Evolution of Multispectral Aerosol Absorption Properties in a Biogenically-Influenced Urban Environment during the CARES Campaign

 ,
, 



Abstract
1. Introduction
2. Experiments and Discussions
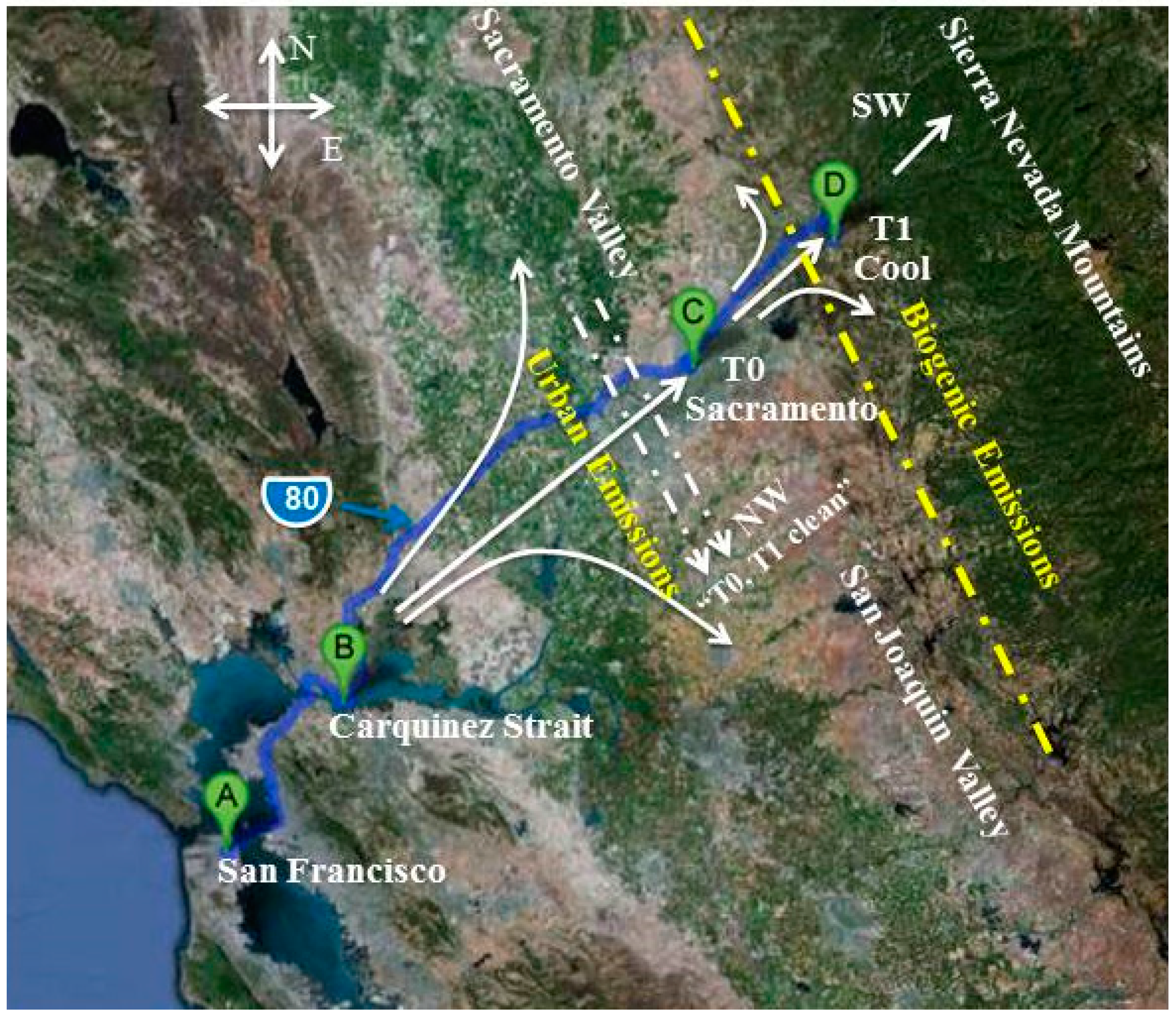
2.1. Ground Sites
2.2. Measurements of Aerosol Optical Properties
2.3. Collocated Measurements Used in This Study
2.4. Error Analysis
3. Results and Discussion
3.1. Daily Average of Aerosol Chemical, Physical, and Optical Properties
3.2. Aerosol Light Absorption Efficiency
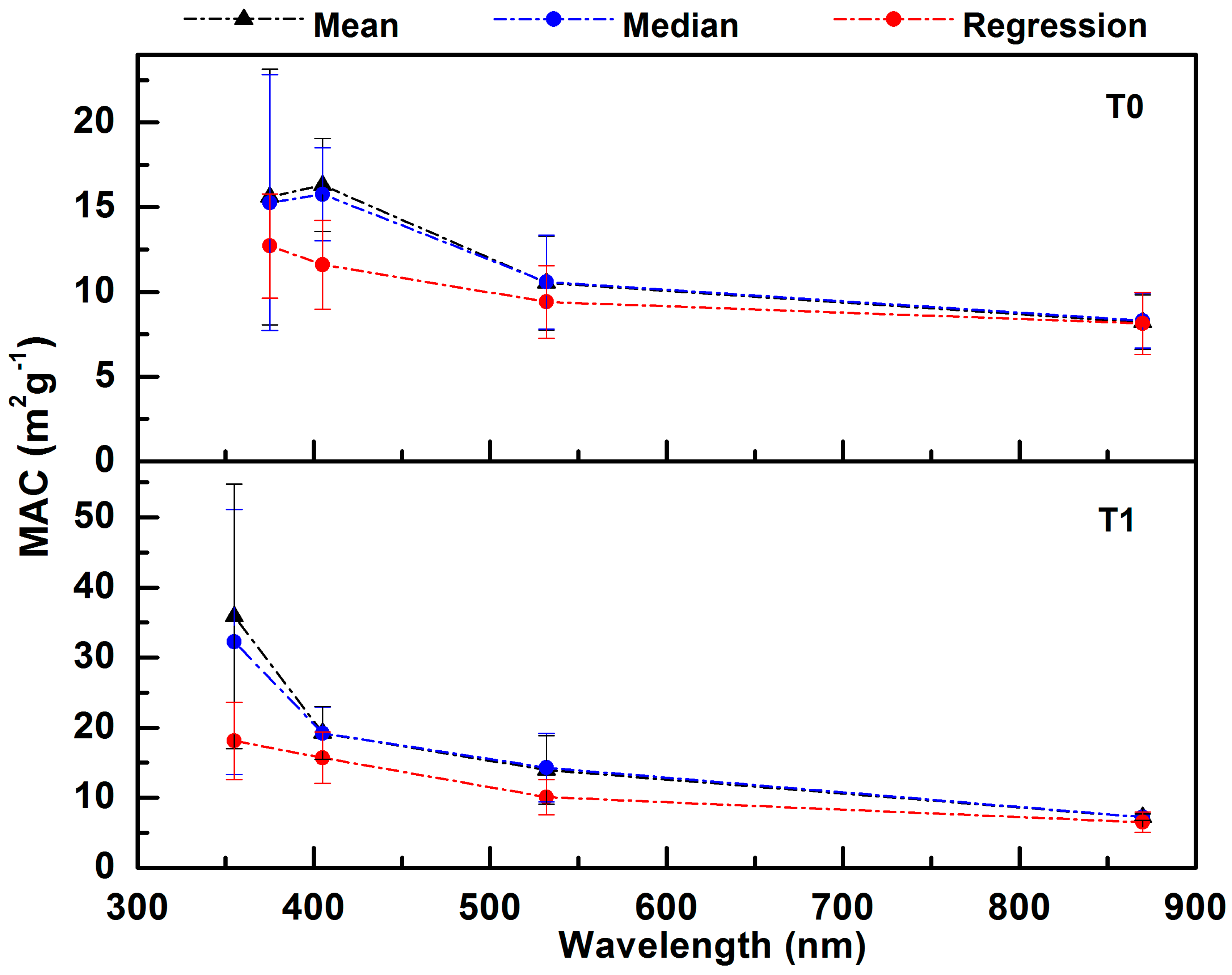
3.2.1. MAC from Linear Regression, Mean, and Median Analyses
3.2.2. MAC Frequency Distribution and Monte Carlo Simulation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Charlson, R.J.; Schwartz, S.E.; Hales, J.M.; Cess, R.D.; Coakley, J.A.; Hansen, J.E.; Hofmann, D.J. Climate Forcing by Anthropogenic Aerosols. Science 1992, 255, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.E. The Whitehouse Effect—Shortwave Radiative Forcing of Climate by Anthropogenic Aerosols: An Overview. J. Aerosol Sci. 1996, 27, 359–382. [Google Scholar] [CrossRef]
- Chýlek, P.; Wong, J. Effect of Absorbing Aerosol on Global Radiation Budget. Geophys. Res. Lett. 1995, 22, 929–931. [Google Scholar] [CrossRef]
- Hassan, T.; Moosmüller, H.; Chung, C.E. Coefficients of an Analytical Aerosol Forcing Equation Determined with a Monte-Carlo Radiation Model. J. Quant. Spectrosc. Radiat. Transf. 2015, 164, 129–136. [Google Scholar] [CrossRef]
- Moosmüller, H.; Ogren, J.A. Parameterization of the Aerosol Upscatter Fraction as Function of the Backscatter Fraction and Their Relationships to the Asymmetry Parameter for Radiative Transfer Calculations. Atmosphere 2017, 8. [Google Scholar] [CrossRef]
- Moosmüller, H.; Sorensen, C.M. Small and Large Particle Limits of Single Scattering Albedo for Homogeneous, Spherical Particles. J. Quant. Spectrosc. Radiat. Transf. 2018, 204, 250–255. [Google Scholar] [CrossRef]
- Coakley, J.A.; Bernstein, R.L.; Durkee, P.A. Effect of Ship-Stack Effluents on Cloud Reflectivity. Science 1987, 237, 1020–1022. [Google Scholar] [CrossRef] [PubMed]
- Twomey, S. The Influence of Pollution on the Shortwave Albedo of Clouds. J. Atmos. Sci. 1977, 34, 1149–1152. [Google Scholar] [CrossRef]
- Haywood, J.M.; Ramaswamy, V. Global Sensitivity Studies of the Direct Radiative Forcing due to Anthropogenic Sulfate and Black Carbon Aerosol. J. Geophys. Res. 1998, 103, 6043–6058. [Google Scholar] [CrossRef]
- Lack, D.A.; Moosmüller, H.; McMeeking, G.R.; Chakrabarty, R.K.; Baumgardner, D. Characterizing Elemental, Equivalent Black, and Refractory Black Carbon Aerosol Particles: A Review of Techniques, Their Limitations and Uncertainties. Anal. Bioanal. Chem. 2014, 406, 99–122. [Google Scholar] [CrossRef] [PubMed]
- Bond, T.; Bergstrom, R. Light Absorption by Carbonaceous Particles: An Investigative Review. Aerosol Sci. Tech. 2006, 40, 27–67. [Google Scholar] [CrossRef]
- Ramanathan, V.; Chung, C.; Kim, D.; Bettge, T.; Buja, L.; Kiehl, J.T.; Washington, W.M.; Fu, Q.; Sikka, D.R.; Wild, M. Atmospheric Brown Clouds: Impacts on South Asian Climate and Hydrological Cycle. Proc. Natl. Acad. Sci. USA 2005, 102, 5326–5333. [Google Scholar] [CrossRef] [PubMed]
- Bond, T.C.; Doherty, S.J.; Fahey, D.W.; Forster, P.M.; Berntsen, T.; DeAngelo, B.J.; Flanner, M.G.; Ghan, S.; Kärcher, B.; Koch, D.; et al. Bounding the Role of Black Carbon in the Climate System: A Scientific Assessment. J. Geophys. Res. 2013, 118, 5380–5552. [Google Scholar] [CrossRef]
- Andreae, M.O.; Gelencsér, A. Black Carbon or Brown Carbon? The Nature of Light-Absorbing Carbonaceous Aerosols. Atmos. Chem. Phys. 2006, 6, 3131–3148. [Google Scholar]
- Barnard, J.C.; Volkamer, R.; Kassianov, E.I. Estimation of the Mass Absorption Cross Section of the Organic Carbon Component of Aerosols in the Mexico City Metropolitan Area. Atmos. Chem. Phys. 2008, 8, 6665–6679. [Google Scholar] [CrossRef]
- Chakrabarty, R.K.; Moosmüller, H.; Chen, L.-W.A.; Lewis, K.; Arnott, W.P.; Mazzoleni, C.; Dubey, M.K.; Wold, C.E.; Hao, W.M.; Kreidenweis, S.M. Brown Carbon in Tar Balls from Smoldering Biomass Combustion. Atmos. Chem. Phys. 2010, 10, 6363–6370. [Google Scholar] [CrossRef]
- Flowers, B.A.; Dubey, M.K.; Mazzoleni, C.; Stone, E.A.; Schauer, J.J.; Kim, S.-W.; Yoon, S.C. Optical-Chemical-Microphysical Relationships and Closure Studies for Mixed Carbonaceous Aerosols Observed at Jeju Island; 3-Laser Photoacoustic Spectrometer, Particle Sizing, and Filter Analysis. Atmos. Chem. Phys. 2010, 10, 10387–10398. [Google Scholar] [CrossRef]
- Gyawali, M.; Arnott, W.P.; Lewis, K.; Moosmüller, H. In Situ Aerosol Optics in Reno, NV, USA during and after the Summer 2008 California Wildfires and the Influence of Absorbing and Non-Absorbing Organic Coatings on Spectral Light Absorption. Atmos. Chem. Phys. 2009, 9, 8007–8015. [Google Scholar] [CrossRef]
- Kirchstetter, T.W.; Novakov, T.; Hobbs, P.V. Evidence that the Spectral Dependence of Light Absorption by Aerosols is Affected by Organic Carbon. J. Geophys. Res. 2004, 109. [Google Scholar] [CrossRef]
- Lack, D.A.; Langridge, J.M.; Bahreini, R.; Cappa, C.D.; Middlebrook, A.M.; Schwarz, J.P. Brown Carbon and Internal Mixing in Biomass Burning Particles. Proc. Natl. Acad. Sci. USA 2012, 109, 14802–14807. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.; Arnott, W.P.; Moosmüller, H.; Wold, C.E. Strong Spectral Variation of Biomass Smoke Light Absorption and Single Scattering Albedo Observed with a Novel Dual-Wavelength Photoacoustic Instrument. J. Geophys. Res. 2008, 113. [Google Scholar] [CrossRef]
- Zhang, Q.; Jimenez, J.L.; Canagaratna, M.R.; Allan, J.D.; Coe, H.; Ulbrich, I.; Alfarra, M.R.; Takami, A.; Middlebrook, A.M.; Sun, Y.L.; et al. Ubiquity and Dominance of Oxygenated Species in Organic Aerosols in Anthropogenically-Influenced Northern Hemisphere Midlatitudes. Geophys. Res. Lett. 2007, 34. [Google Scholar] [CrossRef]
- IPCC. Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Stocker, T.F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S.K., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P.M., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2013. [Google Scholar]
- Zaveri, R.A.; Shaw, W.J.; Cziczo, D.J.; Schmid, B.; Ferrare, R.A.; Alexander, M.L.; Alexandrov, M.; Alvarez, R.J.; Arnott, W.P.; Atkinson, D.B.; et al. Overview of the 2010 Carbonaceous Aerosols and Radiative Effects Study (CARES). Atmos. Chem. Phys. 2012, 12, 7647–7687. [Google Scholar] [CrossRef]
- Fast, J.D.; Gustafson, W.I., Jr.; Berg, L.K.; Shaw, W.J.; Pekour, M.; Shrivastava, M.; Barnard, J.C.; Ferrare, R.A.; Hostetler, C.A.; Hair, J.A.; et al. Transport and Mixing Patterns over Central California during the Carbonaceous Aerosol and Radiative Effects Study (CARES). Atmos. Chem. Phys. 2012, 12, 1759–1783. [Google Scholar] [CrossRef]
- Shilling, J.E.; Zaveri, R.A.; Fast, J.D.; Kleinman, L.; Alexander, M.L.; Canagaratna, M.R.; Fortner, E.; Hubbe, J.M.; Jayne, J.T.; Sedlacek, A.; et al. Enhanced SOA Formation from Mixed Anthropogenic and Biogenic Emissions during the CARES Campaign. Atmos. Chem. Phys. 2013, 13, 2091–2113. [Google Scholar] [CrossRef]
- Setyan, A.; Zhang, Q.; Merkel, M.; Knighton, W.B.; Sun, Y.; Song, C.; Shilling, J.E.; Onasch, T.B.; Herndon, S.C.; Worsnop, D.R.; et al. Characterization of Submicron Particles Influenced by Mixed Biogenic and Anthropogenic Emissions Using High-Resolution Aerosol Mass Spectrometry: Results from CARES. Atmos. Chem. Phys. 2012, 12, 8131–8156. [Google Scholar] [CrossRef]
- Setyan, A.; Song, C.; Merkel, M.; Knighton, W.B.; Onasch, T.B.; Canagaratna, M.R.; Worsnop, D.R.; Wiedensohler, A.; Shilling, J.E.; Zhang, Q. Chemistry of New Particle Growth in Mixed Urban and Biogenic Emissions—Insights from CARES. Atmos. Chem. Phys. 2014, 14, 6477–6494. [Google Scholar] [CrossRef]
- Cappa, C.D.; Onasch, T.B.; Massoli, P.; Worsnop, D.R.; Bates, T.S.; Cross, E.S.; Davidovits, P.; Hakala, J.; Hayden, K.L.; Jobson, B.T.; et al. Radiative Absorption Enhancements Due to the Mixing State of Atmospheric Black Carbon. Science 2012, 337, 1078–1081. [Google Scholar] [CrossRef] [PubMed]
- Kassianov, E.; Pekour, M.; Barnard, J. Aerosols in Central California: Unexpectedly Large Contribution of Coarse Mode to Aerosol Radiative Forcing. Geophys. Res. Lett. 2012, 39. [Google Scholar] [CrossRef]
- Moffet, R.C.; Prather, K.A. In-Situ Measurements of the Mixing State and Optical Properties of Soot with Implications for Radiative Forcing Estimates. Proc. Natl. Acad. Sci. USA 2009, 106, 11872–11877. [Google Scholar] [CrossRef] [PubMed]
- China, S.; Scarnato, B.; Owen, R.C.; Zhang, B.; Ampadu, M.T.; Kumar, S.; Dzepina, K.; Dziobak, M.P.; Fialho, P.; Perlinger, J.A.; et al. Morphology and Mixing State of Aged Soot Particles at a Remote Marine Free Troposphere Site: Implications for Optical Properties. Geophys. Res. Lett. 2015, 42, 1243–1250. [Google Scholar] [CrossRef]
- Liu, S.; Aiken, A.C.; Gorkowski, K.; Dubey, M.K.; Cappa, C.D.; Williams, L.R.; Herndon, S.C.; Massoli, P.; Fortner, E.C.; Chhabra, P.S.; et al. Enhanced Light Absorption by Mixed Source Black and Brown Carbon Particles in UK Winter. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Lack, D.A.; Cappa, C.D. Impact of Brown and Clear Carbon on Light Absorption Enhancement, Single Scatter Albedo and Absorption Wavelength Dependence of Black Carbon. Atmos. Chem. Phys. 2010, 10, 4207–4220. [Google Scholar] [CrossRef]
- Cross, E.S.; Onasch, T.B.; Ahern, A.; Wrobel, W.; Slowik, J.G.; Olfert, J.; Lack, D.A.; Massoli, P.; Cappa, C.D.; Schwarz, J.P.; et al. Soot Particle Studies—Instrument Inter-Comparison—Project Overview. Aerosol Sci. Technol. 2010, 44, 592–611. [Google Scholar] [CrossRef]
- Arnott, W.P.; Moosmüller, H.; Rogers, C.F.; Jin, T.; Bruch, R. Photoacoustic Spectrometer for Measuring Light Absorption by Aerosol: Instrument Description. Atmos. Environ. 1999, 33, 2845–2852. [Google Scholar] [CrossRef]
- Moosmüller, H.; Chakrabarty, R.K.; Arnott, W.P. Aerosol Light Absorption and its Measurement: A Review. J. Quant. Spectrosc. Radiat. Transf. 2009, 110, 844–878. [Google Scholar] [CrossRef]
- Abu-Rahmah, A.; Arnott, W.P.; Moosmüller, H. Integrating Nephelometer with a Low Truncation Angle and an Extended Calibration Scheme. Meas. Sci. Technol. 2006, 17, 1723–1732. [Google Scholar] [CrossRef]
- Moosmüller, H.; Arnott, W.P. Angular Truncation Errors in Integrating Nephelometry. Rev. Sci. Instrum. 2003, 74, 3492–3501. [Google Scholar] [CrossRef]
- Gyawali, M.; Arnott, W.P.; Zaveri, R.A.; Song, C.; Moosmüller, H.; Liu, L.; Mishchenko, M.I.; Chen, L.-W.A.; Green, M.C.; Watson, J.G.; et al. Photoacoustic Optical Properties at UV, VIS, and Near IR Wavelengths for Laboratory Generated and Winter Time Ambient Urban Aerosols. Atmos. Chem. Phys. 2012, 12, 2587–2601. [Google Scholar] [CrossRef]
- Moosmüller, H.; Chakrabarty, R.K. Technical Note: Simple Analytical Relationships between Ångström Coefficients of Aerosol Extinction, Scattering, Absorption, and Single Scattering Albedo. Atmos. Chem. Phys. 2011, 11, 10677–10680. [Google Scholar] [CrossRef]
- Moosmüller, H.; Chakrabarty, R.K.; Ehlers, K.M.; Arnott, W.P. Absorption Ångström Coefficient, Brown Carbon, and Aerosols: Basic Concepts, Bulk Matter, and Spherical Particles. Atmos. Chem. Phys. 2011, 11, 1217–1225. [Google Scholar] [CrossRef]
- Schwarz, J.P.; Gao, R.S.; Fahey, D.W.; Thomson, D.S.; Watts, L.A.; Wilson, J.C.; Reeves, J.M.; Darbeheshti, M.; Baumgardner, D.G.; Kok, G.L.; et al. Single-Particle Measurements of Midlatitude Black Carbon and Light-Scattering Aerosols from the Boundary Layer to the Lower Stratosphere. J. Geophys. Res. 2006, 111. [Google Scholar] [CrossRef]
- Subramanian, R.; Kok, G.L.; Baumgardner, D.; Clarke, A.; Shinozuka, Y.; Campos, T.L.; Heizer, C.G.; Stephens, B.B.; De Foy, B.; Voss, P.B.; et al. Black Carbon over Mexico: The Effect of Atmospheric Transport on Mixing State, Mass Absorption Cross-Section, and BC/CO Ratios. Atmos. Chem. Phys. 2010, 10, 219–237. [Google Scholar] [CrossRef]
- Laborde, M.; Mertes, P.; Zieger, P.; Dommen, J.; Baltensperger, U.; Gysel, M. Sensitivity of the Single Particle Soot Photometer to Different Black Carbon Types. Atmos. Meas. Tech. 2012, 5, 1031–1043. [Google Scholar] [CrossRef]
- Moteki, N.; Kondo, Y. Dependence of Laser-Induced Incandescence on Physical Properties of Black Carbon Aerosols: Measurements and Theoretical Interpretation. Aerosol Sci. Technol. 2010, 44, 663–675. [Google Scholar] [CrossRef]
- China, S.; Salvadori, N.; Mazzoleni, C. Effect of Traffic and Driving Characteristics on Morphology of Atmospheric Soot Particles at Freeway On-Ramps. Environ. Sci. Technol. 2014, 48, 3128–3135. [Google Scholar] [CrossRef] [PubMed]
- Bevington, P.R. Data Reduction and Error Analysis for the Physical Sciences; McGraw-Hill: New York, NY, USA, 1969. [Google Scholar]
- Lewis, K.A. Development of a Dual-Wavelength Photoacoustic Instrument for Measurement of Light Absorption and Scattering by Aerosol and Gases in Atmospheric Science Program. Doctoral Thesis, University of Nevada, Reno, NV, USA, May 2007. [Google Scholar]
- Arnott, W.P.; Moosmüller, H.; Walker, J.W. Nitrogen Dioxide and Kerosene-Flame Soot Calibration of Photoacoustic Instruments for Measurement of Light Absorption by Aerosols. Rev. Sci. Instrum. 2000, 71, 4545–4552. [Google Scholar] [CrossRef]
- China, S.; Mazzoleni, C.; Gorkowski, K.; Aiken, A.C.; Dubey, M.K. Morphology and Mixing State of Individual Freshly Emitted Wildfire Carbonaceous Particles. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Andreae, M.O.; Crutzen, P.J. Atmospheric Aerosols: Biogeochemical Sources and Role in Atmospheric Chemistry. Science 1997, 276, 1052–1058. [Google Scholar] [CrossRef]
- Worton, D.R.; Goldstein, A.H.; Farmer, D.K.; Docherty, K.S.; Jimenez, J.L.; Gilman, J.B.; Kuster, W.C.; De Gouw, J.; Williams, B.J.; Kreisberg, N.M.; et al. Origins and Composition of Fine Atmospheric Carbonaceous Aerosol in the Sierra Nevada Mountains, California. Atmos. Chem. Phys. 2011, 11, 10219–10241. [Google Scholar] [CrossRef]
- Reid, J.S.; Hobbs, P.V. Physical and Optical Properties of Young Smoke from Individual Biomass Fires in Brazil. J. Geophys. Res. 1998, 103, 32013–32030. [Google Scholar] [CrossRef]
- Chakrabarty, R.K.; Arnold, I.J.; Francisco, D.M.; Hatchett, B.; Hosseinpour, F.; Loria, M.; Pokharel, A.; Woody, B.M. Black and Brown Carbon Fractal Aggregates from Combustion of Two Fuels Widely Used in Asian Rituals. J. Quant. Spectrosc. Radiat. Transf. 2013, 122, 25–30. [Google Scholar] [CrossRef]
- Doran, C. Corrigendum to “The T1-T2 Study: Evolution of Aerosol Properties Downwind of Mexico City” published in Atmos. Chem. Phys., 7, 1585–1598, 2007. Atmos. Chem. Phys. 2007, 7, 2197–2198. [Google Scholar]
- Doran, J.C.; Barnard, J.C.; Arnott, W.P.; Cary, R.; Coulter, R.; Fast, J.D.; Kassianov, E.I.; Kleinman, L.; Laulainen, N.S.; Martin, T.; et al. The T1-T2 Study: Evolution of Aerosol Properties Downwind of Mexico City. Atmos. Chem. Phys. 2007, 7, 1585–1598. [Google Scholar] [CrossRef]
- Yang, M.; Howell, S.G.; Zhuang, J.; Huebert, B.J. Attribution of Aerosol Light Absorption to Black Carbon, Brown Carbon, and Dust in China—Interpretations of Atmospheric Measurements during EAST-AIRE. Atmos. Chem. Phys. 2009, 9, 2035–2050. [Google Scholar] [CrossRef]
- Engelbrecht, J.P.; Moosmüller, H.; Pincock, S.; Jayanty, P.K.M.; Lersch, T.; Casuccio, G.S. Technical Note: Mineralogical, Chemical, Morphological, and Optical Interrelationships of Mineral Dust Re-Suspensions. Atmos. Chem. Phys. 2016, 16, 10809–10830. [Google Scholar] [CrossRef]
- Moosmüller, H.; Engelbrecht, J.P.; Skiba, M.; Frey, G.; Chakrabarty, R.K.; Arnott, W.P. Single Scattering Albedo of Fine Mineral Dust Aerosols Controlled by Iron Concentration. J. Geophys. Res. 2012, 117. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regression MAC (m2 g−1) | Mean and Median MAC (m2 g−1) | |||||||
|---|---|---|---|---|---|---|---|---|
| λ (nm) | Slope | Slope Error | PA Calibration Error | SP2 Calibration Error | Precision Error | Mean | Median | Standard Deviation |
| T0 Site | ||||||||
| 375 | 12.71 | 1.12 | 0.76 | 3.36 | 1.17 | 15.61 | 15.26 | 7.55 |
| 405 | 11.61 | 0.26 | 0.79 | 3.47 | 1.42 | 16.30 | 15.76 | 2.74 |
| 532 | 9.42 | 0.32 | 0.53 | 2.33 | 0.35 | 10.53 | 10.58 | 2.77 |
| 870 | 8.13 | 0.17 | 0.42 | 1.83 | 0.14 | 8.21 | 8.30 | 1.62 |
| T1 Site | ||||||||
| 355 | 18.10 | 3.95 | 1.61 | 7.09 | 3.79 | 35.88 | 32.24 | 18.90 |
| 405 | 15.71 | 0.97 | 0.96 | 4.22 | 1.95 | 19.25 | 19.19 | 3.73 |
| 532 | 10.12 | 1.03 | 0.72 | 3.15 | 0.49 | 13.97 | 14.30 | 4.88 |
| 870 | 6.51 | 0.11 | 0.36 | 1.59 | 0.10 | 7.24 | 7.23 | 0.46 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gyawali, M.; Arnott, W.P.; Zaveri, R.A.; Song, C.; Flowers, B.; Dubey, M.K.; Setyan, A.; Zhang, Q.; China, S.; Mazzoleni, C.; et al. Evolution of Multispectral Aerosol Absorption Properties in a Biogenically-Influenced Urban Environment during the CARES Campaign. Atmosphere 2017, 8, 217. https://doi.org/10.3390/atmos8110217
Gyawali M, Arnott WP, Zaveri RA, Song C, Flowers B, Dubey MK, Setyan A, Zhang Q, China S, Mazzoleni C, et al. Evolution of Multispectral Aerosol Absorption Properties in a Biogenically-Influenced Urban Environment during the CARES Campaign. Atmosphere. 2017; 8(11):217. https://doi.org/10.3390/atmos8110217
Chicago/Turabian StyleGyawali, Madhu, W. Patrick Arnott, Rahul A. Zaveri, Chen Song, Bradley Flowers, Manvendra K. Dubey, Ari Setyan, Qi Zhang, Swarup China, Claudio Mazzoleni, and et al. 2017. "Evolution of Multispectral Aerosol Absorption Properties in a Biogenically-Influenced Urban Environment during the CARES Campaign" Atmosphere 8, no. 11: 217. https://doi.org/10.3390/atmos8110217
APA StyleGyawali, M., Arnott, W. P., Zaveri, R. A., Song, C., Flowers, B., Dubey, M. K., Setyan, A., Zhang, Q., China, S., Mazzoleni, C., Gorkowski, K., Subramanian, R., & Moosmüller, H. (2017). Evolution of Multispectral Aerosol Absorption Properties in a Biogenically-Influenced Urban Environment during the CARES Campaign. Atmosphere, 8(11), 217. https://doi.org/10.3390/atmos8110217