
Importance of Gaseous Elemental Mercury Fluxes in Western Maryland


Abstract
:1. Introduction
2. Experimental Section
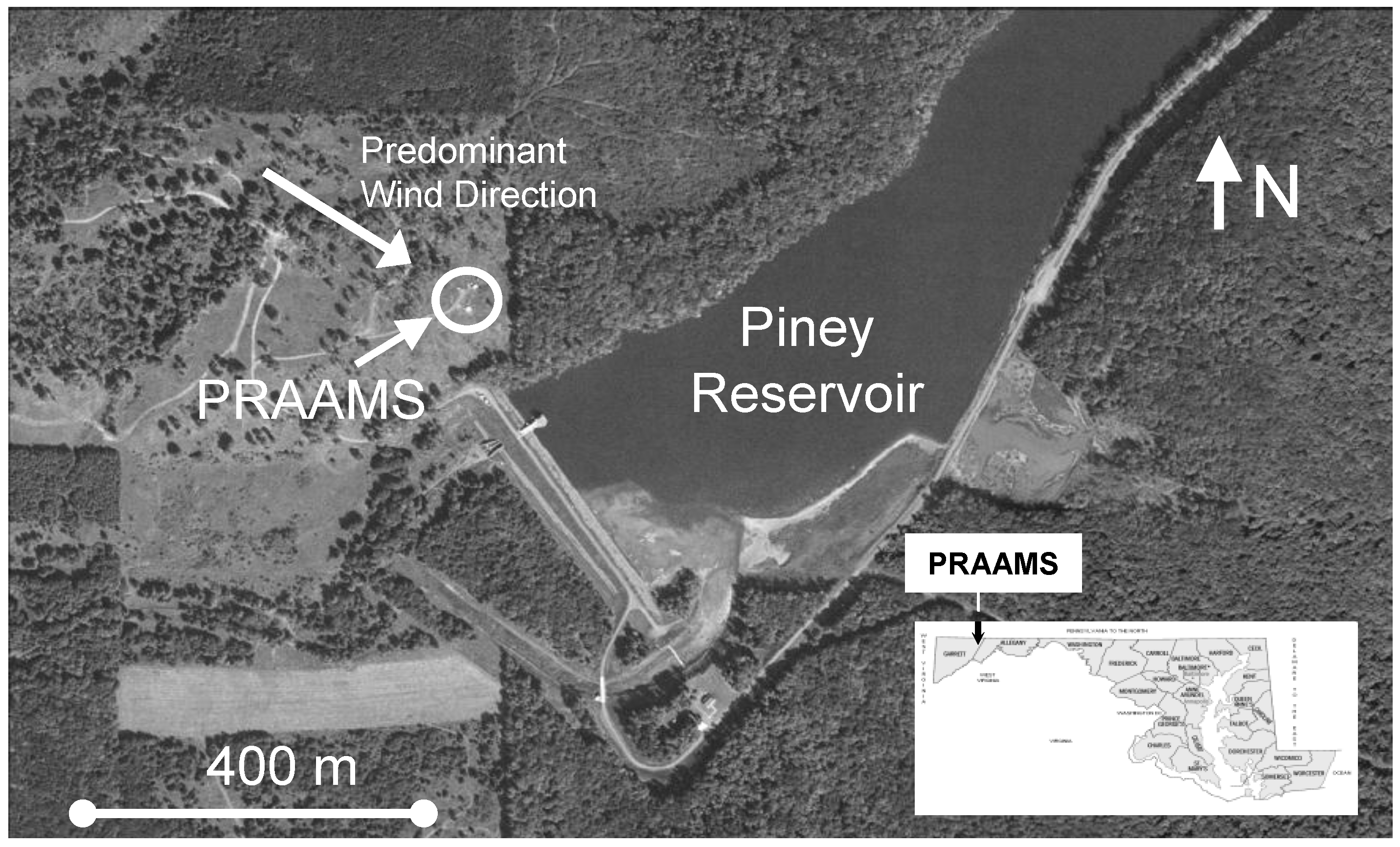
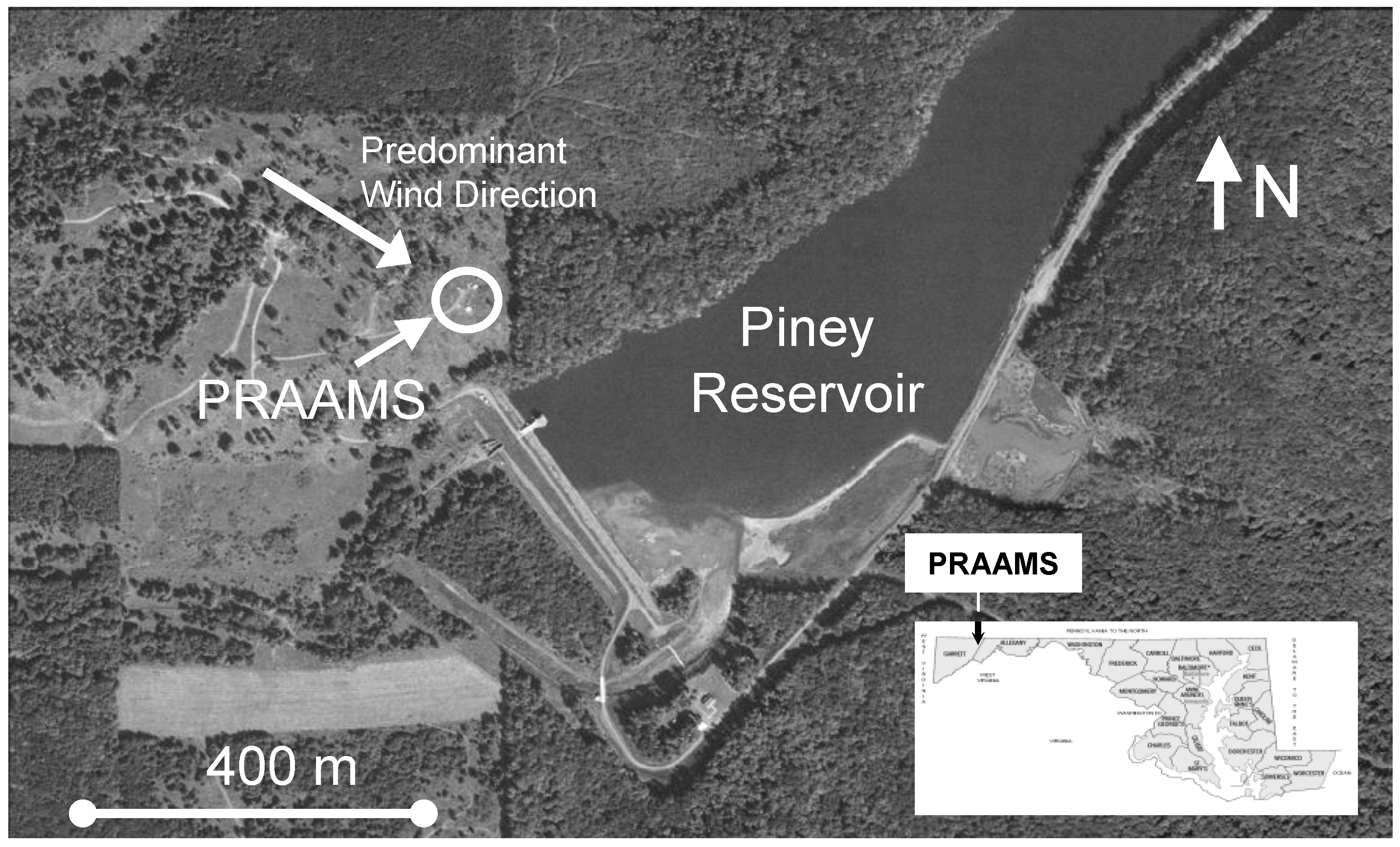
2.1. Site Description
2.2. Modified Bowen Ratio
2.3. GEM Measurements
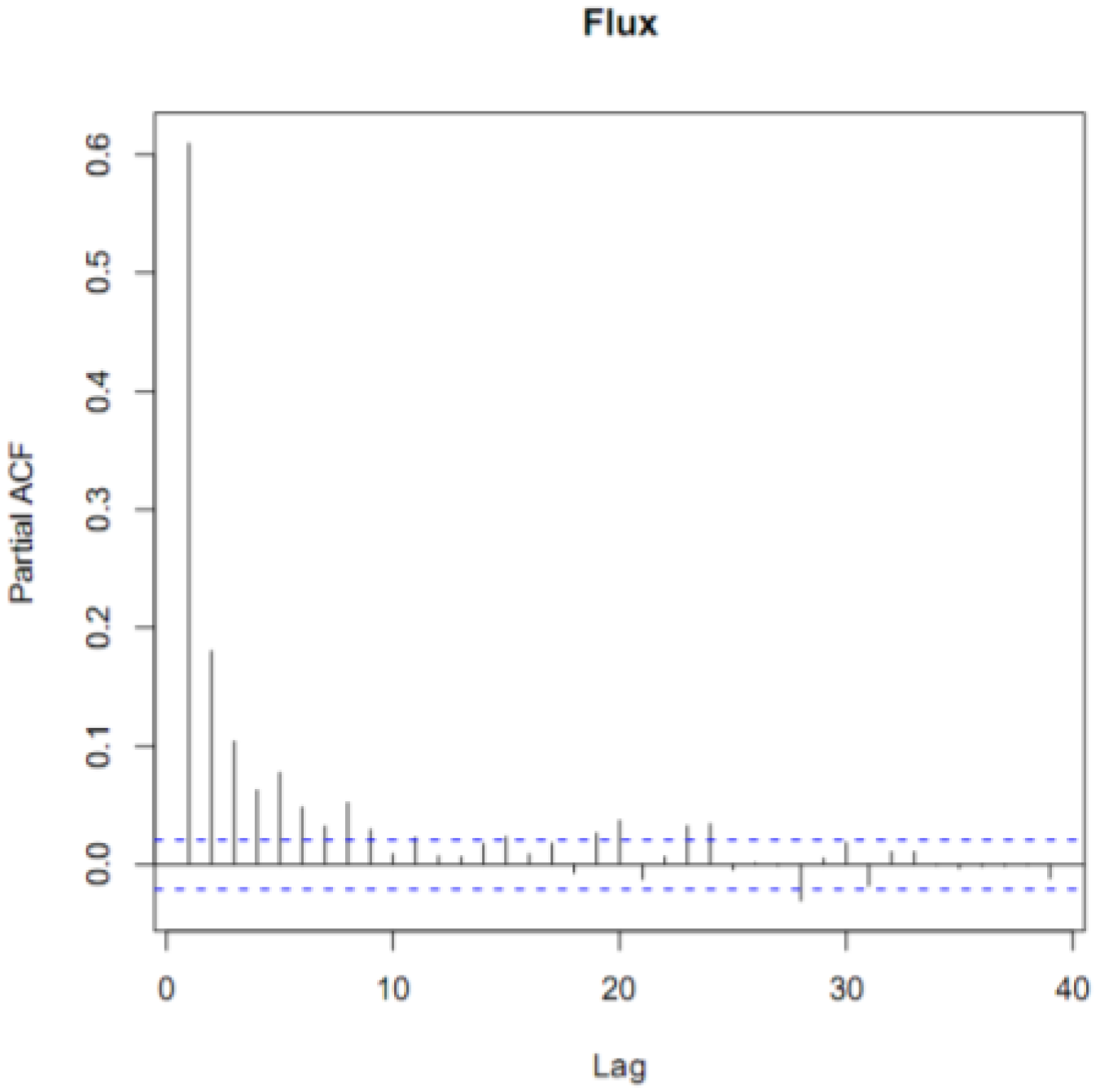
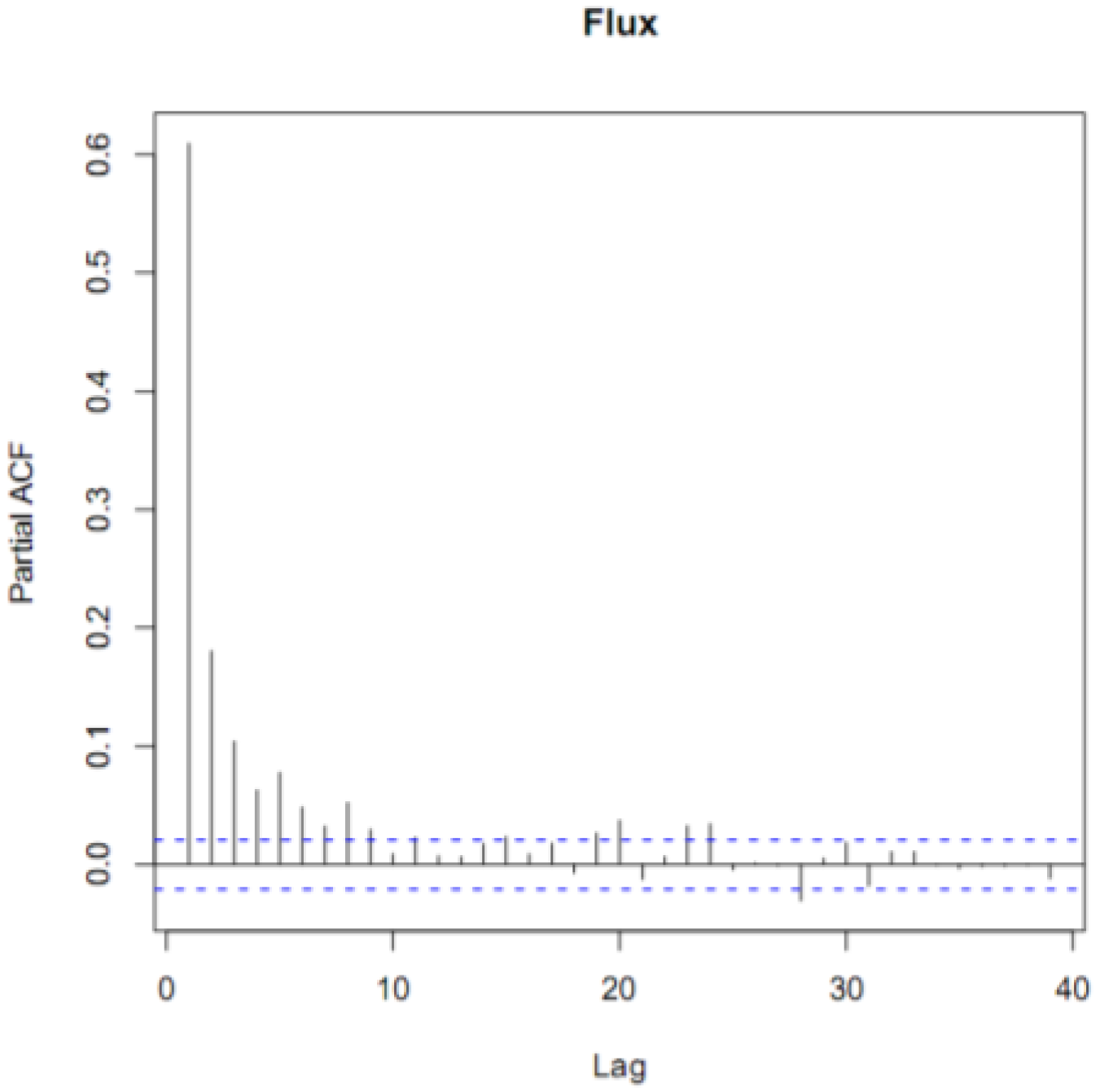
2.4. Statistical Analyses
2.5. Flux Footprint
2.6. GEM Flux Validation
3. Results and Discussion
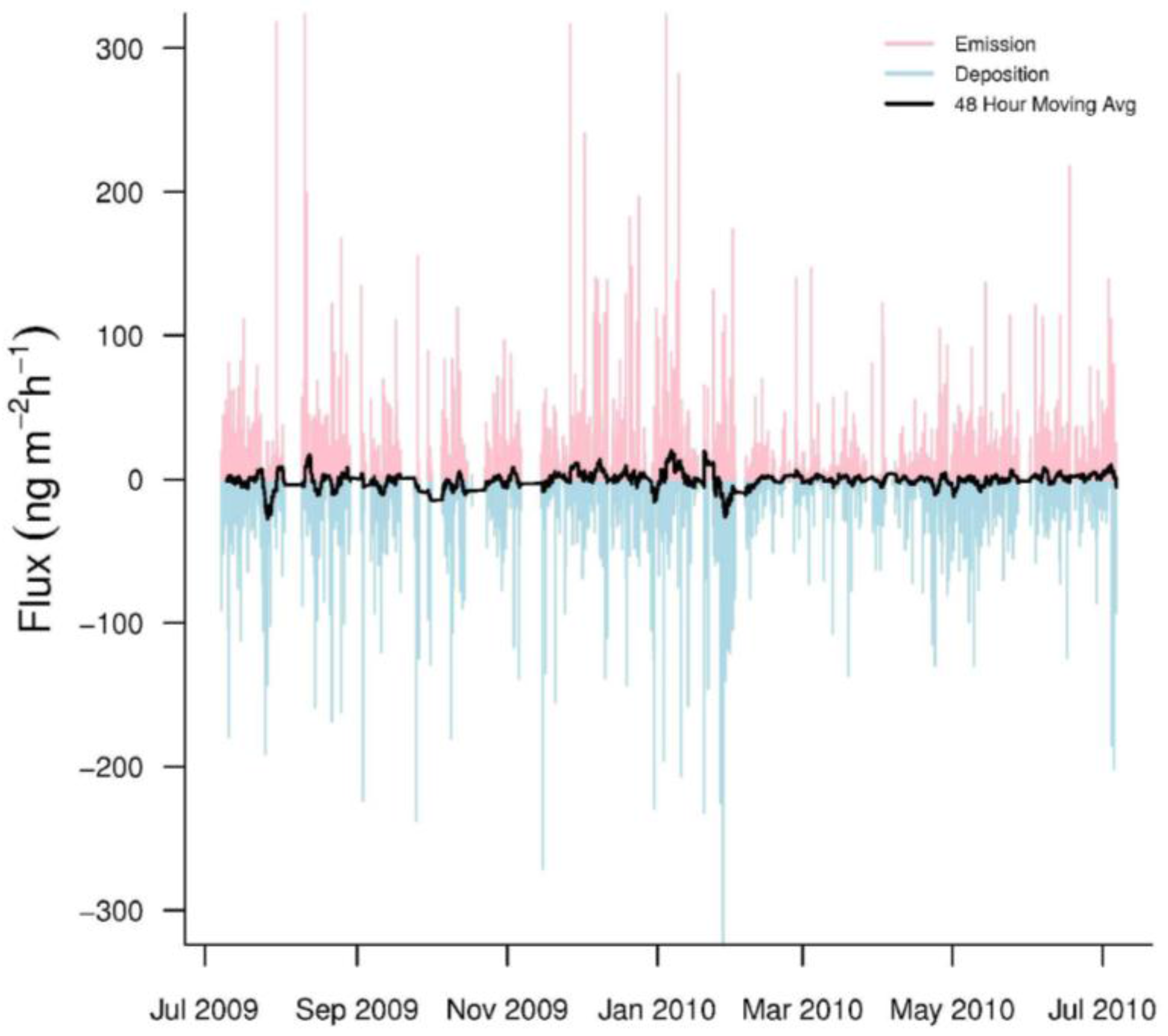
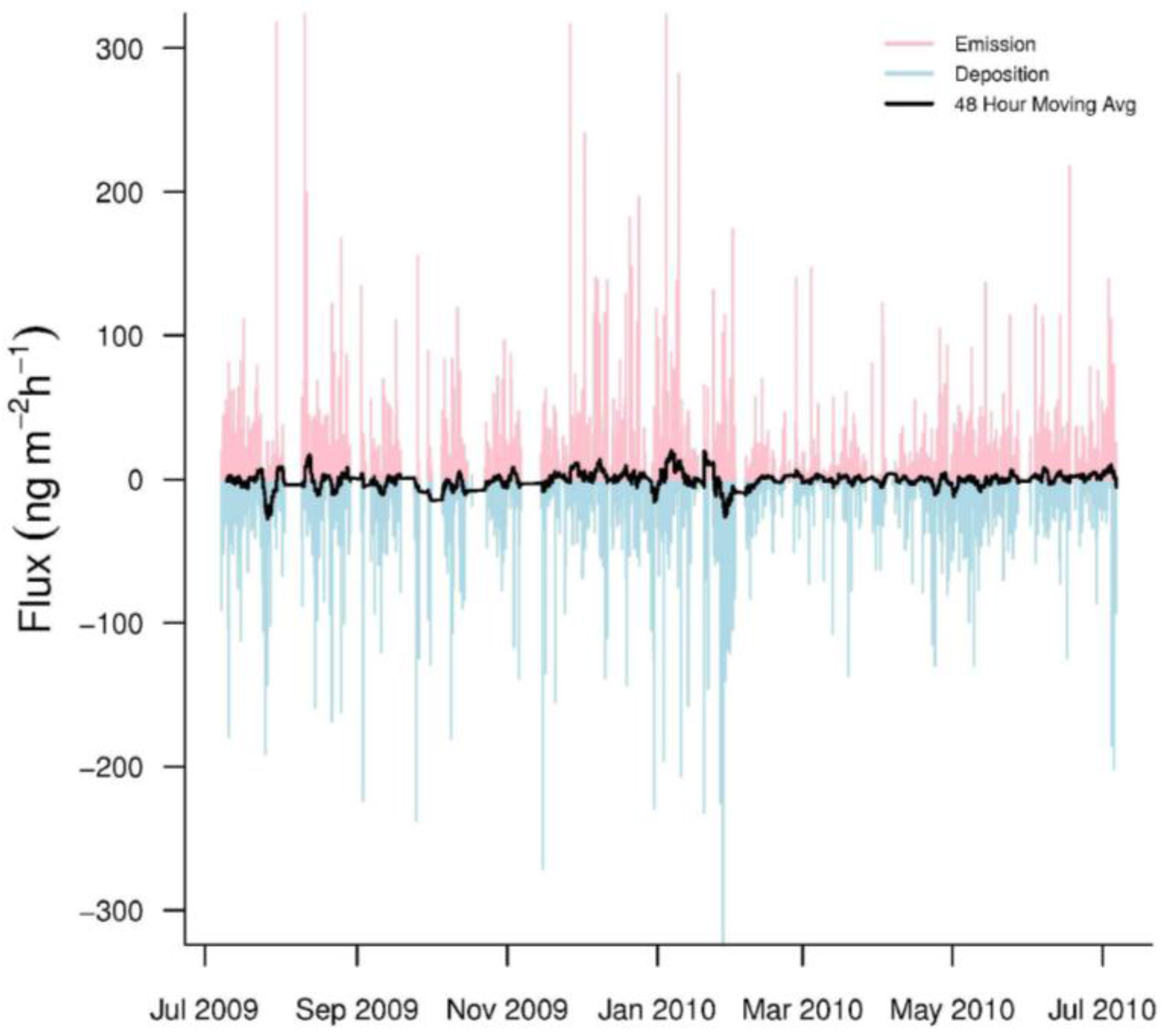
3.1. GEM Fluxes
3.2. GEM Emission and Deposition
3.3. Comparison with Other Measurements at PRAAMS
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lindberg, S.; Bullock, R.; Ebinghaus, R.; Engstrom, D.; Feng, X.B.; Fitzgerald, W.; Pirrone, N.; Prestbo, E.; Seigneur, C. A synthesis of progress and uncertainties in attributing the sources of mercury in deposition. Ambio 2007, 36, 19–32. [Google Scholar] [CrossRef]
- Gustin, M.S.; Lindberg, S.E.; Weisberg, P.J. An update on the natural sources and sinks of atmospheric mercury. Appl. Geochem. 2008, 23, 482–493. [Google Scholar] [CrossRef]
- Gustin, M.; Jaffe, D. Reducing the uncertainty in measurement and understanding of mercury in the atmosphere. Environ. Sci. Technol. 2010, 44, 2222–2227. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.H.; Moore, C.W.; Wohlfahrt, G.; HÖrtang, L.; Kljun, N.; Obrist, D. Eddy covariance flux measurements of gaseous elemental mercury using cavity ring-down spectroscopy. Environ. Sci. Technol. 2015, 49, 1559–1568. [Google Scholar] [CrossRef] [PubMed]
- Poissant, L.; Pilote, M.; Beauvais, C.; Constant, P.; Zhang, H.H. A year of continuous measurements of three atmospheric mercury species (GEM, RGM and Hgp) in southern Quebec, Canada. Atmos. Environ. 2005, 39, 1275–1287. [Google Scholar] [CrossRef]
- Bash, J.O.; Miller, D.R. A relaxed eddy accumulation system for measuring surface fluxes of total gaseous mercury. J. Atmos. Ocean. Technol. 2008, 25, 244–257. [Google Scholar] [CrossRef]
- Converse, A.D.; Riscassi, A.L.; Scanlon, T.M. Seasonal variability in gaseous mercury fluxes measured in a high-elevation meadow. Atmos. Environ. 2010, 44, 2176–2185. [Google Scholar] [CrossRef]
- Baya, A.P.; Van Heyst, B. Assessing the trends and effects of environmental parameters on the behaviour of mercury in the lower atmosphere over cropped land over four seasons. Atmos. Chem. Phys. 2010, 10, 8617–8628. [Google Scholar] [CrossRef]
- Choi, H.D.; Holsen, T.M. Gaseous mercury emissions from unsterilized and sterilized soils: The effect of temperature and UV radiation. Environ. Pollut. 2009, 157, 1673–1678. [Google Scholar] [CrossRef] [PubMed]
- Engle, M.A.; Gustin, M.S.; Lindberg, S.E.; Gertler, A.W.; Ariya, P.A. The influence of ozone on atmospheric emissions of gaseous elemental mercury and reactive gaseous mercury from substrates. Atmos. Environ. 2005, 39, 7506–7517. [Google Scholar] [CrossRef]
- Gustin, M.S.; Taylor, G.E.; Maxey, R.A. Effect of temperature and air movement on the flux of elemental mercury from substrate to the atmosphere. J. Geophys. Res. Atmos. 1997, 102, 3891–3898. [Google Scholar] [CrossRef]
- Moore, C.; Carpi, A. Mechanisms of the emission of mercury from soil: Role of UV radiation. J. Geophys. Res. Atmos. 2005, 110, D24302. [Google Scholar] [CrossRef]
- Kim, K.H.; Lindberg, S.E.; Meyers, T.P. Micrometeorological measurements of mercury-vapor fluxes over background forest soils in Eastern Tennessee. Atmos. Environ. 1995, 29, 267–282. [Google Scholar] [CrossRef]
- Schluter, K. Review: Evaporation of mercury from soils. An integration and synthesis of current knowledge. Environ. Geol. 2000, 39, 249–271. [Google Scholar] [CrossRef]
- Hall, B. The gas-phase oxidation of elemental mercury by ozone. Water Air Soil Pollut. 1995, 80, 301–315. [Google Scholar] [CrossRef]
- Schroeder, W.H.; Anlauf, K.G.; Barrie, L.A.; Lu, J.Y.; Steffen, A.; Schneeberger, D.R.; Berg, T. Arctic springtime depletion of mercury. Nature 1998, 394, 331–332. [Google Scholar] [CrossRef]
- Steffen, A.; Douglas, T.; Amyot, M.; Ariya, P.; Aspmo, K.; Berg, T.; Bottenheim, J.; Brooks, S.; Cobbett, F.; Dastoor, A.; et al. A synthesis of atmospheric mercury depletion event chemistry in the atmosphere and snow. Atmos. Chem. Phys. 2008, 8, 1445–1482. [Google Scholar] [CrossRef]
- Castro, M.S.; Moore, C.W.; Sherwell, J.S.; Brooks, S.B. Dry deposition of gaseous oxidized mercury. Sci. Total Environ. 2011, 417, 232–240. [Google Scholar]
- Castro, M.S.; Sherwell, J. Effectiveness of emission controls to reduce the atmospheric concentrations of mercury. Envion. Sci. Technol. 2015, 49, 14000–14007. [Google Scholar] [CrossRef] [PubMed]
- SERCC. Southeastern Regional Climate Center. Available online: http://www.sercc.com/climateinfo/historical/historical_md.html (accessed on 22 August 2016).
- USDA. Web Soil Survey. Available online: http://websoilsurvey.nrcs.usda.gov/app/WebSoilSurvey.aspx (accessed on 22 August 2016).
- Moore, C.W.; Castro, M.S.; Heyes, A. Investigatioin of factors influencing gaseous mercury fluxes in background soils of western Maryland. Sci. Total Environ. 2011, 419, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, J.; Obrist, D.; Zeeman, M.J.; Conen, F.; Eugster, W.; Alewell, C. Elemental mercury fluxes over a sub-alpine grassland determined with two micrometeorological methods. Atmos. Environ. 2008, 42, 2922–2933. [Google Scholar] [CrossRef]
- Gustin, M.S.; Lindberg, S.; Marsik, F.; Casimir, A.; Ebinghaus, R.; Edwards, G.; Hubble-Fitzgerald, C.; Kemp, R.; Kock, H.; Leonard, T.; et al. Nevada STORMS project: Measurement of mercury emissions from naturally enriched surfaces. J. Geophys. Res. Atmos. 1999, 104, 21831–21844. [Google Scholar] [CrossRef]
- Lindberg, S.E.; Kim, K.H.; Meyers, T.P.; Owens, J.G. Micrometeorological gradient approach for quantifying air-surface exchange of mercury-vapor—Tests over contaminated doils. Environ. Sci. Technol. 1995, 29, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Tekran. Model 2537A Mercury Vapor Analyzer User Manual. Available online: http://www.tekran.com/products/ambient-air/tekran-model-2537-cvafs-automated-mercury-analyzer/ (accessed on 22 August 2016).
- Hsieh, C.-I.; Katul, G.; Chi, T.W. An approximate analytical model for footprint estimation of scalar fluxes in thermally stratified atmospheric flows. Adv. Water Resour. 2000, 23, 765–772. [Google Scholar] [CrossRef]
- Oishi, A.C.; Oren, R.; Stoy, P.C. Estimating components of forest evapotranspiration: A footprint approach for scaling sap flux measurements. Agric. For. Meterol. 2000, 148, 1719–1732. [Google Scholar] [CrossRef]
- Park, S.J.; Park, S.U.; Ho, C.H.; Mahrt, L. Flux-gradient relationship of water vapor in the surface layer obtained from CASES-99 experiment. J.Geophys. Res. Atmos. 2009, 114, D08115. [Google Scholar] [CrossRef]
- Wohlfahrt, G.; Hortnagl, L.; Hammerle, A.; Graus, M.; Hansel, A. Measuring eddy covariance fluxes of ozone with a slow-response analyser. Atmos. Environ. 2009, 43, 4570–4576. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.P. Introduction to Micrometeorology, 2nd ed.; Academic Press: San Diego, CA, USA, 2001. [Google Scholar]
- Lindberg, S.E.; Hanson, P.J.; Meyers, T.P.; Kim, K.H. Air/surface exchange of mercury vapor over forests—The need for a reassessment of continental biogenic emissions. Atmos. Environ. 1998, 32, 895–908. [Google Scholar] [CrossRef]
- Cobos, D.R.; Baker, J.M.; Nater, E.A. Conditional sampling for measuring mercury vapor fluxes. Atmos. Environ. 2002, 36, 4309–4321. [Google Scholar] [CrossRef]
- Fritsche, J.; Wohlfahrt, G.; Ammann, C.; Zeeman, M.; Hammerle, A.; Obrist, D.; Alewell, C. Summertime elemental mercury exchange of temperate grasslands on an ecosystem-scale. Atmos. Chem. Phys. 2008, 8, 7709–7722. [Google Scholar] [CrossRef] [PubMed]
- Poissant, L.; Pilote, M.; Constant, P.; Beauvais, C.; Zhang, H.H.; Xu, X.H. Mercury gas exchanges over selected bare soil and flooded sites in the bay St. Francois wetlands. Atmos. Environ. 2004, 38, 4205–4214. [Google Scholar] [CrossRef]
- Zhang, L.M.; Wright, L.P.; Blanchard, P. A review of current knowledge concerning dry deposition of atmospheric mercury. Atmos. Environ. 2009, 43, 5853–5864. [Google Scholar] [CrossRef]
- Xin, M.; Gustin, M.; Johnson, D. Laboratory investigation of the potential for re-emission of atmospherically derived Hg from soils. Environ. Sci. Technol. 2007, 41, 4946–4951. [Google Scholar] [CrossRef] [PubMed]
- Jacob, D.J.; Horowitz, L.W.; Munger, J.W.; Heikes, B.G.; Dickerson, R.R.; Artz, R.S.; Keene, W.C. Seasonal transition from NOx- to hydrocarbon-limited conditions for ozone production over the eastern United-States in September. J. Geophys. Res. Atmos. 1995, 100, 9315–9324. [Google Scholar] [CrossRef]
- Boudala, F.S.; Folkins, I.; Beauchamp, S.; Tordon, R.; Neima, J.; Johnson, B. Mercury flux measurements over air and water in Kejimkujik National Park, Nova Scotia. Water Air Soil Pollut. 2000, 122, 183–202. [Google Scholar] [CrossRef]
- Ericksen, J.A.; Gustin, M.S.; Xin, M.; Weisberg, P.J.; Fernandez, G.C.J. Air-soil exchange of mercury from background soils in the United States. Sci. Total Environ. 2006, 366, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Poissant, L.; Casimir, A. Water-air and soil-air exchange rate of total gaseous mercury measured at background sites. Atmos. Environ. 1998, 32, 883–893. [Google Scholar] [CrossRef]
- Risch, M.R.; DeWild, J.F.; Krabbenhoft, D.P.; Kolka, R.K.; Zhang, L. Litterfall mercury dry deposition in the eastern USA. Environ. Pollut. 2012, 161, 284–290. [Google Scholar] [CrossRef] [PubMed]
- NADP. National Atmospheric Deposition Program 2009 Annual Summary; NADP Data Report 2009-01, Illinois State Water Survey; University of Illionois at Urbana-Champaign: Charpaign, IL, USA, 2010. [Google Scholar]
- Ryaboshapko, A.; Bullock, O.R.; Christensen, J.; Cohen, M.; Dastoor, A.; Ilyin, I.; Petersen, G.; Syrakov, D.; Artz, R.S.; Davignon, D.; et al. Intercomparison study of atmospheric mercury models: Comparison of models with short-term measurements. Sci. Total Environ. 2007, 376, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.K.; Vanarsdale, A.; Keeler, G.J.; Chalmers, A.; Poissant, L.; Kamman, N.C.; Brulotte, R. Estimation and mapping of wet and dry mercury deposition across northeastern North America. Ecotoxicology 2005, 14, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Engle, M.A.; Tate, M.T.; Krabbenhoft, D.P.; Schauer, J.J.; Kolker, A.; Shanley, J.B.; Bothner, M.H. Comparison of atmospheric mercury speciation and deposition at nine sites across central and eastern North America. J. Geophys. Res. Atmos. 2010, 115, D18306. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Parameter | Frequency | Equipment |
|---|---|---|
| Soil Temperature | 10 Minutes | Decagon Devices 5TE |
| Soil Moisture | 10 Minutes | Decagon Devices 5TE |
| Soil Redox | 10 Minutes | Platinum wire probe |
| Total UV | 10 Minutes | Apogee Instruments SU-100 |
| UVB | 10 Minutes | Skye Instruments SKU 430 |
| Net Solar Radiation | 10 Minutes | Kipp and Zonen CNR-1 |
| Albedo | 10 Minutes | Kipp and Zonen CNR-1 |
| Ozone | 1 Hour | Thermo Electron 49C |
| CO | 1 Hour | Thermo Electron 58i-Tle |
| OCEC | 1 Hour | Sunset Labs 3F |
| SO2 | 1 Hour | Eco Tek EC9850t |
| NO | 1 Hour | Eco Tek EC9843 |
| NO2 | 1 Hour | Eco Tek EC9843 |
| NOy | 1 Hour | Eco Tek EC9843 |
| SO4 | 1 Hour | 5020 SPA Thermo |
| PM2.5 | 1 Hour | Met One Instruments BAM 1020 PM2.5 |
| Wind Speed (10 m) | 1 Hour | Vaisala WXT 520 |
| Wind Direction (10 m) | 1 Hour | Vaisala WXT 520 |
| Relative Humidity (RH) | 1 Hour | Vaisala WXT 520 |
| Air Temp (10 m) | 1 Hour | Vaisala WXT 520 |
| Dew point | 1 Hour | Calculated from Air Temp (10m) and RH |
| Rain | 1 Hour | Vaisala WXT 520 |
| Barometric Pressure | 1 Hour | Vaisala WXT 520 |
| Surface Wetness | 1 Hour | Cambell Scientific 237-L |
| Wind Speed (3 m) | 1 Hour | RM Young 05103 Wind Monitor |
| Wind Direction (3 m) | 1 Hour | RM Young 05103 Wind Monitor |
| GEM | 1 Hour | Tekran 2537 |
| GOM | 2 Hour | Tekran 1130 |
| Particulate Mercury | 2 Hour | Tekran 1135 |
| Start | End | Mean (GEM) Inlet 1 (ng·m−3) | Mean (GEM) Inlet 2 (ng·m−3) | Mean Difference | Maximum Gradient Magnitude | Minimum Gradient Magnitude | |
|---|---|---|---|---|---|---|---|
| Collocated | 26 June 2009 | 2 July 2009 | 1.199 | 1.190 | 0.009 | 0.086 | 0.000 |
| Collocated | 2 August 2009 | 9 August 2009 | 1.093 | 1.094 | 0.001 | 0.134 | 0.000 |
| Collocated | 29 August 2009 | 2 September 2009 | 1.024 | 1.008 | 0.016 | 0.125 | 0.000 |
| Collocated | 18 September 2009 | 23 September 2009 | 1.029 | 1.028 | 0.001 | 0.122 | 0.000 |
| Collocated | 6 November 2009 | 14 November 2009 | 1.532 | 1.534 | 0.008 | 0.110 | 0.000 |
| Collocated | 1 February 2010 | 5 February 2010 | 1.809 | 1.809 | 0.000 | 0.098 | 0.002 |
| Separated (above 0.009 threshold) | 2 July 2009 | 6 July 2010 | - | - | −0.004 | 0.570 | 0.009 |
| Mean TGM Flux (ng·m−2·hr−1) | Min Flux (ng·m−2·hr−1) | Max Flux (ng·m−2·hr−1) | Time of Year | Ecosystem | Study |
|---|---|---|---|---|---|
| PRAAMS: | |||||
| −1.21 ± 29.3 | −224.0 | 353.6 | Summer | Upland Meadow surrounded with deciduous forest | This study |
| −0.31 ± 30.9 | −271.3 | 316.8 | Fall | Upland Meadow surrounded with deciduous forest | This study |
| −0.23 ± 33.3 | −346.1 | 379.8 | Winter | Upland Meadow surrounded with deciduous forest | This study |
| −0.84 ± 22.3 | −129.7 | 217.7 | Spring | Upland Meadow surrounded with deciduous forest | This study |
| Other Studies: | |||||
| 2.5 ± 19.1 | −124.8 | 82.4 | 6–12 August 2009 | High elevation meadow | [7] |
| 0.3 ± 16.8 | −77.1 | 67.6 | 7–14 November 2008 | High elevation meadow | [7] |
| 4.1 ± 25.7 | −112.0 | 119.1 | 11–17 February 2009 | High elevation meadow | [7] |
| −4.8 ± 25.5 | −125.7 | 71.0 | 11–19 May 2009 | High elevation meadow | [7] |
| NR | −5.4 | 4.2 | 2–10 June 1994 | Forest Floor | [32] |
| −4.3 ± NR | −42.0 | 20.0 | 26 August–23 November 2005 | Grassland | [23] |
| −1.7 ± NR | −35.0 | 34.0 | 27 March–30 August 2006 | Grassland | [23] |
| 0.3 ± NR | −34.0 | 29.0 | 24 November 2005–26 March 2006 | Grassland with snow | [23] |
| 9.67 ± NR | −91.7 | 190.5 | 7–14 May and 31 May–8 June 2001 | Agricultural Field | [33] |
| −4.3 ± NR | −27.0 | 14.0 | 7 June–20 July 2006 | Grassland | [34] |
| −1.6 ± NR | −14.0 | 14.0 | 7 June–20 July 2006 | Grassland | [34] |
| −2.1 ± NR | −41.0 | 26.0 | 14–29 June 2006 | Grassland | [34] |
| −0.5 ± NR | −76.0 | 37.0 | 14–29 June 2006 | Grassland | [34] |
| 0.2 ± NR | −33.0 | 29.0 | 14–26 September 2006 | Managed Farmland | [34] |
| 0.3 ± NR | −18.0 | 30.0 | 14–26 September 2006 | Managed Farmland | [34] |
| 32.1 ± 55.6 | −110.0 | 278.0 | 23 August–3 September 2002 | Wetlands, open water, mixed vegetation | [35] |
| Emission | Deposition | |||
|---|---|---|---|---|
| Season | Parameter | Correlation Coefficient | Parameters | Correlation Coefficient |
| Summer | Relative Humidity | 0.44 | ||
| UVB | 0.47 | WS (3 m) | −0.42 | |
| Relative Humidity | −0.36 | Ozone | −0.40 | |
| Ozone | 0.31 | Total UV | −0.40 | |
| WS (3 m) | 0.27 | UVB | −0.38 | |
| Total UV | 0.26 | Surface Wetness | 0.35 | |
| Albedo | 0.28 | |||
| Fall | UVB | 0.28 | WS (3 m) | −0.29 |
| Winter | WS (10 m) | 0.24 | WS (10 m) | −0.26 |
| Spring | Net Solar Radiation | −0.47 | ||
| Net Solar Radiation | 0.37 | UVB | −0.41 | |
| UVB | 0.31 | Total UV | −0.28 | |
| WS (10 m) | −0.28 | |||
| Oe-A soil Horizon | WS (3 m) | −0.26 | ||
| Soil Temperature | 0.30 | Relative Humidity | 0.25 | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro, M.S.; Moore, C.W. Importance of Gaseous Elemental Mercury Fluxes in Western Maryland. Atmosphere 2016, 7, 110. https://doi.org/10.3390/atmos7090110
Castro MS, Moore CW. Importance of Gaseous Elemental Mercury Fluxes in Western Maryland. Atmosphere. 2016; 7(9):110. https://doi.org/10.3390/atmos7090110
Chicago/Turabian StyleCastro, Mark S., and Christopher W. Moore. 2016. "Importance of Gaseous Elemental Mercury Fluxes in Western Maryland" Atmosphere 7, no. 9: 110. https://doi.org/10.3390/atmos7090110
APA StyleCastro, M. S., & Moore, C. W. (2016). Importance of Gaseous Elemental Mercury Fluxes in Western Maryland. Atmosphere, 7(9), 110. https://doi.org/10.3390/atmos7090110