Jacob, D.J.; Zhang, Y.; Dibble, T.S.; Slemr, F.; Amos, H.M.; Schmidt, J.A.; Corbitt, E.S.; Marais, E.A.; Sunderland, E.M. A new mechanism for atmospheric mercury redox chemistry: Implications for the global mercury budget. Atmos. Chem. Phys. 2017, 17, 6353–6371. [Google Scholar] [CrossRef]
- Si, L.; Ariya, P.A. Photochemical Reactions of Divalent Mercury with Thioglycolic Acid: Formation of Mercuric Sulfide Particles. Chemosphere 2015, 119, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Prather, K.A.; Hatch, C.D.; Grassian, V.H. Analysis of atmospheric aerosols. Ann. Rev. Anal. Chem. 2008, 1, 485–514. [Google Scholar] [CrossRef] [PubMed]
- Finlayson-Pitts, B.J. Reactions at surfaces in the atmosphere: Integration of experiments and theory as necessary (but not necessarily sufficient) for prediction the physical chemistry of aerosols. Phys. Chem. Chem. Phys. 2009, 11, 7760–7779. [Google Scholar] [CrossRef] [PubMed]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006. [Google Scholar]
- Lin, C.-J.; Pehkonen, S.O. Aqueous free radical chemistry of mercury in the presence of iron oxides and ambient aerosol. Atmos. Environ. 1997, 31, 4125–4137. [Google Scholar] [CrossRef]
- Tong, Y.; Eichhorst, T.; Olson, M.R.; McGinnis, J.E.; Turner, I.; Rutter, A.P.; Shafer, M.M.; Wanga, X.; Schauer, J.J. Atmospheric photolytic reduction of Hg(II) in dry aerosols. Environ.Sci. Processes Impacts 2013, 15, 1883–1888. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Eichhorst, T.; Olson, M.R.; Rutter, A.P.; Shafer, M.M.; Wang, X.; Schauer, J.J. Comparison of heterogeneous photolytic reduction of Hg(II) in the coal fly ashes and synthetic aerosols. Atmos. Res. 2014, 138, 324–329. [Google Scholar] [CrossRef]
- Tacey, S.A.; Xu, L.; Mavrikakis, M.; Schauer, J.J. Heterogeneous Reduction Pathways for Hg(II) Species on Dry Aerosols: A First-Principles Computational Study. J. Phys. Chem. A 2016, 120, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Kurien, U.; Hu, Z.; Lee, H.; Dastoor, A.P.; Ariya, P.A. Radiation Enhanced Uptake of Hg0(g) on Iron (Oxyhydr)Oxide Nanoparticles. RSC Adv. 2017, 7, 45010–45021. [Google Scholar] [CrossRef]
- Deeds, D.; Ghoshdastidar, A.; Raofie, F.; Guerette, E.-A.; Tessier, A.; Ariya, P. Development of a Particle-Trap Preconcentration-Soft Ionization Mass Spectrometric Technique for the Quantification of mercury halides in air. Anal. Chem. 2015, 87, 5109–5116. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Gas Phase Reaction | Diluent gas a (T = 298 K) | Rate Coefficient b (cm3 molec−1 s−1) | References |
|---|---|---|---|
| Air, N2, 1 atm | (3.2 ± 0.3) × 10−12 | [19] | |
| Air, NO, 1 atm | 9 × 10−13 | [20] | |
| N/A, 1 atmc | 1.01 × 10−12exp(209.03/T) | [3] | |
| N/A, 1 atm | 2.07 × 10−12 | ||
| N/A (180–400 K) | 1.1 × 10−12(T/298K)−2.37 | [21] | |
| N/A, 1 atm | 1.1 × 10−12 | ||
| N2, 1atm (243–298 K) | (1.46 ± 0.34) × 10−32 × (T/298)−(1.86 ± 1.49) cm6/molec2/s | [22] | |
| N2, 1 atm | (3.6 ± 0.9) × 10−13 | ||
| Ar, 1 atm (260 K)c | 1.2 × 10−12 | [23] | |
| Air, 1 atm | (1.6 ± 0.8) × 10−12 | [24] | |
M = NO2 or HO2 | N/A (220–320 K) | k ([M], T)d | [25] |
| CF3Br, 0.26 atm (397 K) | 7 × 10−17 | [26] | |
| N/A, 1 atm (180–400 K) | 2.5 × 10−10(T/298K)−0.57 | [21] | |
| N/A, 1 atm | 2.5 × 10−10 | ||
| Ar, 0.93 atm (383–443 K) | (3.2 ± 1.7) × 10−11 | [27] | |
| Air, NO, 1 atm | 6.4 × 10−11 | [20] | |
| Air, N2, 1 atm | (1.0 ± 0.2) × 10−11 | [19] | |
| N/A, 1 atmc | 1.38 × 10−12exp(208.02/T) | [3] | |
| N/A, 1 atm | 2.81 × 10−12 | ||
| N2 (243–298 K) | (2.2 ± 0.5) × 10−32 × exp [(680 ± 400)(1/T − 1/298)] cm6/molec2/s | [28] | |
| N2, 1 atm | 5.4 × 10−13 | ||
| N2, 1 atm | 1.2 × 10−10 | [29] | |
| Air, 1 atm | (1.8 ± 0.5) × 10−11 | [24] | |
| N/A, 1 atm (293 K) | 4.2 × 10−19 | [30,31] | |
| N/A, 1 atm (293 K) | 4.9 × 10−18 | [31,32] | |
| Air, 1 atm (293 K) | 1.7 × 10−18 | [33] | |
| N2/O2, 1 atm (293 K) | (3 ± 2) × 10−20 | [34] | |
| N2, 1 atm | (7.5 ± 0.9) × 10−19 | [35] | |
| Air, 1 atm | (6.4 ± 2.3) × 10−19 | [36] | |
| N2, 1 atm | (6.2 ± 1.1) × 10−19 | [37] | |
| Air, 1 atm | (7.4 ± 0.5) × 10−19 | [38] | |
| Air, 1 atm | (8.7 ± 2.8) × 10−14 | [39] | |
| N/A, 1 atm (343 K) | (1.6 ± 0.2) × 10−11 | [40] | |
| Air, 1 atm | <1.2 × 10−13 | [41] | |
| N/A, 1 atm (180–400 K) | 3.2 × 10−13(T/298K)−3.06 | [21] | |
| N/A, 1 atm | 3.2 × 10−13 | ||
| Air/N2, 1 atm | (9.0 ± 1.3) × 10−14 | [42] | |
| N/A, 1 atmc | 9.2 × 10−13exp(206.81/T) | [3] | |
| N/A, 1 atm | 1.86 × 10−12 | ||
| N/A, 1 atm (180–400 K) | 4.0 × 10−13(T/298 K)−2.38 | [21] | |
| N/A, 1 atm | 4.0 × 10−13 | ||
| Air, 1 atm | (1.8 ± 0.4) × 10−15 | [36] | |
| N2, 1 atm | ≤(1.27 ± 0.58) × 10−19 | [43] | |
| Air, N2, 1 atm | (2.6 ± 0.2) × 10−18 | [19] | |
| Air, 1 atm | (2.5 ± 0.9) × 10−18 | [36] | |
| N2, 1 atm | 4.3 × 10−15 | [29] | |
| Air, N2, 1 atm | <(0.9 ± 0.2) × 10−16 | [19] | |
| N2, 1 atm | 1.1 × 10−11 | [29] | |
| Air, NO, 1 atm | (3.0–6.4) × 10−14 | [20] | |
| N2, 1 atm | (1–100) × 10−15 | [44] | |
| N2, (5–10) × 10−3atm(294 K) | <4 × 10−15 | [39] | |
| Air, 1 atm | <7 × 10−15 | [36] | |
| N/A, 1 atm | ≤4.1 × 10−16 | [45] | |
| N2, N/A (293 K) | <8.5 × 10−19 | [46] |
| Reactant(s) | Rate Constants | T(K) | pH | Potential Mechanism | Reference |
|---|---|---|---|---|---|
| Identified Aqueous Reduction Pathways of Hg2+ | |||||
| Hg2+ + sulfite (aq) | 0.6 s−1 | 299 | 3.0–4.84 | [70] | |
| 0.0106 ± 0.0009 s−1 | 298 | 3 | [71] | ||
| 0.013 ± 0.007 s−1 | 298 | 7 | Same as above | [72] | |
| <10−4 s−1 | 299 | 3.0–4.84 | [70] | ||
| Hg(OH)2 | 3 × 10−7 s−1 | 293 | 7 | [73] | |
| HgS22− | ~10−7 s−1 | 298 | Not available | [73] | |
| Hg2+ + HO2 | 1.7 × 104 M−1 s−1 | 298 | [74] | ||
| Not available | Intramolecular 2e− transfer via Hg2+-oxalate complex | [75] | |||
| Hg2++Dicarboxylic acids (C2–C4) | (1.2 ± 0.2) × 104 M−1 s−1(Oxalic) (4.9 ± 0.8) × 103 M−1 s−1(Malonic); (2.8 ± 0.5) × 103 M−1 s−1(Succinic) | 296 | 3.0 | Mainly intramolecular 2e− transfer via Hg2+-dicarboxylate complexes | [76] |
| Identified Aqueous Oxidation Pathways of Hg0 | |||||
| Hg0 + O3 | (4.7 ± 2.2) × 107 M−1 s−1 | 298 | 4.5–9.5 | [77] | |
| Hg0 + •OH | 2.0 × 109 M−1 s−1 | 298 | [74] | ||
| (2.4 ± 0.3) × 109 M−1 s–1 | 298 | [78] | |||
| 5.5 × 109 M–1 s–1 | Not available | [79] | |||
| Hg0 + aqueous bromine | 0.28 ± 0.02 M–1 s–1 0.27 ± 0.04 M–1 s–1 0.2 ± 0.03 M–1 s–1 | 294–296 | 2, 6.8, 11.7 | [80] | |
| Hg0 + HOCl/OCl− | (2.09 ± 0.06) × 106 M–1 s–1 | Ambient | [81] | ||
| (1.99 ± 0.05) × 106 M–1 s–1 | |||||
| Reactants | Surfaces | Major Findings | References |
|---|---|---|---|
| Hg2+ + organic acids | 0.1 g/L iron oxides particles or 0.01 g/L ambient aerosols |
| [96] |
| HgCl2 | Synthetic NaCl aerosols |
| [97] |
| HgCl2 | Coal fly ash or synthetic aerosols |
| [98] |
| Hg2+ + sulfite | Fly ash |
| [72] |
| HgCl2, HgBr2, Hg(NO3)2, HgSO4 | Fe(110), NaCl(100) and NaCl(111)Na |
| [99] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Si, L.; Ariya, P.A. Recent Advances in Atmospheric Chemistry of Mercury. Atmosphere 2018, 9, 76. https://doi.org/10.3390/atmos9020076
Si L, Ariya PA. Recent Advances in Atmospheric Chemistry of Mercury. Atmosphere. 2018; 9(2):76. https://doi.org/10.3390/atmos9020076
Chicago/Turabian StyleSi, Lin, and Parisa A. Ariya. 2018. "Recent Advances in Atmospheric Chemistry of Mercury" Atmosphere 9, no. 2: 76. https://doi.org/10.3390/atmos9020076
APA StyleSi, L., & Ariya, P. A. (2018). Recent Advances in Atmospheric Chemistry of Mercury. Atmosphere, 9(2), 76. https://doi.org/10.3390/atmos9020076