Atmospheric Black Carbon along a Cruise Path through the Arctic Ocean during the Fifth Chinese Arctic Research Expedition

Abstract
:1. Introduction
2. Experimental Methods
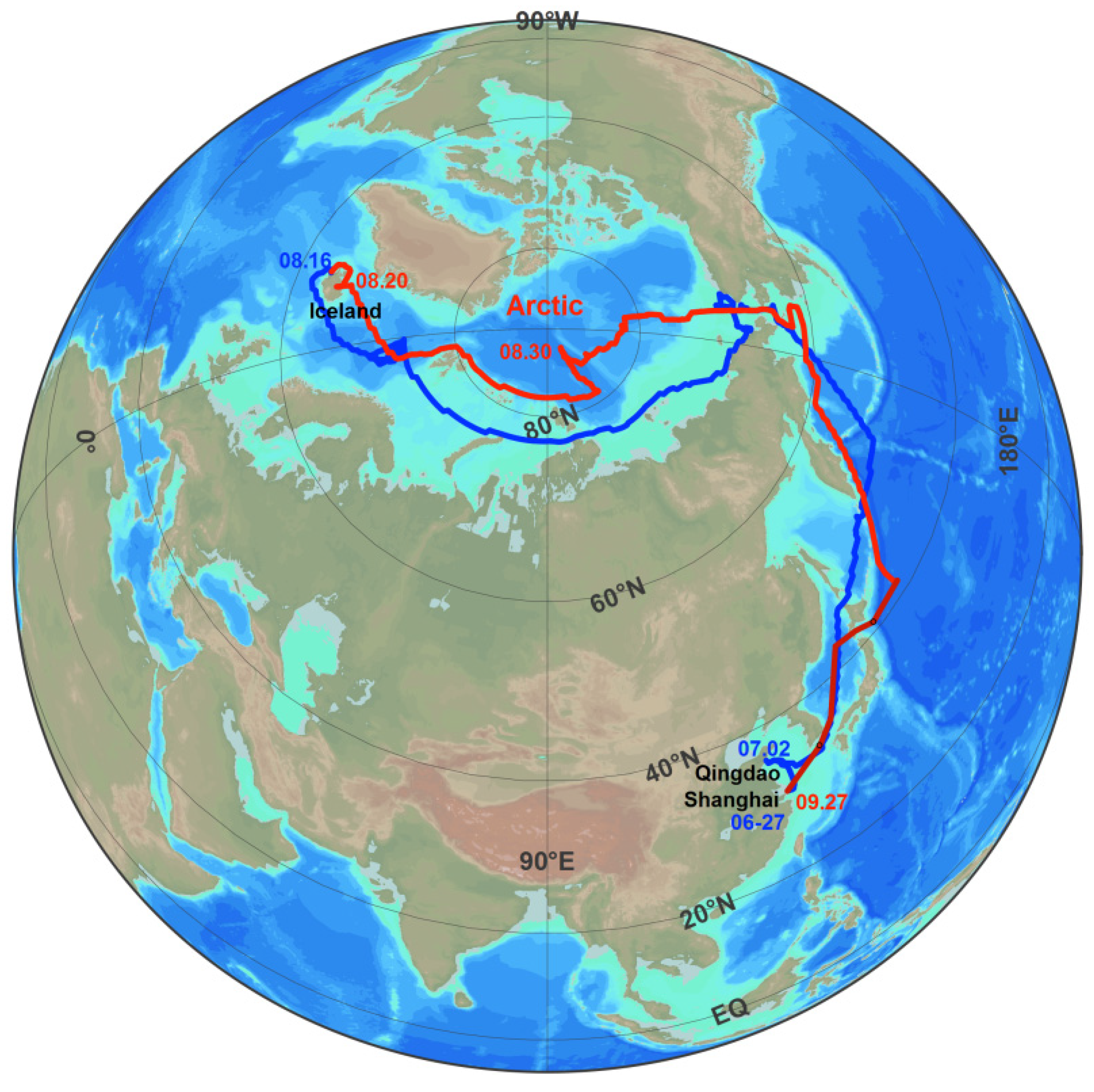
2.1. The Cruise Route
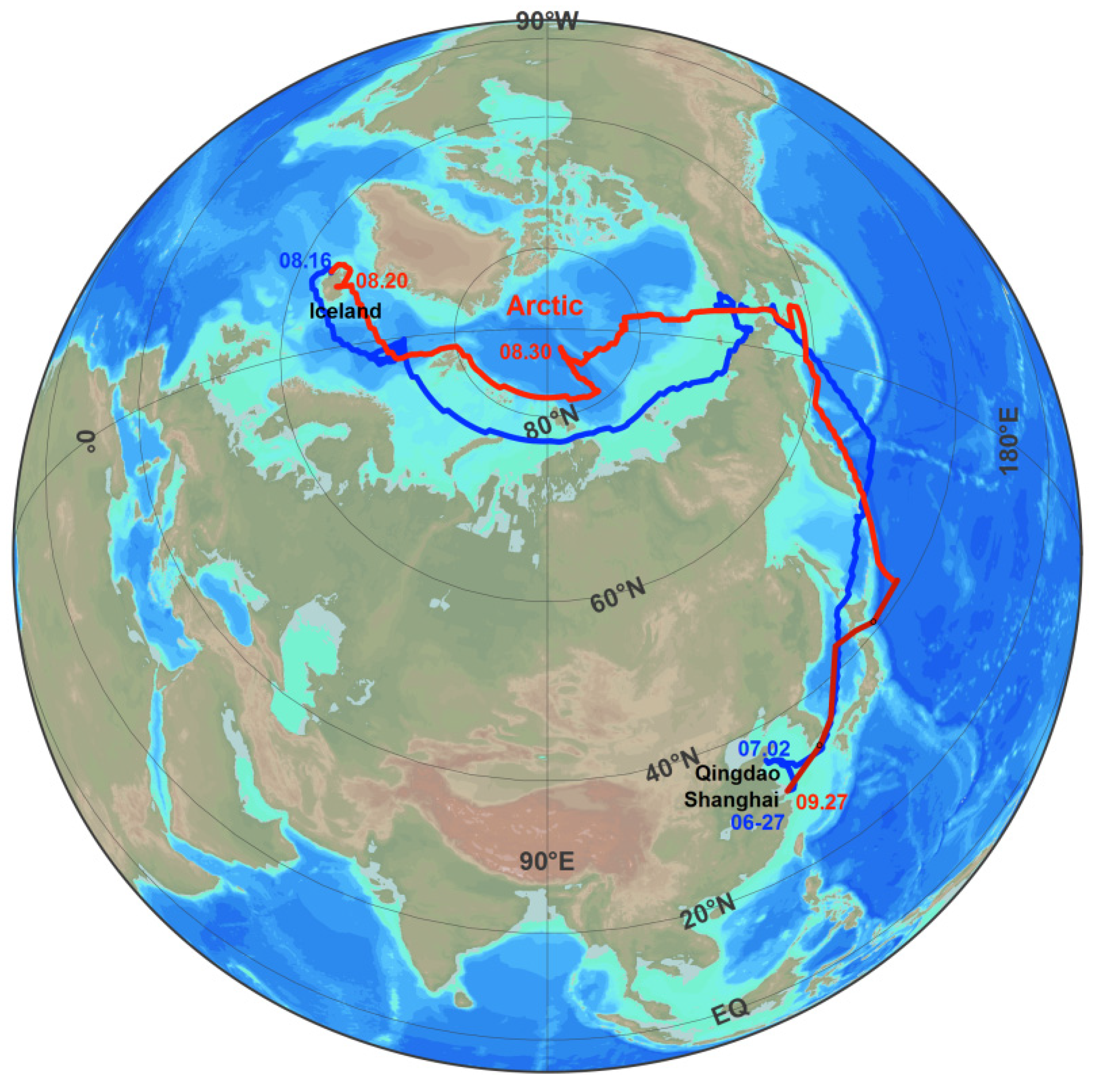
2.2. Observation and the Data Processing
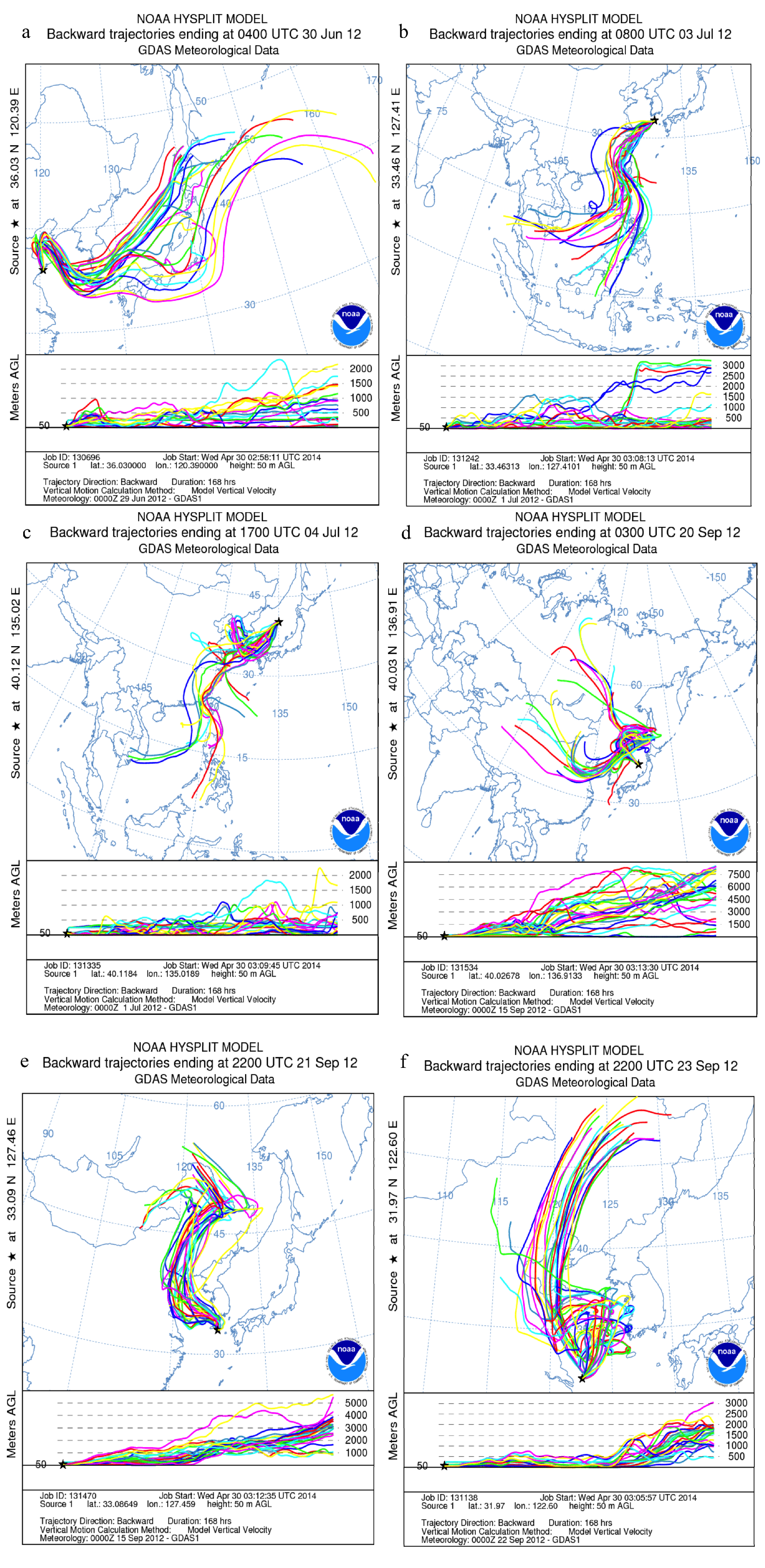
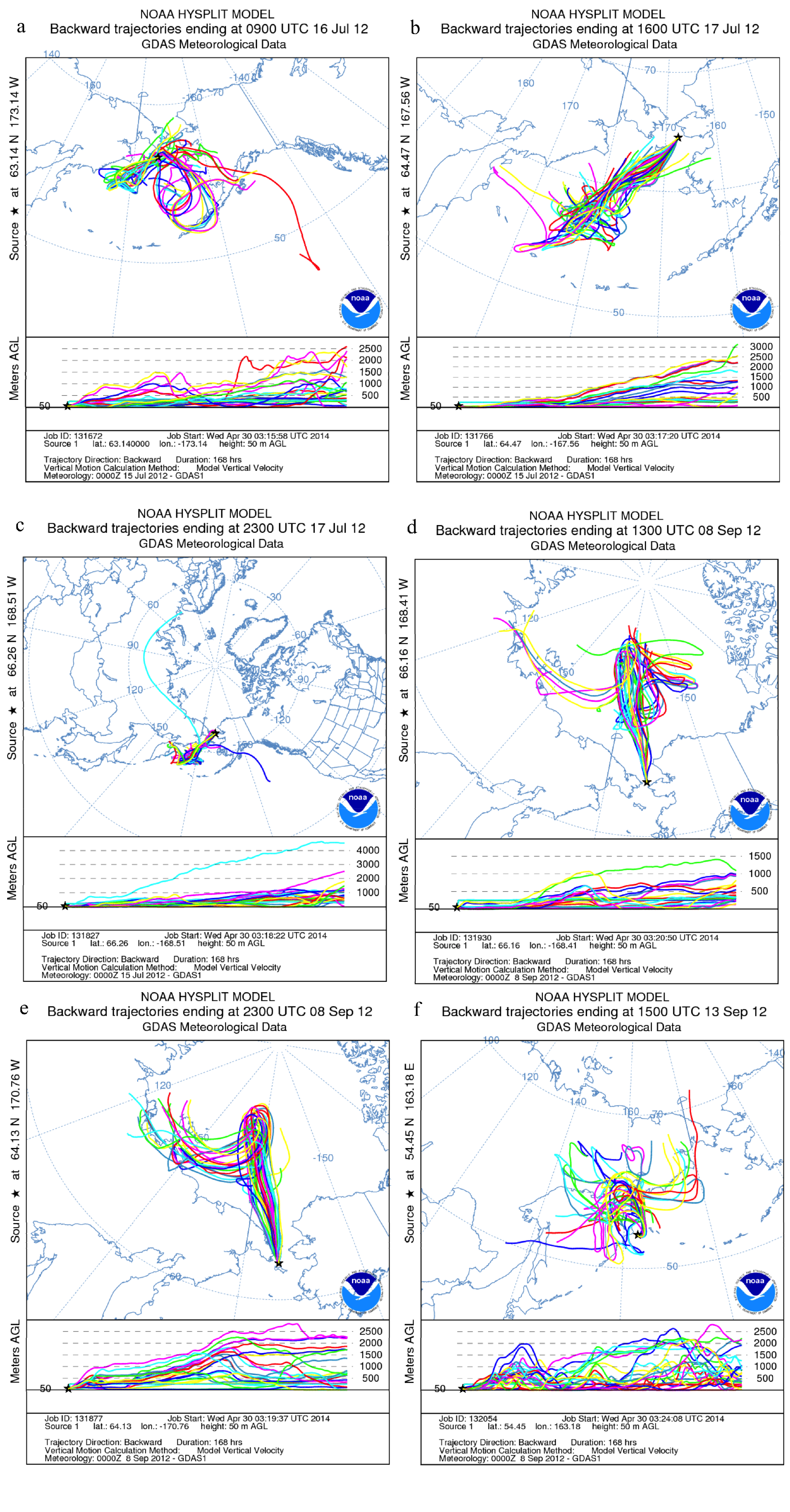
2.3. Air Mass Back Trajectories
3. Results and Discussion
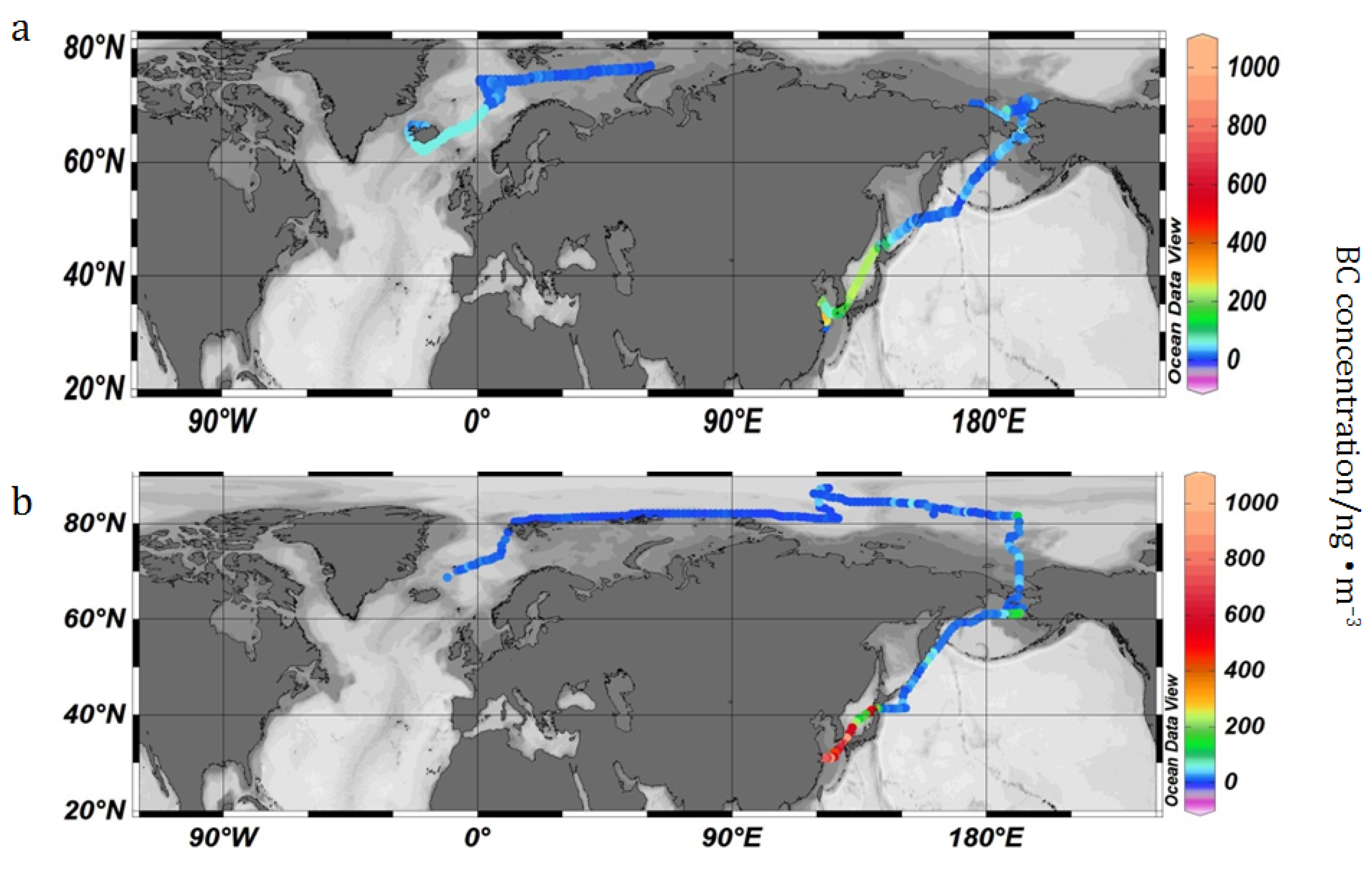
3.1. Temporal and Spatial Variation of Hourly Average Concentration
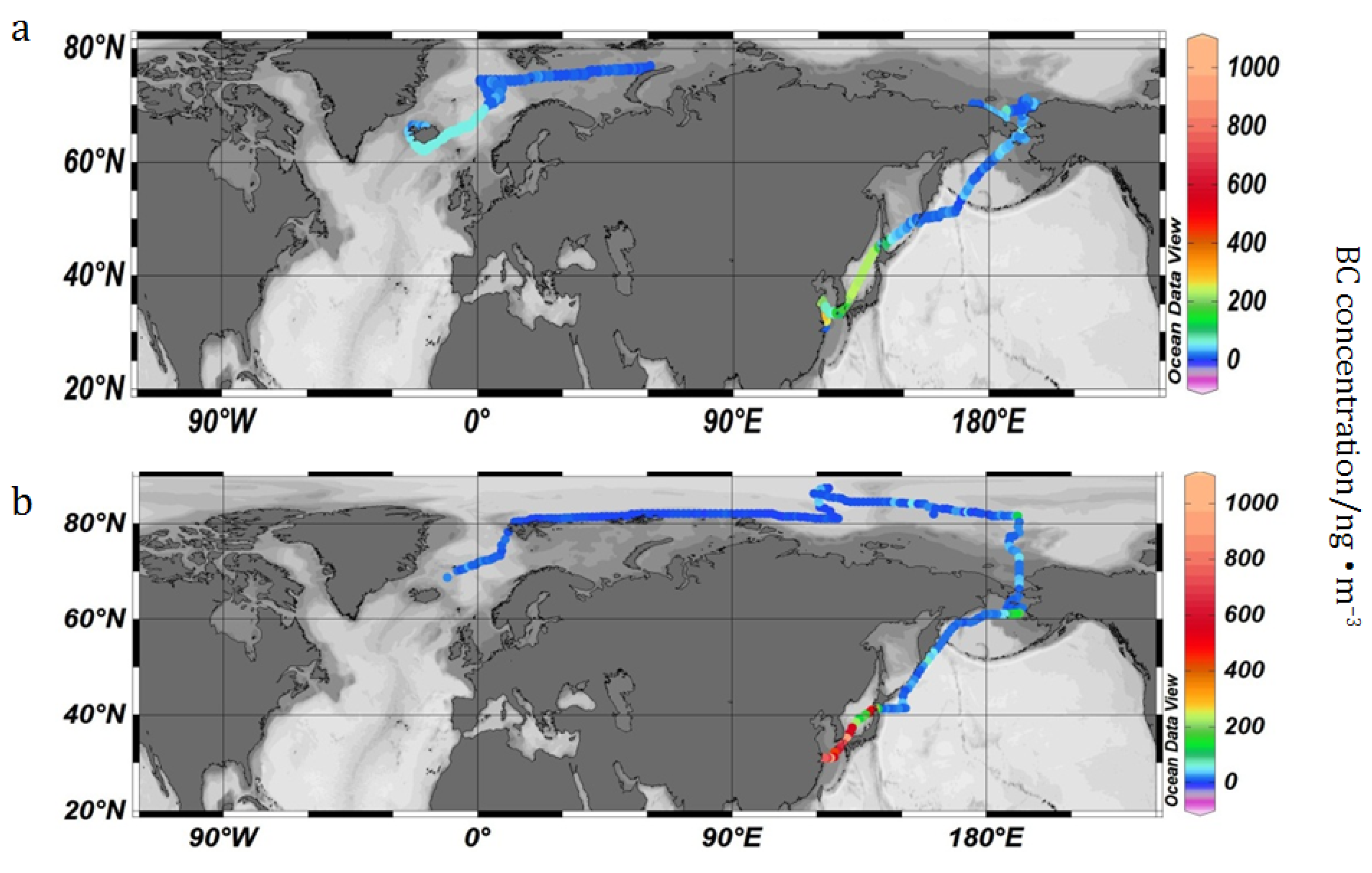

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Sea Names | Time | Average/ng∙m3 | SD/ng·m3 | Median/ng∙m3 |
|---|---|---|---|---|---|
| 1 | Offshore China | 6.27–7.3.2012 | 192.81 | 102.80 | 203.27 |
| 2 | Sea of Japan | 7.3–7.6.2012 | 170.89 | 69.19 | 170.75 |
| 3 | Sea of Okhotsk | 7.6–7.8.2012 | 32.81 | 23.64 | 25.69 |
| 4 | Northwest Pacific Ocean | 7.8–7.11.2012 | 15.18 | 7.95 | 13.00 |
| 5 | Bering Sea and Bering Strait | 7.11–7.19.2012 | 21.89 | 12.98 | 21.50 |
| 6 | Chukchi and East Siberian Sea | 7.19–7.24.2012 | 21.81 | 15.67 | 15.11 |
| 7 | Barents Sea | 7.31–8.2.2012 | 9.40 | 5.39 | 7.88 |
| 8 | Norwegian Sea | 8.2–8.14.2012 | 14.66 | 9.98 | 11.56 |
| 9 | Iceland | 8.16–8.20.2012 | 142.61 | 114.26 | 136.04 |
| 10 | Greenland Sea | 8.22–8.24.2012 | 11.03 | 4.53 | 10.19 |
| 11 | High latitude Arctic Ocean | 8.24–9.6.2012 | 5.96 | 4.58 | 4.87 |
| 12 | Chukchi and East Siberian Sea | 9.6–9.7.2012 | 19.51 | 9.70 | 16.95 |
| 13 | Bering Sea and Bering Strait | 9.8–9.14.2012 | 18.23 | 13.45 | 13.49 |
| 14 | Northwest Pacific Ocean | 9.14–9.19.2012 | 24.24 | 16.47 | 19.26 |
| 15 | Sea of Japan | 9.19–9.21.2012 | 443.87 | 266.09 | 460.61 |
| 16 | Offshore China | 9.21–9.23.2012 | 643.44 | 152.15 | 603.33 |
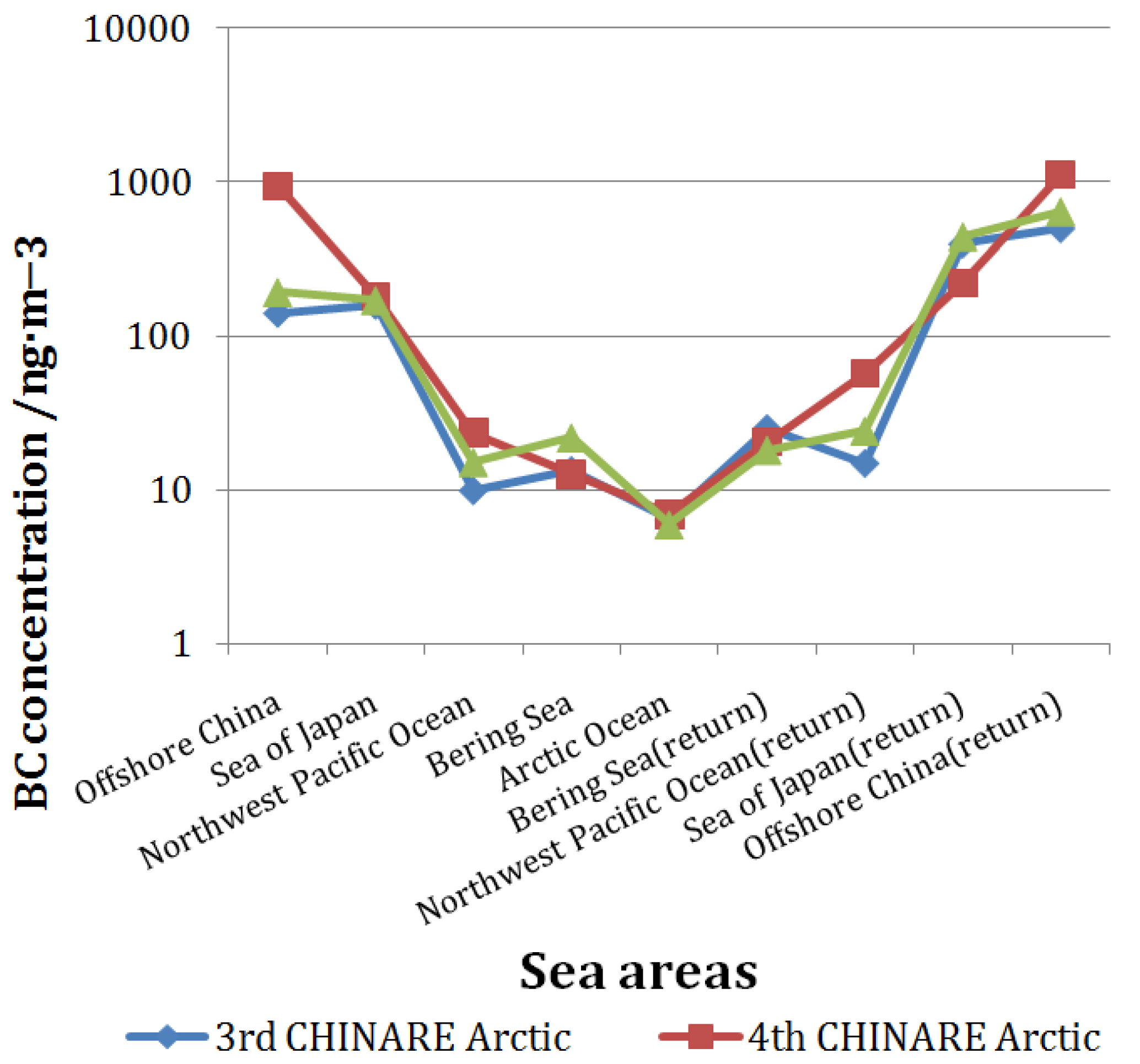
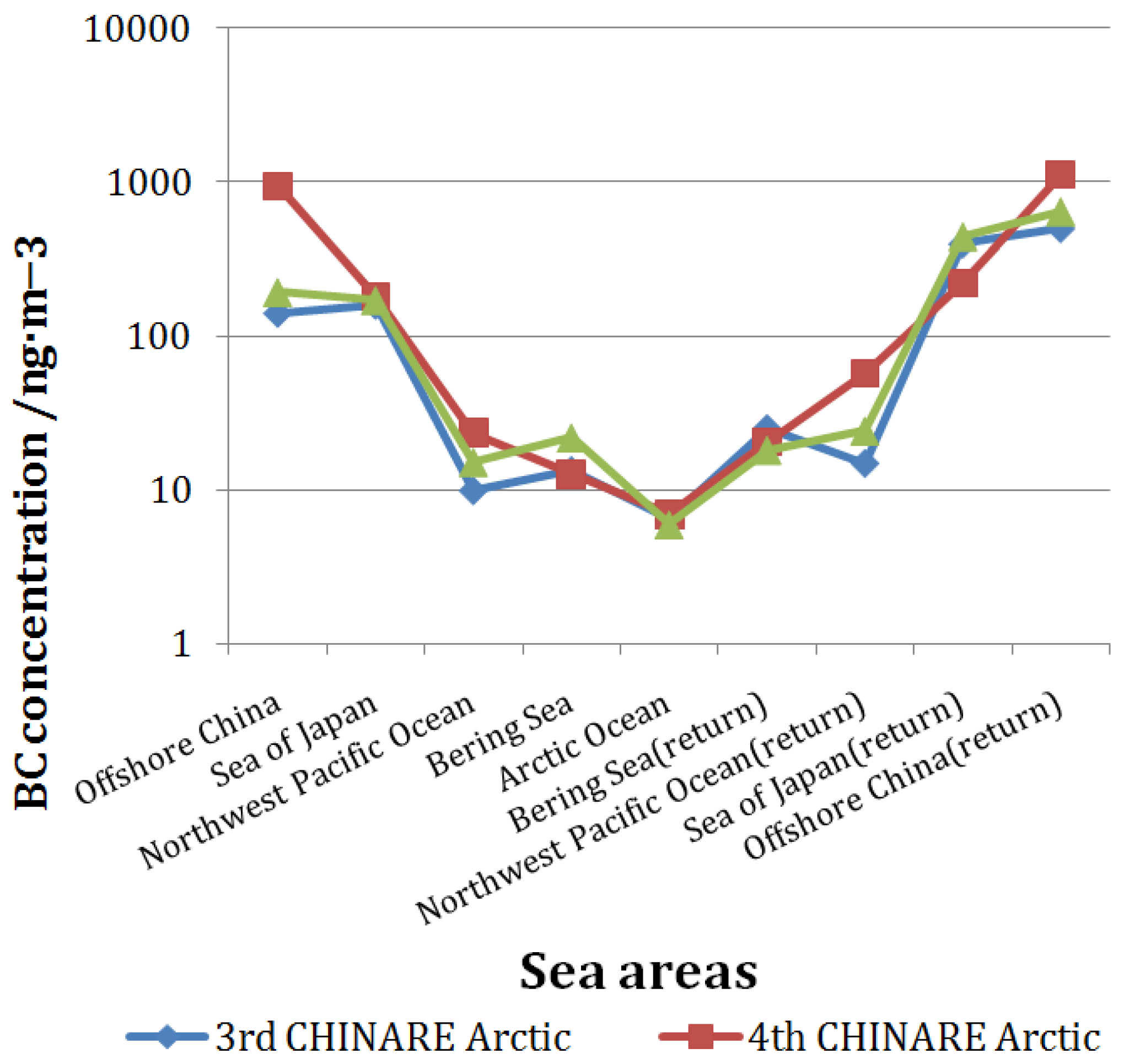
3.2. BC Concentration in the Arctic Region
3.3. Comparison with Previous CHINARE Results
| Site or Route | Period | BC Concentration/ng∙m−3 | Ref. |
|---|---|---|---|
| Ny-Ålesund (78.9°N, 11.9°E) | April 1977–May 1979 | 68 (3~174) | [26] |
| 1990–1992 | 80 | [27] | |
| Zeppelin, Ny-Ålesund (78.9°N, 11.9°E) | 1998–2007 | 39 (0~80) | [22] |
| June–September 1998–2007 | 0~10 | ||
| Yellow River Station, Ny-Ålesund (78.9°N, 11.9°E) | Summer 2005–2008 | 15 | [23] |
| Norwegian part of the Arctic | June–September 1980 | 5 | [28] |
| Barrow (71.2°N, 156.6°W) | 1986–1995 | 10~5,000 | [29] |
| 1988–1993 | 26 (1~300) | [5] | |
| 1989–2003 | 20~40 | [21] | |
| January 1989–December 1990 | 70 (16~320) | [30] | |
| 2006–2007a | 14.1 | [33] | |
| 2006–August 2007a | 3.4 | ||
| Alert (82.4°N, 62.3°W) | 1989–2002 | 8~96 | [31] |
| 1989–2003 | 25~73 | [21] | |
| 2004–2005 | 52.2 | [33] | |
| August 2004–2005 | 14.3 | ||
| Arctic Ocean | August 2008 | 6.4 (3.0~10.7) | [32] |
| August 2010 | 6.9 | ||
| High latitude Arctic Ocean (>72°N) | August 2012 | 5.96 | This work |
3.4. Impact of Terrestrial Transport
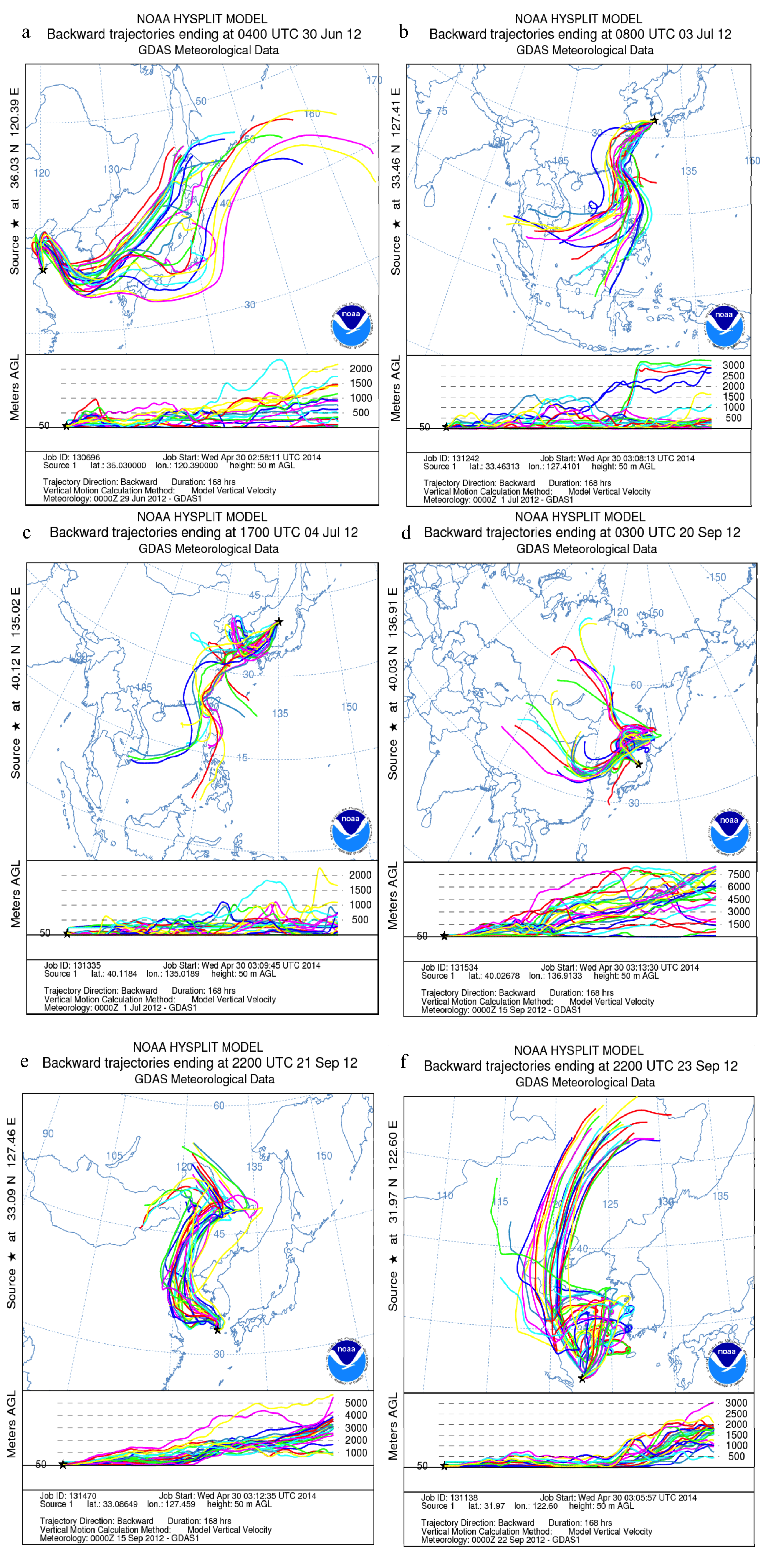
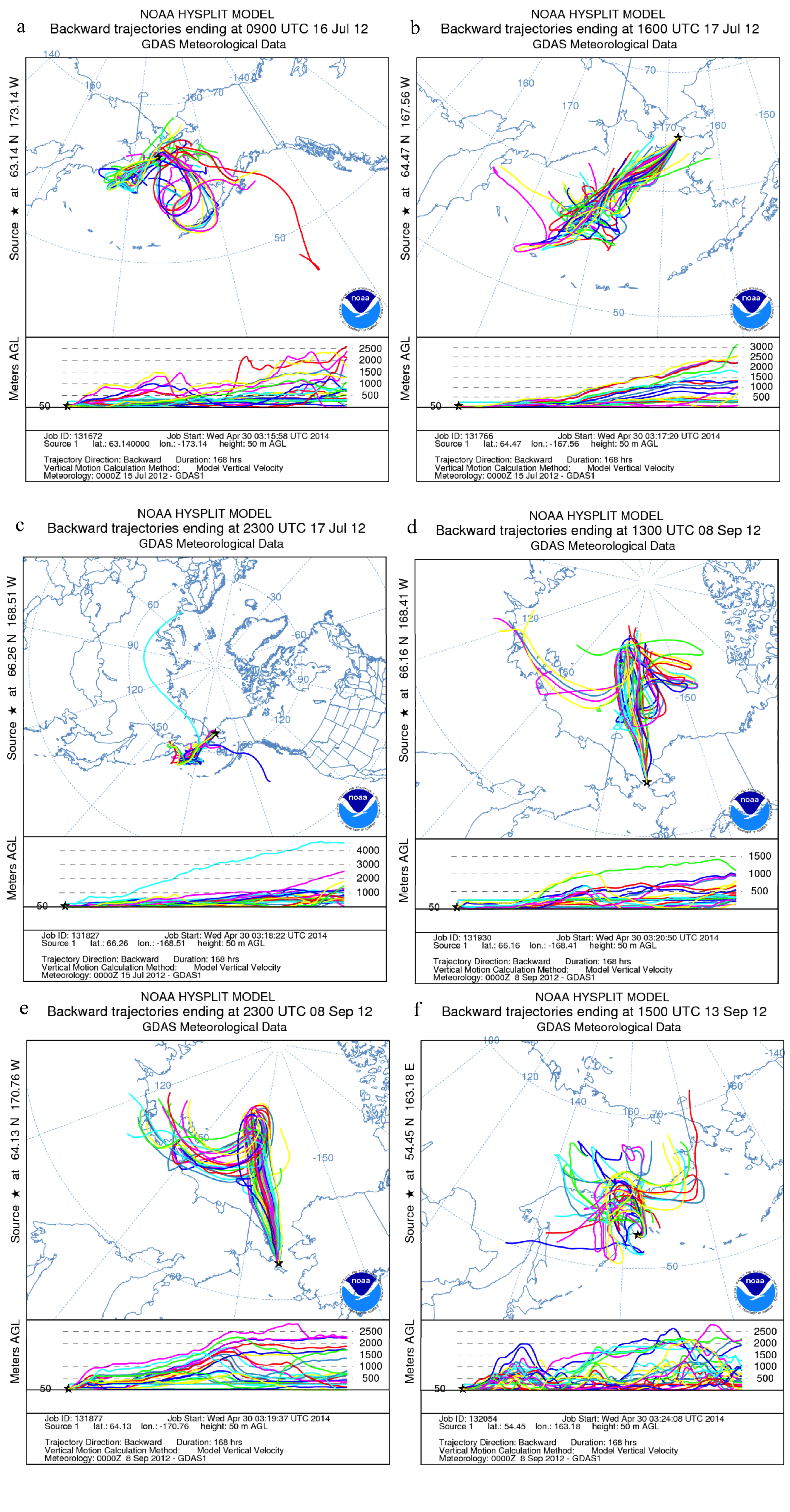
4. Summary
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Andreae, M.O.; Crutzen, P.J. Atmospheric aerosols: Biogeochemical sources and role in atmospheric chemistry. Science 1997, 276, 1052–1058. [Google Scholar] [CrossRef]
- Penner, J.E.; Novakov, T. Carbonaceous particles in the atmosphere: A historical perspective to the Fifth International Conference on Carbonaceous Particles in the Atmosphere. J. Geophys. Res.: Atmos. 1996, 101, 19373–19378. [Google Scholar] [CrossRef]
- Ramanathan, V.; Carmichael, G. Global and regional climate changes due to black carbon. Nat. Geosci. 2008, 1, 221–227. [Google Scholar] [CrossRef]
- Chung, S.H.; Seinfeld, J.H. Global distribution and climate forcing of carbonaceous aerosols. J. Geophys. Res.: Atmos. 2002. [Google Scholar] [CrossRef]
- Arctic Climate Impact Assessment (ACIA). Arctic Climate Impact Assessment; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Forster, P.; Ramaswamy, V.; Artaxo, P.; Berntsen, T.; Betts, R.; Fahey, D.W.; Haywood, J.; Lean, J.; Lowe, D.C.; Myhre, G. Changes in atmospheric constituents and in radiative forcing. In Climate Change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2007; pp. 163–167. [Google Scholar]
- Quinn, P.; Shaw, G.; Andrews, E.; Dutton, E.; Ruoho-Airola, T.; Gong, S. Arctic haze: Current trends and knowledge gaps. Tellus B 2007, 59, 99–114. [Google Scholar] [CrossRef]
- Rosen, H.; Novakov, T.; Bodhaine, B. Soot in the Arctic. Atmos. Environ. 1981, 15, 1371–1374. [Google Scholar] [CrossRef]
- Clarke, A.D.; Noone, K.J. Soot in the Arctic snowpack: A cause for perturbations in radiative transfer. Atmos. Environ. 1985, 19, 2045–2053. [Google Scholar] [CrossRef]
- Jacobson, M.Z. Strong radiative heating due to the mixing state of black carbon in atmospheric aerosols. Nature 2001, 409, 695–697. [Google Scholar] [CrossRef]
- Tang, J.; Bian, L.; Yan, P.; Lu, C. The observational study of black carbon aerosol over the cruise during the Third National Arctic Research Expedition of China. Acta Oceanol. Sin. 2011, 33, 60–68. (in Chinese). [Google Scholar]
- Draxler, R.; Rolph, G. HYSPLIT (HYbrid Single-Particle Lagrangian Integrated Trajectory). 2011. Available online: http://ready.arl.noaa.gov/HYSPLIT.php (accessed on 22 April 2014).
- Koch, D.; Hansen, J. Distant origins of Arctic Black Carbon: A Goddard Institute for Space Studies ModelE experiment. J. Geophys. Res. 2005. [Google Scholar] [CrossRef]
- Barrie, L.A. Arctic air pollution: An overview of current knowledge. Atmos. Environ. 1986, 20, 643–663. [Google Scholar] [CrossRef]
- Hansen, A.; Rosen, H.; Novakov, T. The aethalometer—An instrument for the real-time measurement of optical absorption by aerosol particles. Sci. Total Environ. 1984, 36, 191–196. [Google Scholar] [CrossRef]
- Iversen, T.; Joranger, E. Arctic air pollution and large scale atmospheric flows. Atmos. Environ. 1985, 19, 2099–2108. [Google Scholar] [CrossRef]
- Klonecki, A.; Hess, P.; Emmons, L.; Smith, L.; Orlando, J.; Blake, D. Seasonal changes in the transport of pollutants into the Arctic troposphere-model study. J. Geophys. Res.: Atmos. 2003. [Google Scholar] [CrossRef]
- Flanner, M.G.; Zender, C.S.; Randerson, J.T.; Rasch, P.J. Present-day climate forcing and response from black carbon in snow. J. Geophys. Res.: Atmos. 2007. [Google Scholar] [CrossRef]
- Hansen, J.; Nazarenko, L. Soot climate forcing via snow and ice albedos. Proc. Natl. Acad. Sci. USA 2004, 101, 423–428. [Google Scholar] [CrossRef]
- McConnell, J.R.; Edwards, R.; Kok, G.L.; Flanner, M.G.; Zender, C.S.; Saltzman, E.S.; Banta, J.R.; Pasteris, D.R.; Carter, M.M.; Kahl, J.D. 20th-century industrial black carbon emissions altered arctic climate forcing. Science 2007, 317, 1381–1384. [Google Scholar] [CrossRef]
- Sharma, S.; Andrews, E.; Barrie, L.; Ogren, J.; Lavoue, D. Variations and sources of the equivalent black carbon in the high Arctic revealed by long-term observations at Alert and Barrow: 1989–2003. J. Geophys. Res.: Atmos. 2006. [Google Scholar] [CrossRef]
- Eleftheriadis, K.; Vratolis, S.; Nyeki, S. Aerosol black carbon in the European Arctic: Measurements at Zeppelin station, Ny-Ålesund, Svalbard from 1998–2007. Geophys. Res. Lett. 2009. [Google Scholar] [CrossRef]
- Zhan, J.; Chen, L.; Zhang, Y.; Xulin, Y.; Wei, L. Prospect of research on Arctic black carbon aerosol. Chin. J. Polor Res. 2010, 22, 56–68. (in Chinese). [Google Scholar] [CrossRef]
- Treffeisen, R.; Herber, A.; Ström, J.; Shiobara, M.; Yamanouchi, T.; Yamagata, S.; Holmén, K.; Kriews, M.; Schrems, O. Interpretation of Arctic aerosol properties using cluster analysis applied to observations in the Svalbard area. Tellus B 2004, 56, 457–476. [Google Scholar] [CrossRef]
- Nyeki, S.; Coulson, G.; Colbeck, I.; Eleftheriadis, K.; Baltensperger, U.; Beine, H. Overview of aerosol microphysics at Arctic sunrise: measurements during the NICE renoxification study. Tellus B 2005, 57, 40–50. [Google Scholar] [CrossRef]
- Heintzenberg, J. Size-segregated measurements of particulate elemental carbon and aerosol light absorption at remote Arctic locations. Atmos. Environ. 1982, 16, 2461–2469. [Google Scholar] [CrossRef]
- Heintzenberg, J.; Leck, C. Seasonal variation of the atmospheric aerosol near the top of the marine boundary layer over Spitsbergen related to the Arctic sulphur cycle*. Tellus B 1994, 46, 52–67. [Google Scholar]
- Lannefors, H.; Heintzenberg, J.; HANSSON, H.C. A comprehensive study of physical and chemical parameters of the Arctic summer aerosol; results from the Swedish expedition Ymer-80. Tellus B 1983, 35, 40–54. [Google Scholar]
- Polissar, A.V.; Hopke, P.K.; Paatero, P.; Malm, W.C.; Sisler, J.F. Atmospheric aerosol over Alaska: 2. Elemental composition and sources. J. Geophys. Res.: Atmos. 1998, 103, 19045–19057. [Google Scholar] [CrossRef]
- Hopper, J.; Worthy, D.; Barrie, L.; Trivett, N. Atmospheric observations of aerosol black carbon, carbon dioxide and methane in the high Arctic. Atmos. Environ. 1994, 28, 3047–3054. [Google Scholar] [CrossRef]
- Sharma, S.; Lavoué, D.; Cachier, H.; Barrie, L.; Gong, S. Long-term trends of the black carbon concentrations in the Canadian Arctic. J. Geophys. Res.: Atmos. 2004. [Google Scholar] [CrossRef]
- Tang, J.; Bian, L.; Yan, P.; Lai, X.; Lu, C. On board measurement of black carbon aerosols over the Arctic Ocean in summer. Adv. Polar Sci. 2011, 22, 253–259. [Google Scholar]
- World Meteorological Organization World Data Center for Aerosols. Available online: http://wdca.jrc.it/ (accessed on 29 April 2014).
- Liu, Y.; Zhou, M. Temporal and spatial characteristics of aerosols over the East China Sea. Acta Oceanol. Sin. 1999, 21, 32–40. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xing, J.; Bian, L.; Hu, Q.; Yu, J.; Sun, C.; Xie, Z. Atmospheric Black Carbon along a Cruise Path through the Arctic Ocean during the Fifth Chinese Arctic Research Expedition. Atmosphere 2014, 5, 292-306. https://doi.org/10.3390/atmos5020292
Xing J, Bian L, Hu Q, Yu J, Sun C, Xie Z. Atmospheric Black Carbon along a Cruise Path through the Arctic Ocean during the Fifth Chinese Arctic Research Expedition. Atmosphere. 2014; 5(2):292-306. https://doi.org/10.3390/atmos5020292
Chicago/Turabian StyleXing, Jie, Lingen Bian, Qihou Hu, Juan Yu, Chen Sun, and Zhouqing Xie. 2014. "Atmospheric Black Carbon along a Cruise Path through the Arctic Ocean during the Fifth Chinese Arctic Research Expedition" Atmosphere 5, no. 2: 292-306. https://doi.org/10.3390/atmos5020292
APA StyleXing, J., Bian, L., Hu, Q., Yu, J., Sun, C., & Xie, Z. (2014). Atmospheric Black Carbon along a Cruise Path through the Arctic Ocean during the Fifth Chinese Arctic Research Expedition. Atmosphere, 5(2), 292-306. https://doi.org/10.3390/atmos5020292