Mercury Speciation at a Coastal Site in the Northern Gulf of Mexico: Results from the Grand Bay Intensive Studies in Summer 2010 and Spring 2011

 ,
,
Abstract
:1. Introduction
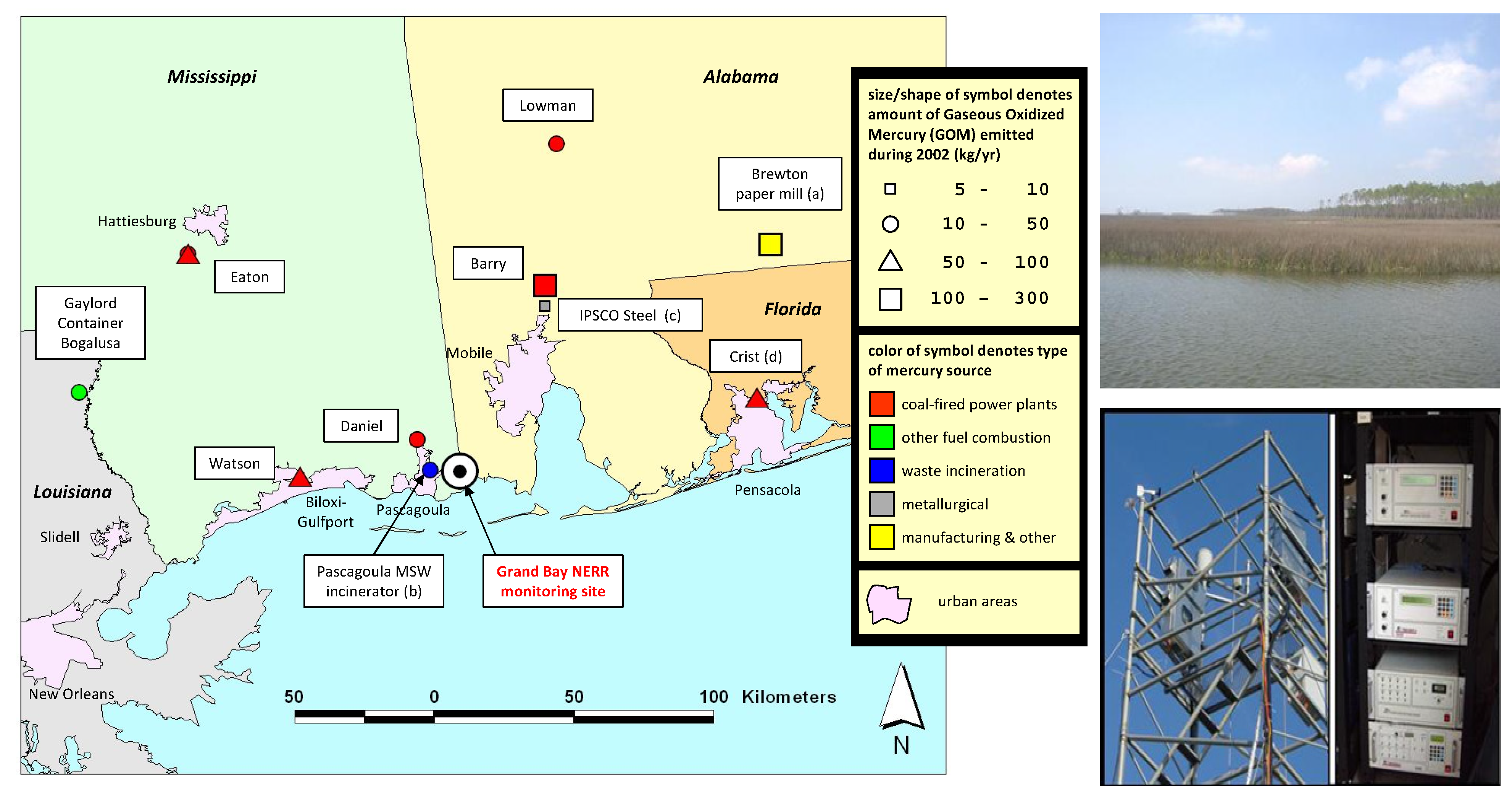
2. Results and Discussion
2.1. Surface Observations
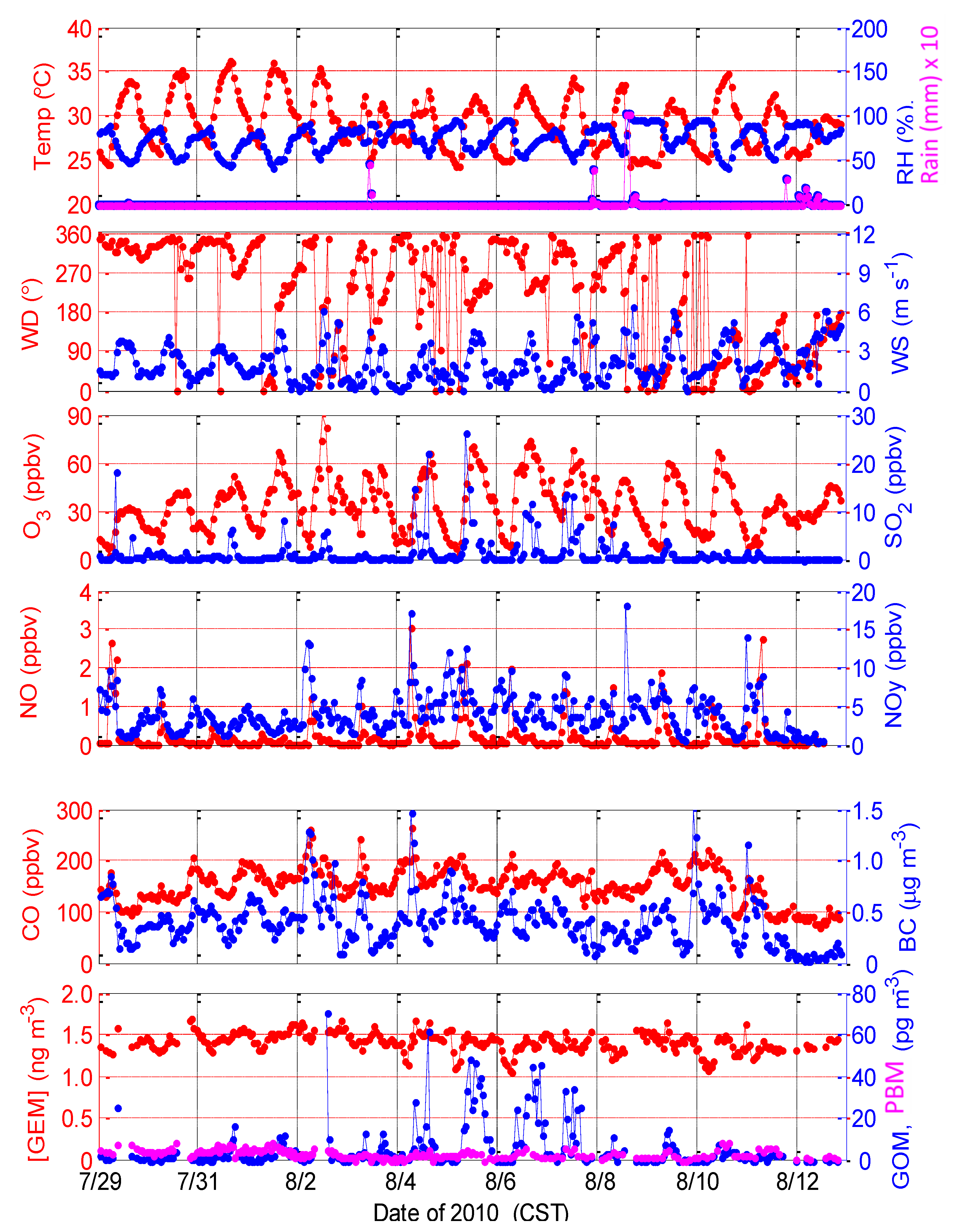
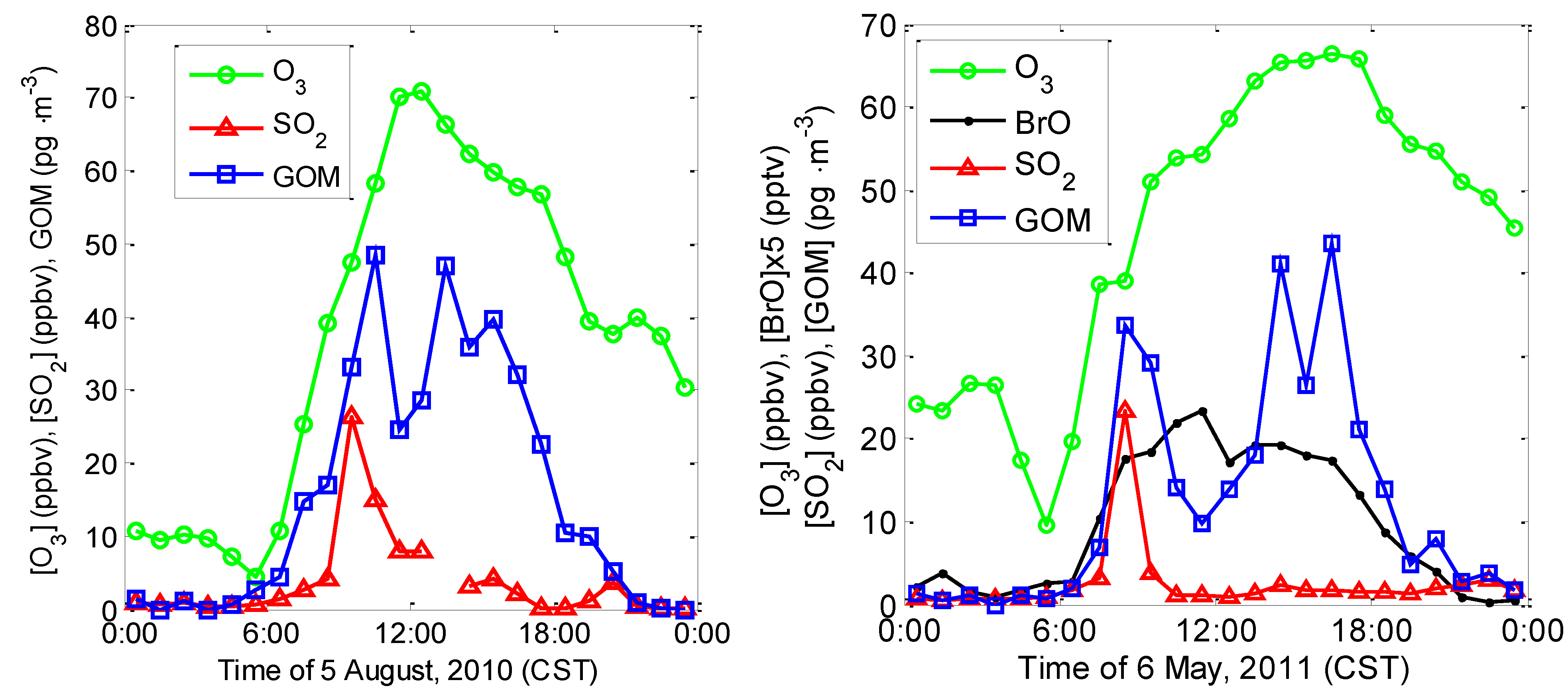
2.1.1. Intensive Study in Summer 2010

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
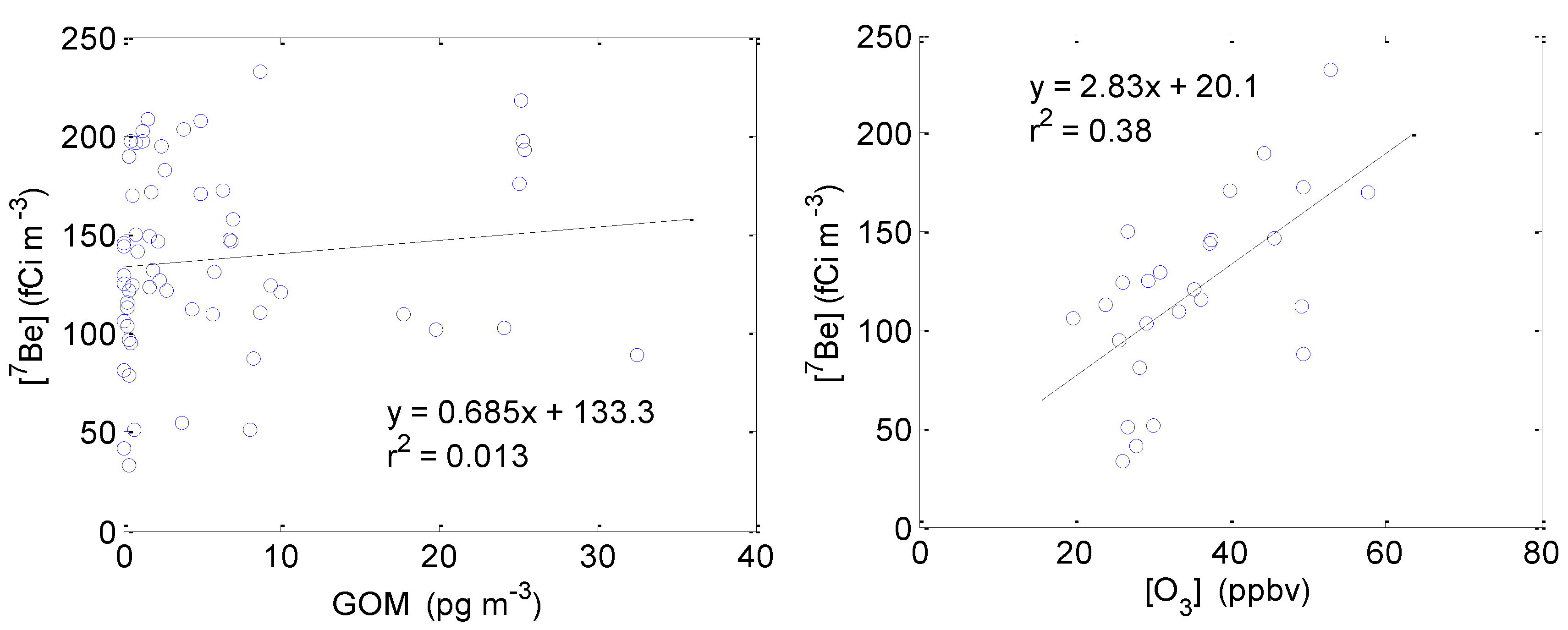{kind=link}
{kind=link}
| Parameter | Mean ± Std | Median | Maximum | Minimum |
|---|---|---|---|---|
| [GEM] (ng∙m−3) | 1.42 ± 0.12, 1.53 ± 0.11 | 1.44, 1.53 | 1.70, 3.12 | 1.06, 1.07 |
| [GOM] (ng∙m−3) | 5.4 ± 10.2, 5.3 ± 10.2 | 1.83, 0.9 | 70.8, 68.7 | 0.0, 0.0 |
| [PBM] (ng∙m−3) | 3.1 ± 1.9, 5.7 ± 6.2 | 2.7, 3.2 | 8.8, 37.0 | 0.0, 0.0 |
| Temperature (°C) | 29.4 ± 3.0, 21.9 ± 4.0 | 29.2, 23.4 | 36.3, 27.4 | 24.3, 8.9 |
| Relative Humidity (%) | 75.1 ± 14.3, 73.9 ± 17.1 | 77.7, 79.5 | 97.2, 96.9 | 41.0, 23.6 |
| Rain (mm, hour) | 0.12 ± 0.85, 0.023 ± 0.38 | 0, 0 | 10.2, 9.0 | 0, 0 |
| Solar Radiation (W∙m−2) | 258 ± 328, 266 ± 332 | 34, 45 | 1037, 983 | 0, 0 |
| Wind Speed (m∙s−1) | 2.2 ± 1.4, 4.7 ± 2.3 | 1.7, 4.8 | 6.5, 10.8 | 0.05, 0.02 |
| [O3] (ppbv) | 34.5 ± 16.5, 38.4 ± 12.5 | 32.6, 36.6 | 91.0, 71.9 | 3.1, 9.5 |
| [NO] (ppbv) | 0.24 ± ,0.43, 0.16 ± 0.32 | 0.08, 0.08 | 3.07, 3.06 | 0.028, 0.029 |
| [NOy] (ppb) | 4.13 ± 2.63, 1.91 ± 2.36 | 3.64, 1.03 | 18.2, 18.4 | 0.30, 0.22 |
| [CO] (ppbv) | 155 ± 35, 139 ± 26 | 156, 141 | 267, 321 | 72, 86 |
| [Black Carbon] (μg∙m−3) | 0.40 ± 0.23, 0.28 ± 0.17 | 0.39, 0.24 | 1.59, 1.35 | 0.03, 0.05 |

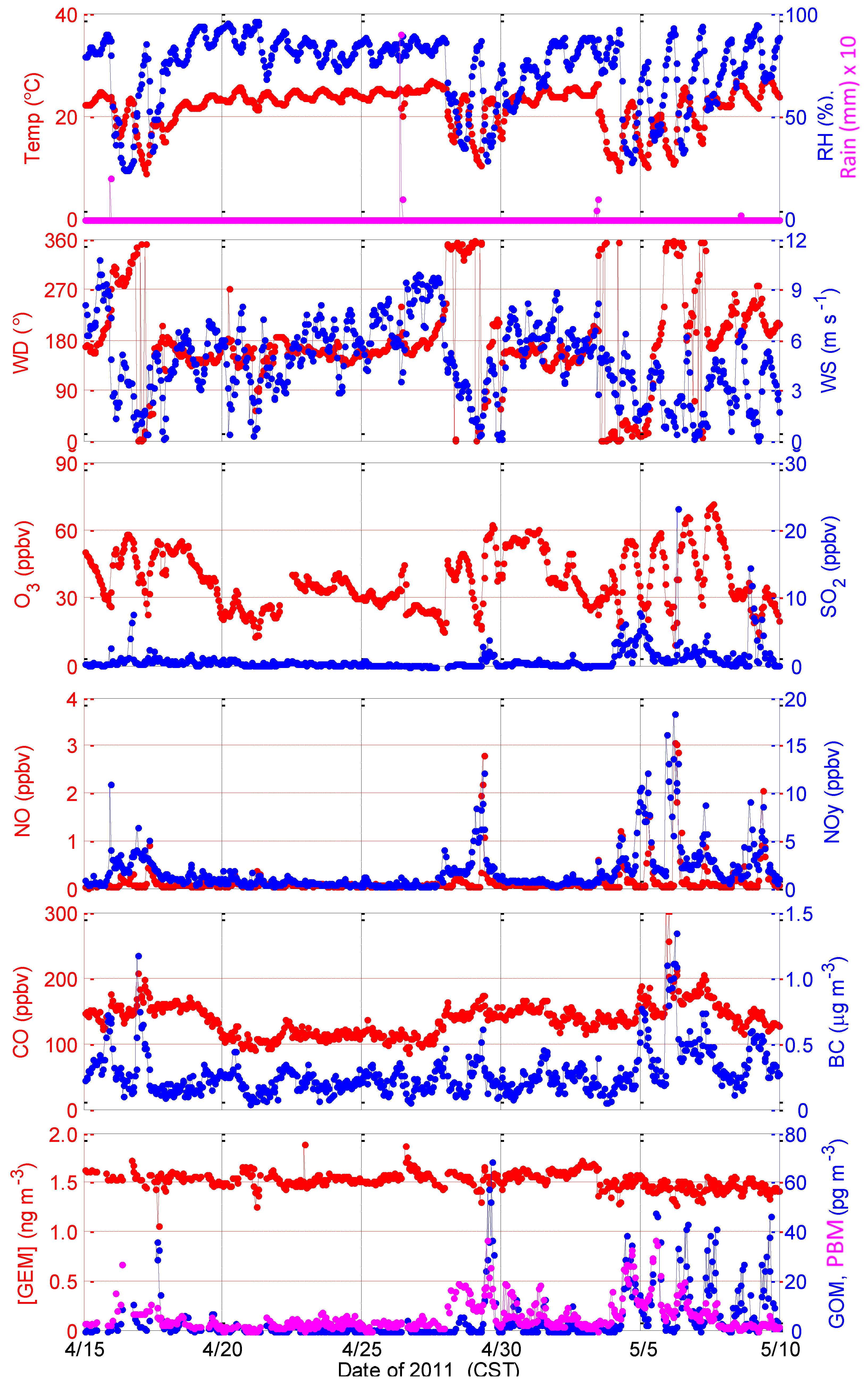
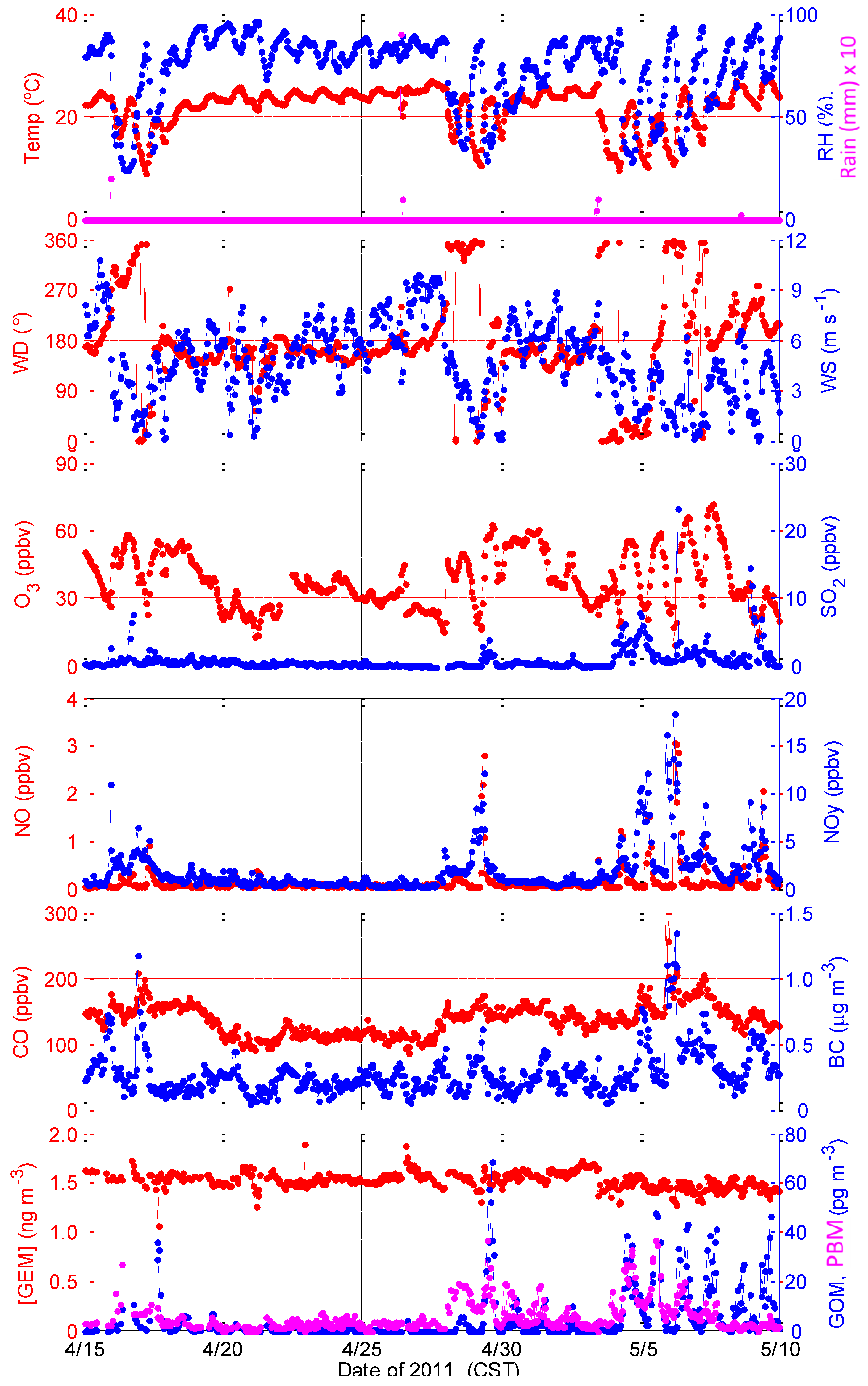
2.1.2. Intensive in Spring 2011
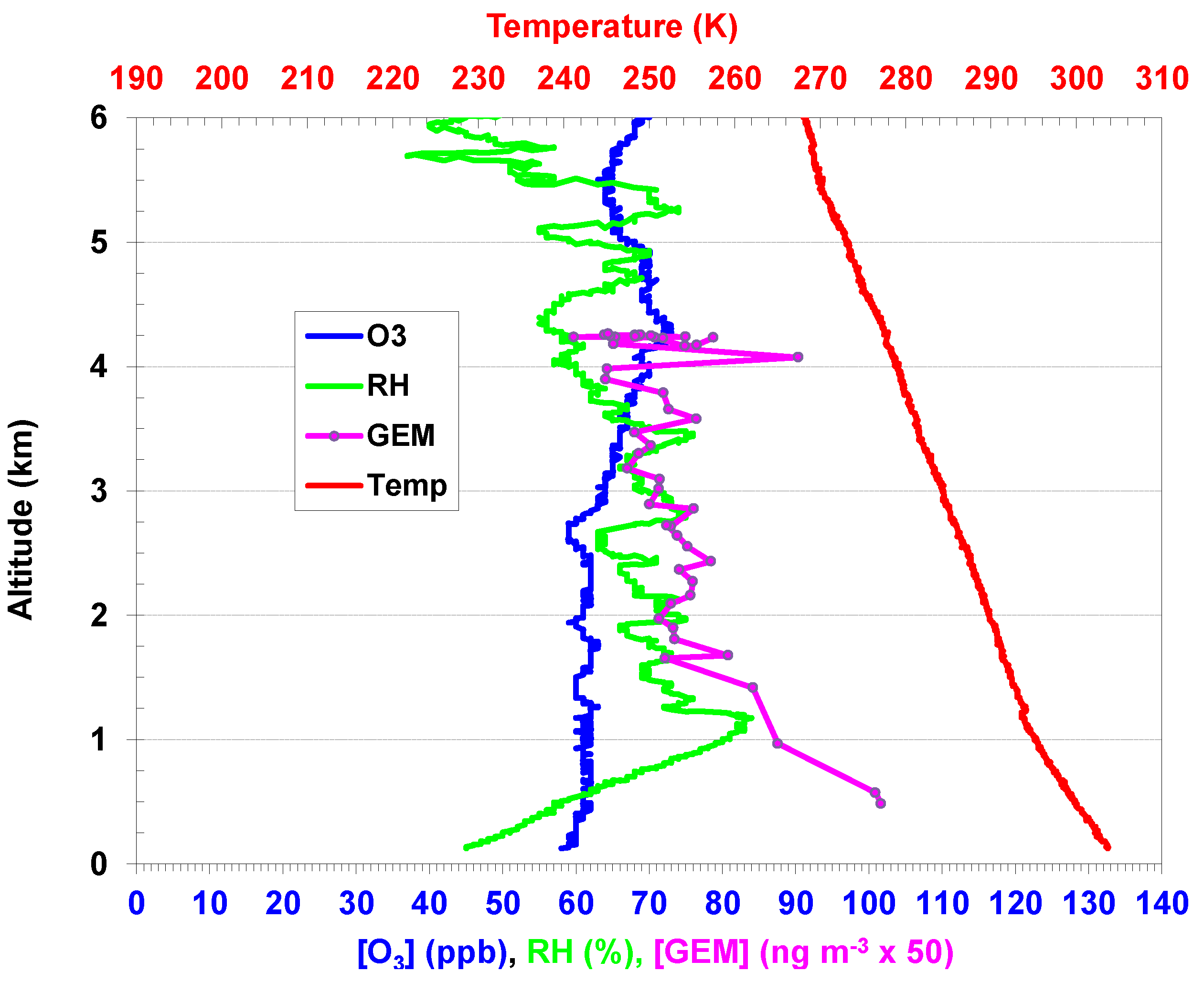
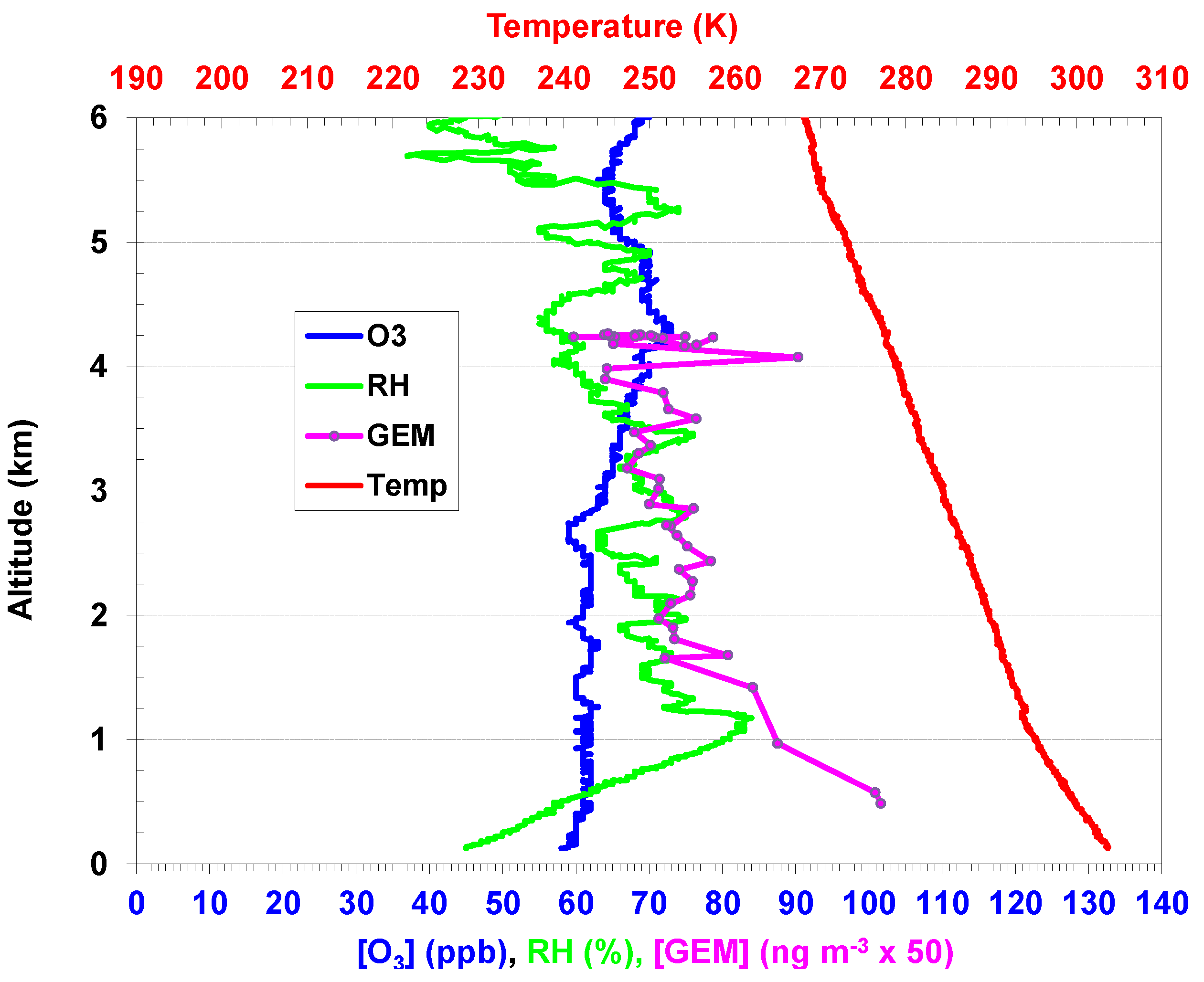
2.2. Aircraft and Ozonesonde Measurements
2.3. Correlation between Mercury Species and Ancillary Measurements
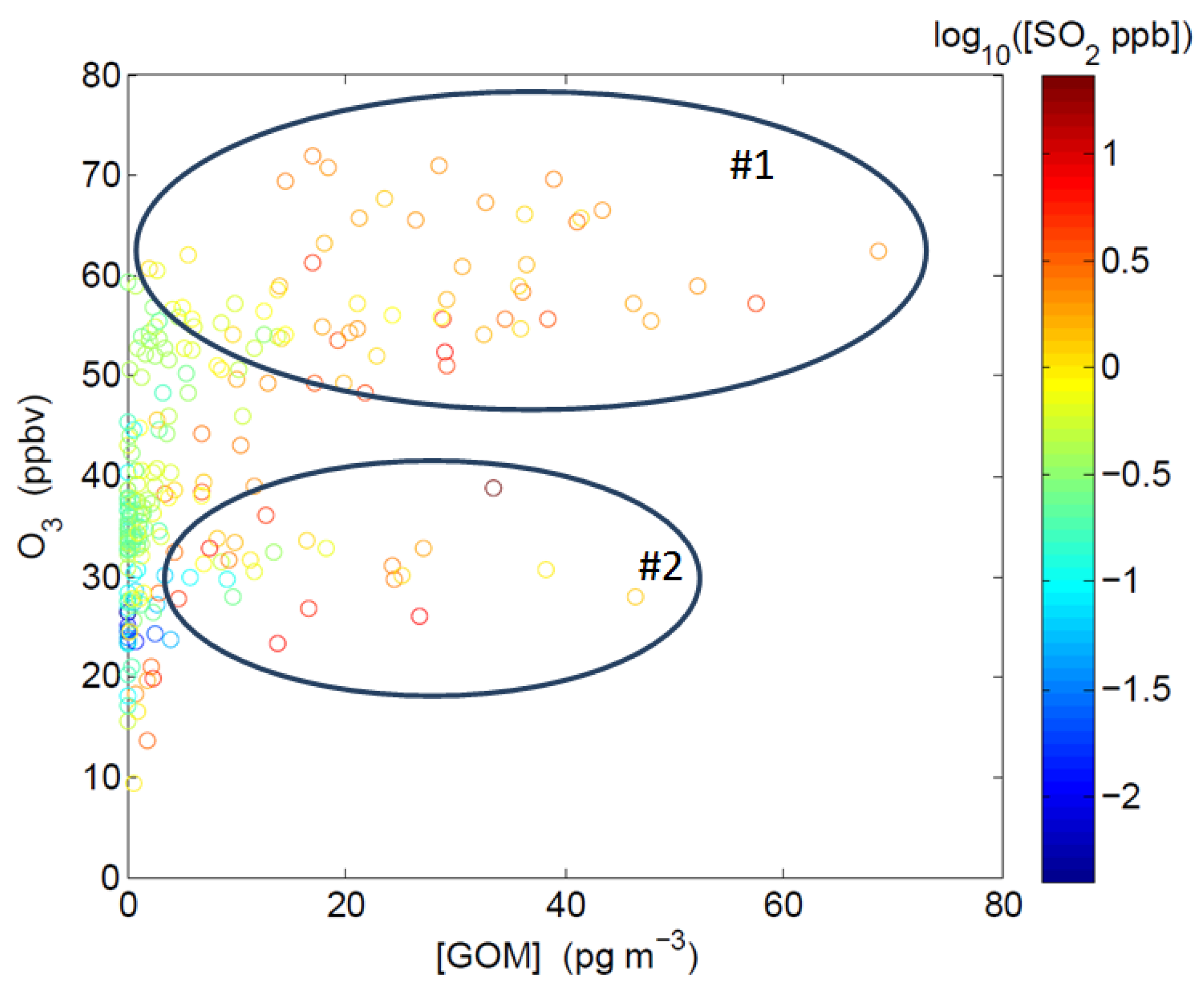
2.3.1. Correlation among GOM, O3, SO2, and BrO
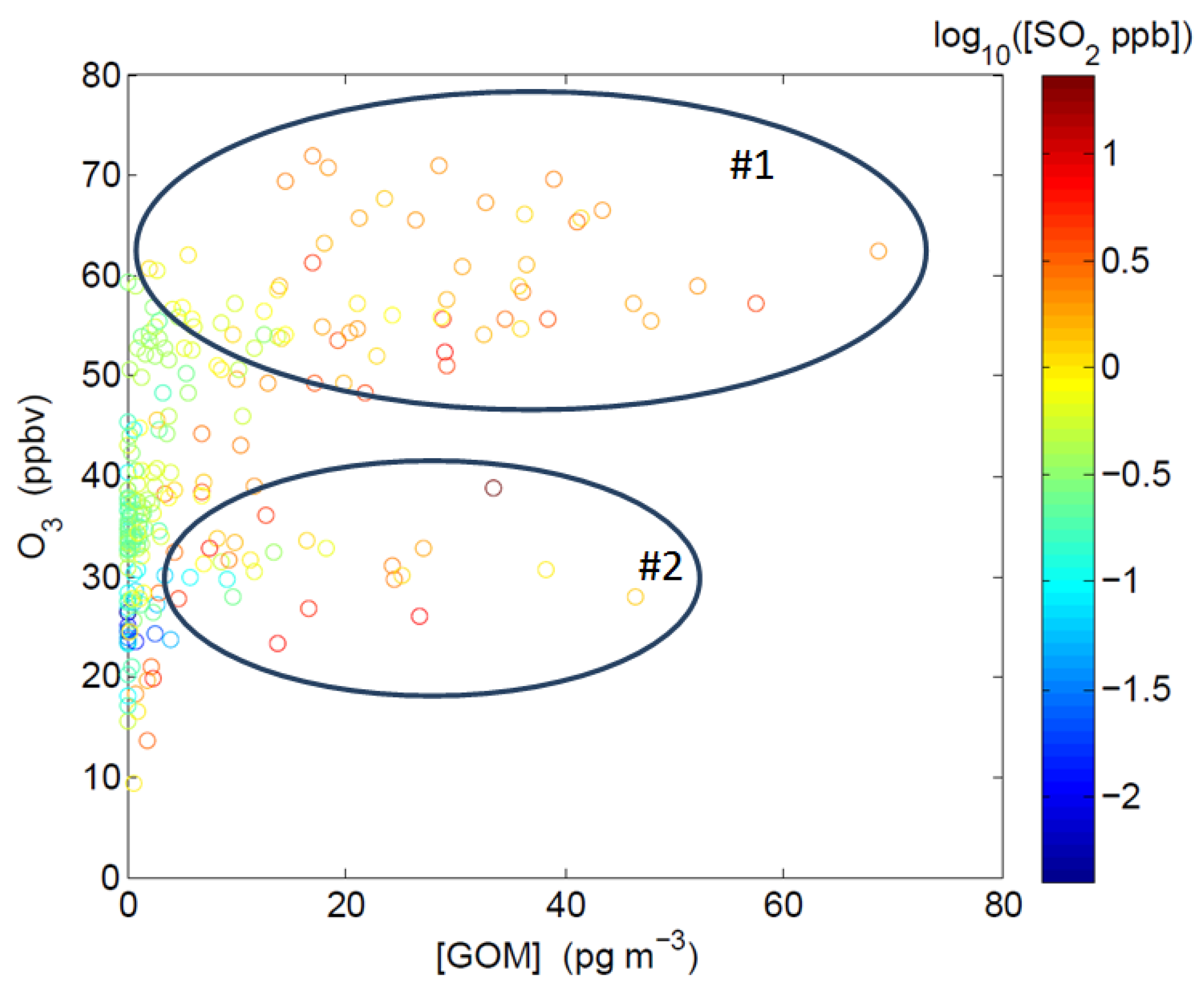
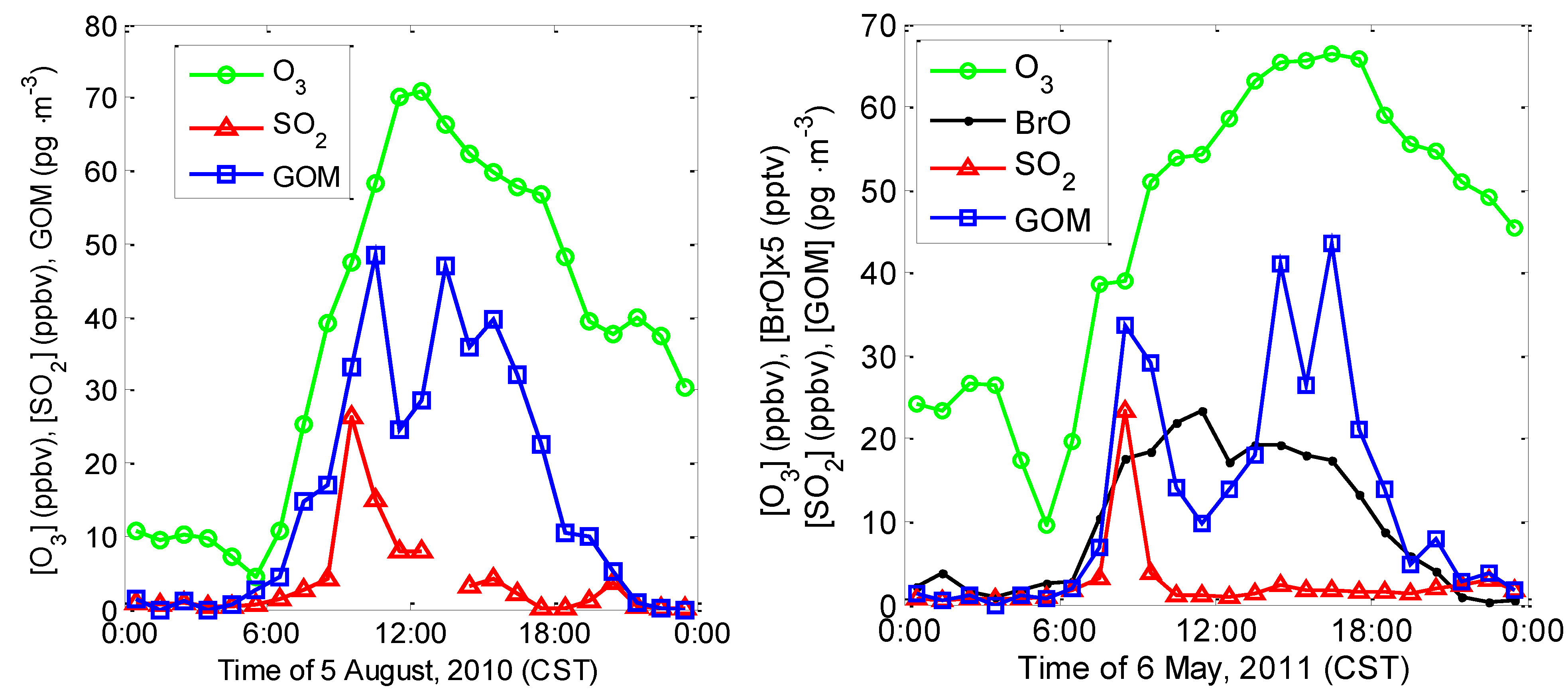
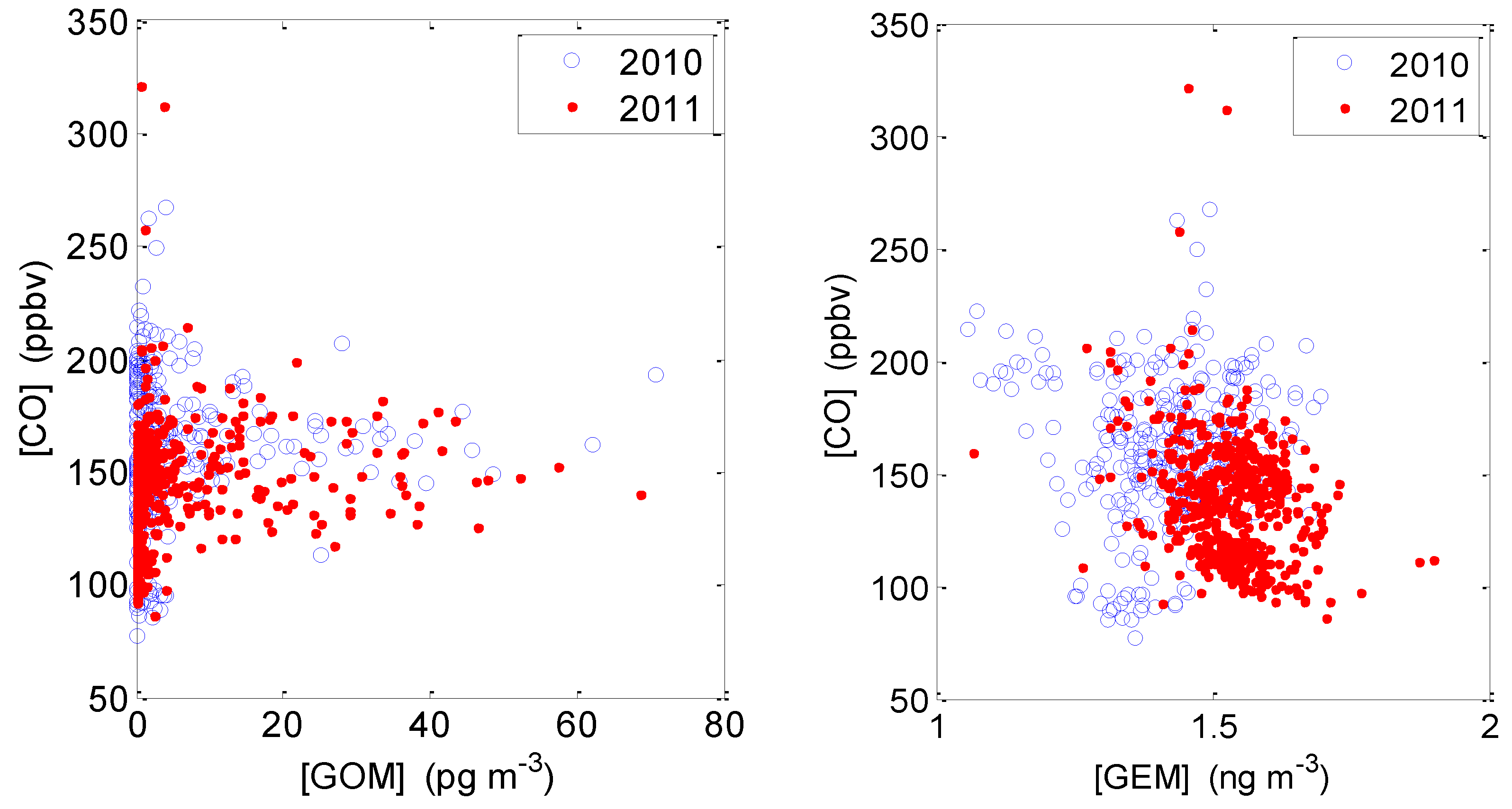
2.3.2. Correlation between GOM/GEM and CO

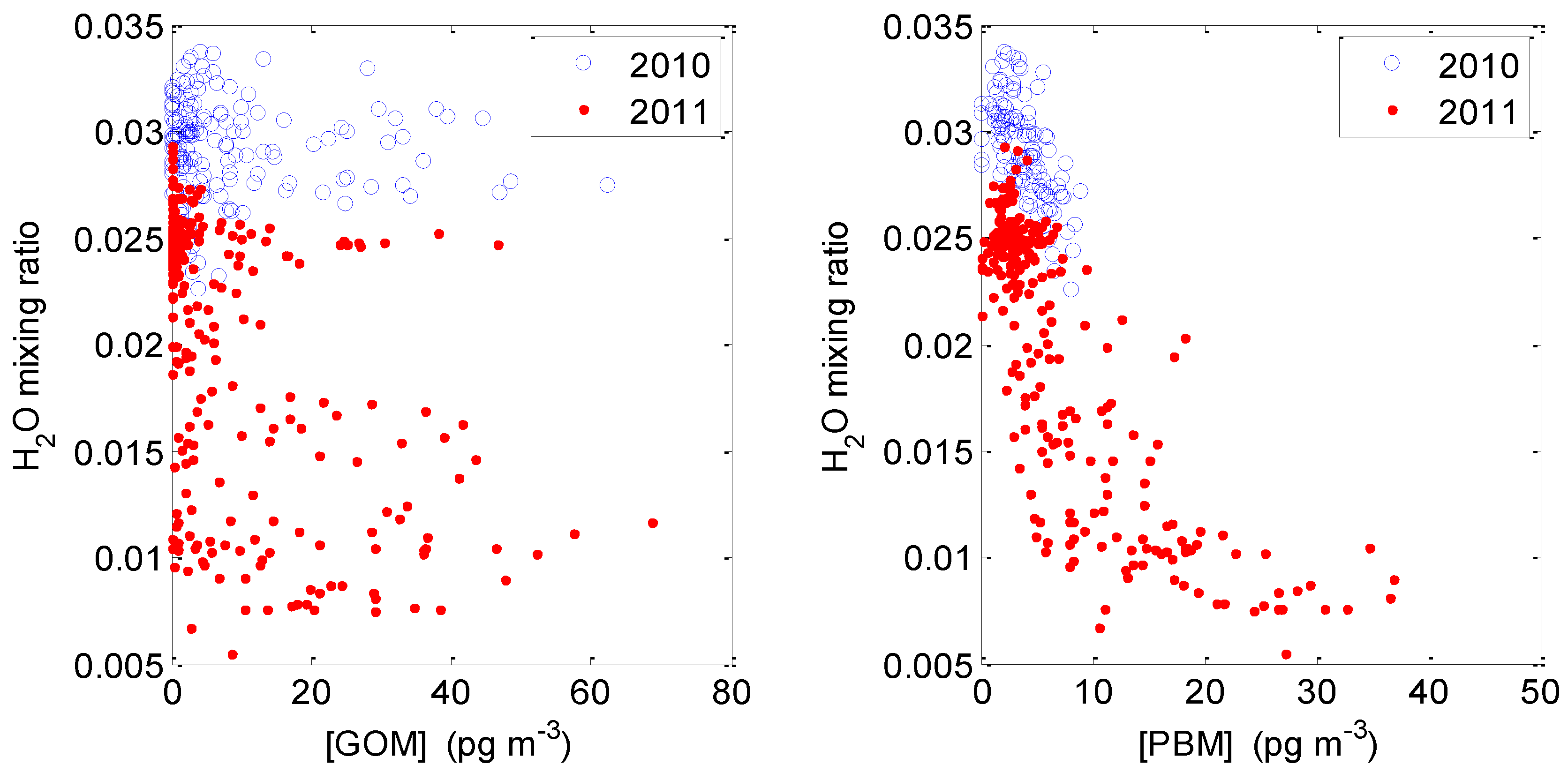
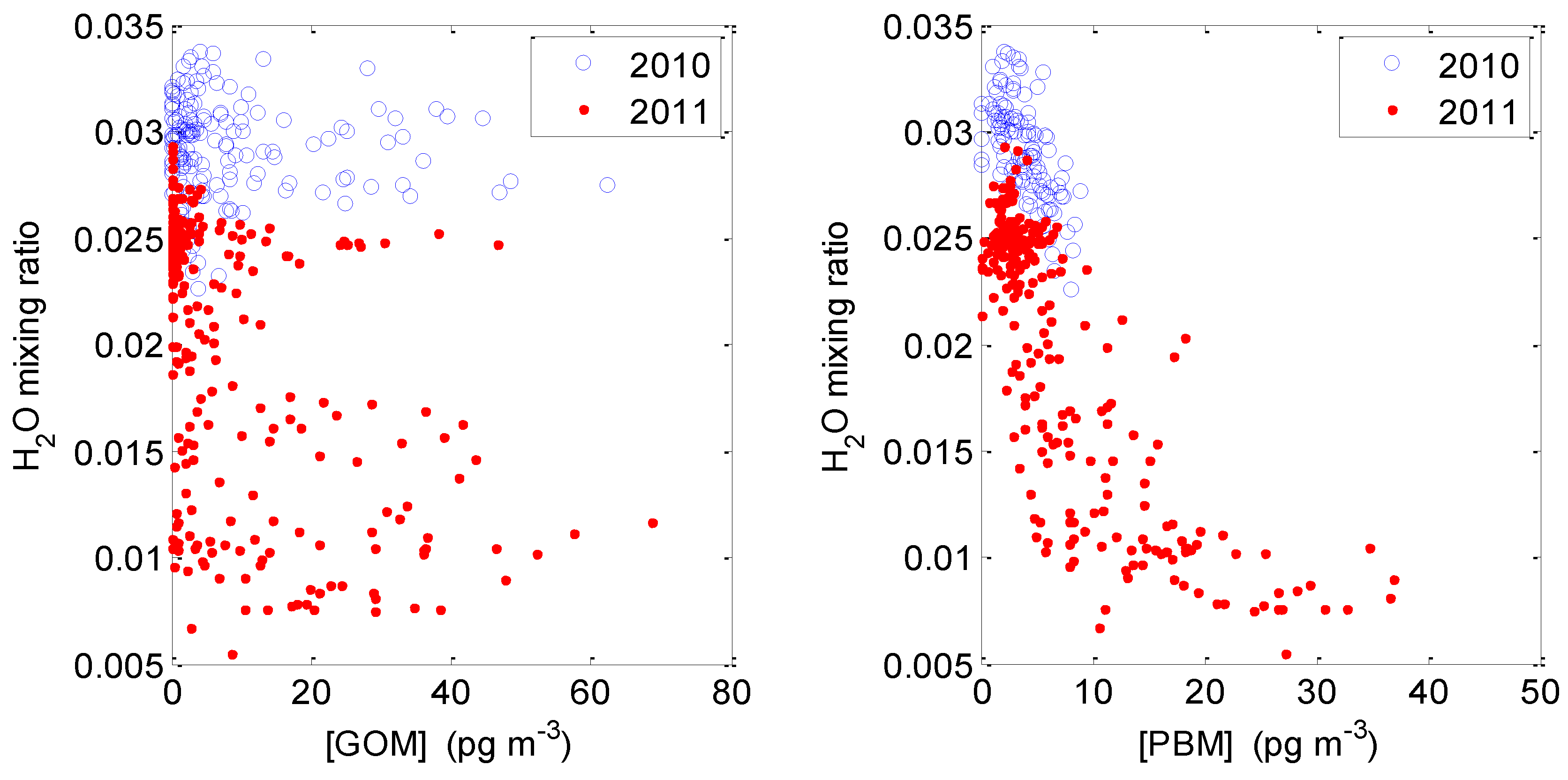
2.3.3. GOM/PBM and Humidity
2.3.4. Correlation between GOM/Ozone and Beryllium-7
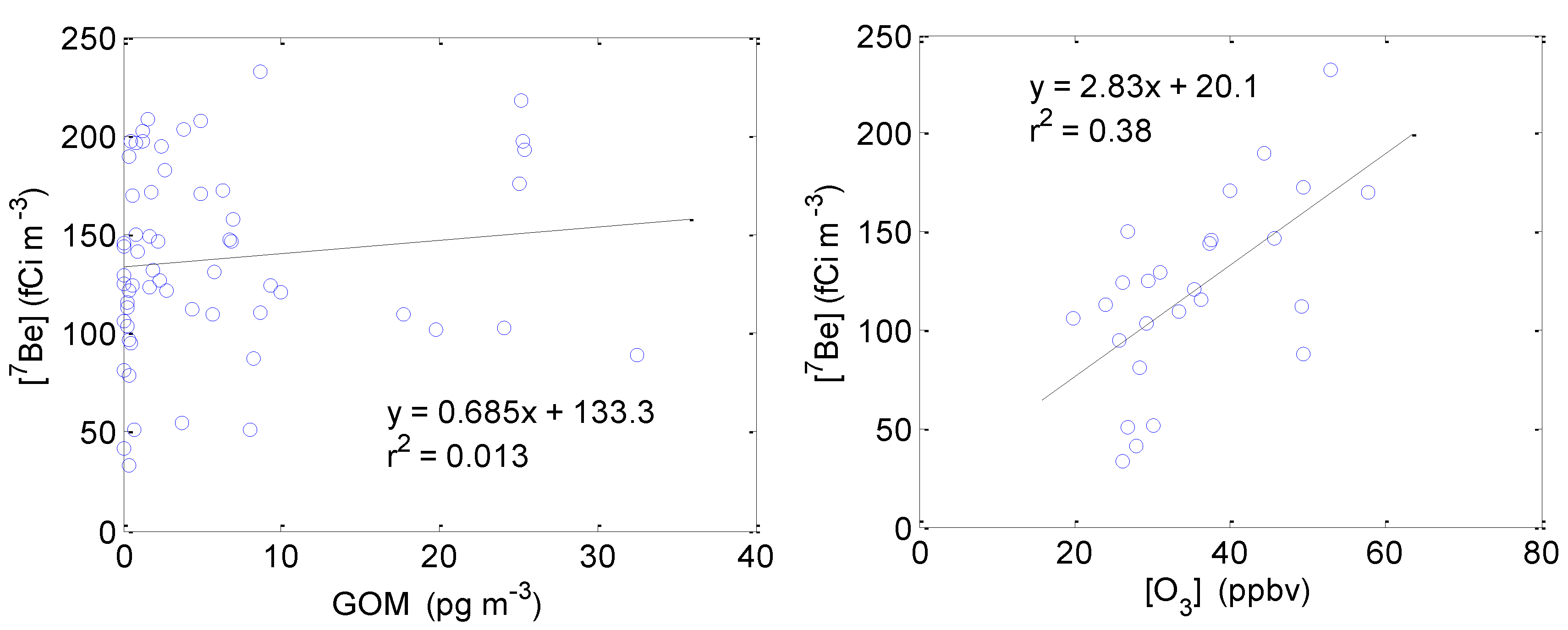
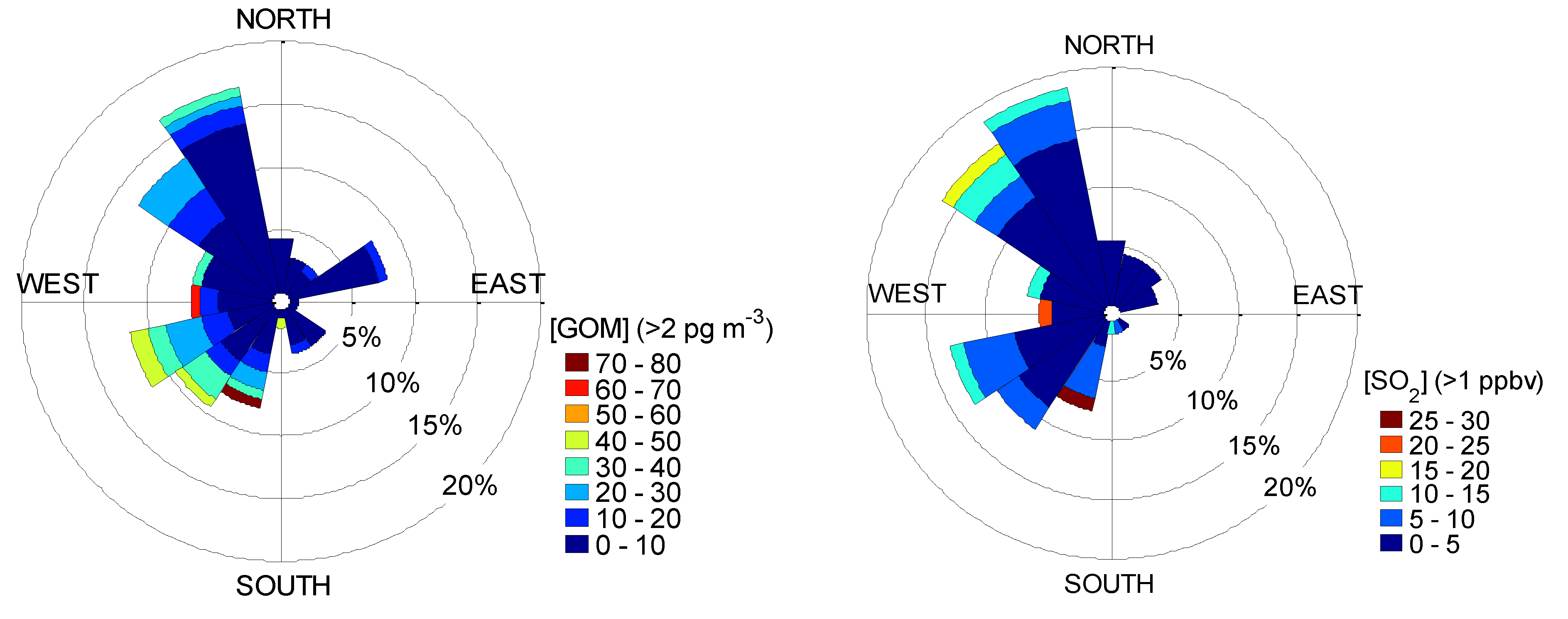
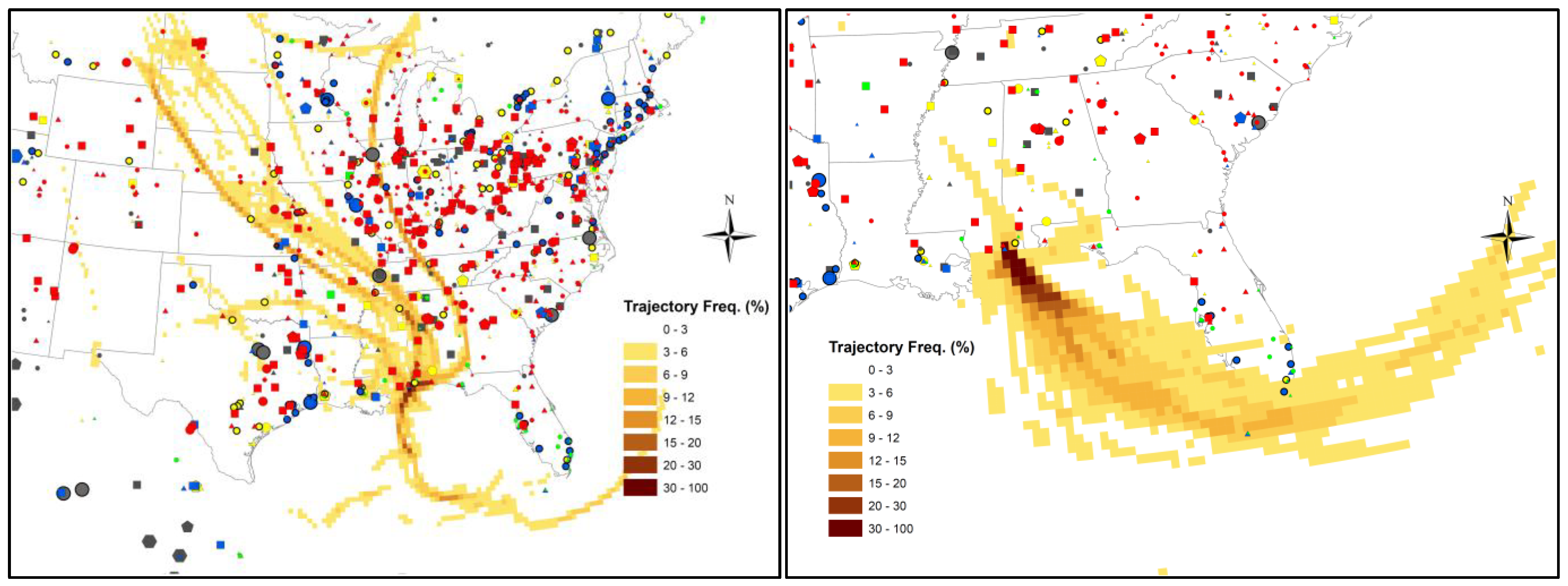
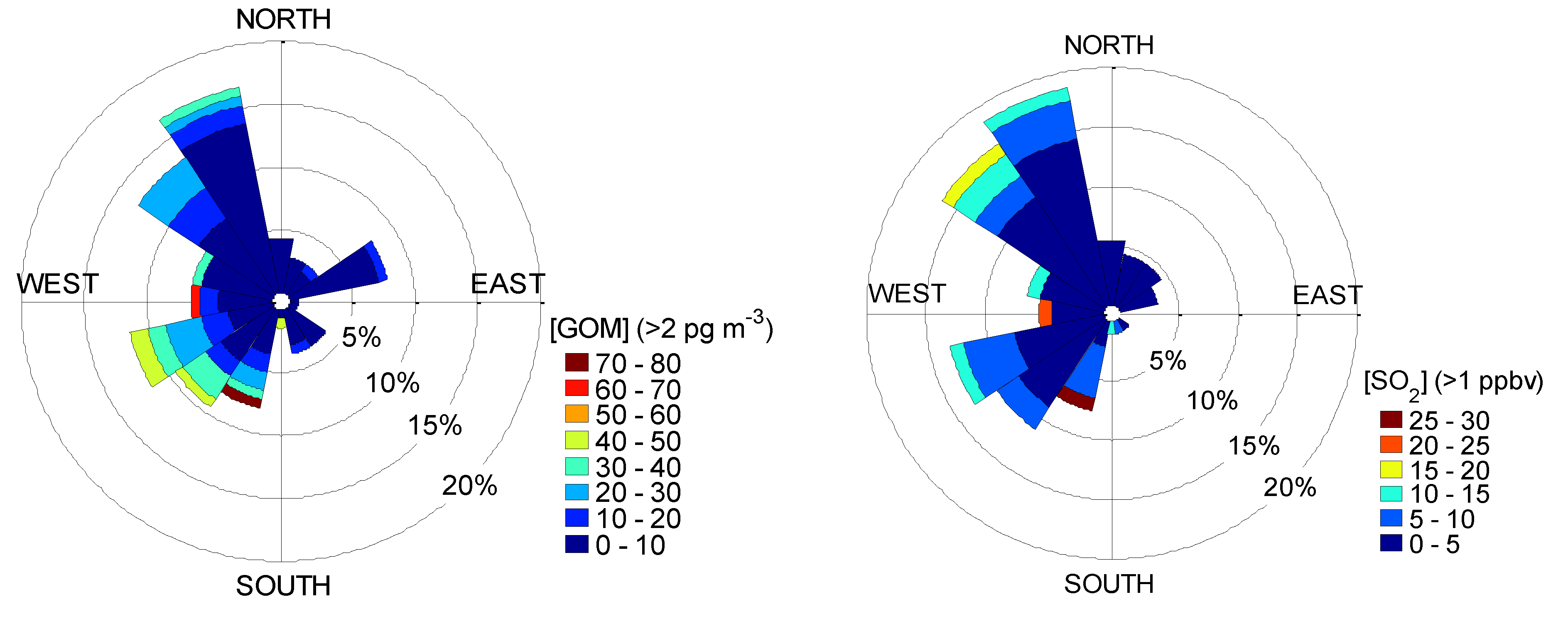
2.4. Back Trajectory Frequency Analysis
2.5. Principal Component Analysis

3. Experimental Section
3.1. Site Description
3.2. Experimental Description
3.2.1. Surface Measurements
3.2.2. Aircraft Measurements
3.2.3. Ozonesonde Launches
3.3. HYSPLIT Back Trajectory Model
3.4. Principal Component Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Mason, R.; Fitzgerald, W.F.; Morel, F.M. The biogeochemical cycling of elemental mercury: Anthropogenic influences. Geochim. Cosmochim. Act. 1994, 58, 3191–3198. [Google Scholar] [CrossRef]
- Schroeder, W.H.; Munthe, J. Atmospheric mercury—An overview. Atmos. Environ. 1998, 32, 809–822. [Google Scholar] [CrossRef]
- Fitzgerald, W.F.; Engstrom, D.R.; Mason, R.P.; Nater, E.A. The case for atmospheric mercury contamination in remote areas. Environ. Sci. Technol. 1998, 3, 1–7. [Google Scholar]
- Lin, C.-J.; Pehkonen, S.O. The chemistry of atmospheric mercury: A review. Atmos. Environ 1999, 33, 2067–2079. [Google Scholar] [CrossRef]
- Selin, N.E. Global Biogeochemical Cycling of Mercury: A Review. Ann. Rev. Environ. Res. 2009, 34, 43–63. [Google Scholar] [CrossRef]
- AMAP/UNEP. Technical Background Report for the Global Mercury Assessment 2013; Arctic Monitoring and Assessment Programme, Oslo, Norway/UNEP Chemicals Branch: Geneva, Switzerland, 2013. [Google Scholar]
- Morel, F.M.M.; Kraepiel, A.M.L.; Amyot, M. The chemical cycle and bioaccumulation of mercury. Annu. Rev. Ecol. Syst. 1998, 29, 543–566. [Google Scholar] [CrossRef]
- Choi, A.L.; Grandjean, P. Methylmercury exposure and health effects in humans. Environ. Chem. 2008, 5, 112–120. [Google Scholar] [CrossRef]
- Sunderland, E. Mercury exposure from domestic and imported estuarine and marine fish in the U.S. Seafood Market. Environ. Health Perspect. 2007, 115, 235–242. [Google Scholar] [CrossRef]
- Gustin, M.S.; Lindberg, S.E.; Weisberg, P.J. An update on the natural sources and sinks of atmospheric mercury. Appl. Geochem. 2008, 23, 482–493. [Google Scholar] [CrossRef]
- UNEP. Global Mercury Assessment 2013: Sources, Emissions, Releases and Environmental Transport. UNEP Chemicals Branch: Geneva, Switzerland, 2013. Available online: http://www.unep.org/PDF/PressReleases/GlobalMercuryAssessment2013.pdf (accessed on 25 February 2014).
- Engle, M.A.; Tate, M.T.; Krabbenhoft, D.P.; Kolker, A.; Olson, M.L.; Edgerton, E.S.; DeWild, J.F.; McPherson, A.K. Characterization and cycling of atmospheric mercury along the central U.S. Gulf Coast. Appl. Geochem. 2008, 23, 419–437. [Google Scholar] [CrossRef]
- National Atmospheric Deposition Program’s Mercury Deposition Network (MDN), Monitoring Mercury Deposition: A Key Tool to Understanding the Link between Emissions and Effects. Available online: http://nadp.sws.uiuc.edu/lib/brochures/mdn.pdf (accessed on 15 February 2014).
- Nair, U.S.; Wu, Y.; Walters, J.; Jansen, J.; Edgerton, E.S. Diurnal and seasonal variation of mercury species at coastal-suburban, urban, and rural sites in the southeastern United States. Atmos. Environ. 2012, 47, 499–508. [Google Scholar] [CrossRef]
- Weiss-Penzias, P.; Jaffe, D.E.; McClintick, A.; Prestbo, E.M.; Landis, M.S. Gaseous elemental mercury in the marine boundary layer: evidence for rapid removal in anthropogenic pollution. Environ. Sci. Technol. 2003, 37, 3755–3763. [Google Scholar] [CrossRef]
- Brunke, E.-G.; Labuschagne, C.; Ebinghaus, R.; Kock, H.H.; Slemr, F. Gaseous elemental mercury depletion events observed at Cape Point during 2007–2008. Atmos. Chem. Phys. 2010, 10, 1121–1131. [Google Scholar] [CrossRef]
- Schroeder, W.H.; Anlauf, K.G.; Barrie, L.A.; Lu, J.Y.; Steffen, A.; Schneeberger, D.R.; Berg, T. Arctic springtime depletion of mercury. Nature 1998, 394, 331–332. [Google Scholar] [CrossRef]
- Ebinghaus, R.; Kock, H.H.; Temme, C.; Einax, J.W.; Löwe, A.G.; Richter, A.; Burrows, J.P.; Schroeder, W.H. Antarctic springtime depletion of atmospheric mercury. Environ. Sci. Technol. 2002, 36, 1238–1244. [Google Scholar] [CrossRef]
- Tas, E.; Obrist, D.; Peleg, M.; Matveev, V.; Faïn, X.; Asaf, D.; Luria, M. Measurement-based modelling of bromine-induced oxidation of mercury above the Dead Sea. Atmos. Chem. Phys. 2012, 12, 2429–2440. [Google Scholar] [CrossRef]
- Ren, X.; Luke, W.T.; Kelley, P.; Cohen, M.; Tong, D.; Artz, R.; Olsen, M.L.; Schmeltz, D. Mercury speciation at a suburban site in the Mid-Atlantic United States: Seasonal and diurnal variations and source-receptor correlationship. Atmos. Chem. Phys. 2014. in preparation. [Google Scholar]
- Gay, D.A.; Schmeltz, D.; Prestbo, E.; Olson, M.; Sharac, T.; Tordon, R. The Atmospheric Mercury Network: measurement and initial examination of an ongoing atmospheric mercury record across North America. Atmos. Chem. Phys. 2013, 13, 11339–11349. [Google Scholar] [CrossRef]
- Tremblay, J.; Servranckx, R. Beryllium-7 as a tracer of stratospheric ozone: A case study. J. Radioanal. Nucl. Chem. 1993, 172, 49–56. [Google Scholar] [CrossRef]
- Dibb, J.E.; Talbot, R.W.; Lefer, B.L.; Scheuer, E.; Gregory, G.L.; Browell, E.V.; Bradshaw, J.D.; Sandholm, S.T.; Singh, H.B. Distributions of beryllium 7 and lead 2109, and soluble aerosol-associated ionic species over the western Pacific: PEM West B, February–March 1994. J. Geophys. Res.: Atmos. 1997, 102, 28287–28302. [Google Scholar] [CrossRef]
- Kikuchi, S.; Sakurai, H.; Gunji, S.; Tokanai, F. Temporal variation of 7Be concentrations in atmosphere for 8 y from 2000 at Yamagata, Japan: Solar influence on the 7Be time series. J. Environ. Radioact. 2009, 100, 515–521. [Google Scholar] [CrossRef]
- Piñero Garcíaa, F.; Ferro Garcíaa, M.A.; Azahrab, M. 7Be behaviour in the atmosphere of the city of Granada January 2005 to December 2009. Atmos. Environ. 2012, 47, 84–91. [Google Scholar] [CrossRef]
- Rolison, J.M.; Landing, W.M.; Luke, W.; Cohen, M.; Salters, V.J.M. Isotopic composition of species-specific atmospheric Hg in a coastal environment. Chem. Geol. 2013, 336, 37–49. [Google Scholar] [CrossRef]
- Thurston, G.D.; Spengler, J.D. A quantitative assessment of source contributions to inhalable particulate matter pollution in metropolitan Boston. Atmos. Environ. 1985, 19, 9–26. [Google Scholar] [CrossRef]
- Buhr, M.; Parrish, D.; Elliot, J.; Holloway, J.; Carpenter, J.; Goldan, P.; Kuster, W.; Trainer, M.; Montzka, S.; McKeen, S.; Fehsenfeld, F. Evaluation of ozone precursor source types using principal component analysis of ambient air measurements in rural Alabama. J. Geophys. Res. 1995, 100, 22853–22860. [Google Scholar] [CrossRef]
- Statheropoulos, M.; Vassiliadis, N.; Pappa, A. Principal component and canonical correlation analysis for examining air pollution and meteorological data. Atmos. Environ. 1998, 32, 1087–1095. [Google Scholar] [CrossRef]
- Guo, H.; Wang, Tao; Louie, P.K.K. Source apportionment of ambient non-methane hydrocarbons in Hong Kong: Application of a principal component analysis/absolute principal component scores (PCA/APCS) receptor model. Environ. Pollut. 2004, 129, 489–498. [Google Scholar] [CrossRef]
- Landis, M.S.; Stevens, R.K.; Schaedlich, F.; Prestbo, E.M. Development and characterization of an annular denuder methodology for the measurement of divalent inorganic reactive gaseous mercury in ambient air. Environ. Sci. Technol. 2002, 36, 3000–3009. [Google Scholar] [CrossRef]
- Lindberg, S.; Brooks, S.; Lin, C.-J.; Scott, K.; Landis, M.; Stevens, R.; Goodsite, M.; Richter, A. Dynamic oxidation of gaseous mercury in the Arctic troposphere at polar sunrise. Environ. Sci. Technol. 2002, 36, 1245–1256. [Google Scholar] [CrossRef]
- Ambrose, J.L.; Lyman, S.N.; Huang, J.; Gustin, M.S.; Jaffe, D.A. Fast time resolution oxidized mercury measurements during the Reno Atmospheric Mercury Intercomparison Experiment (RAMIX). Environ. Sci. Technol. 2013, 47, 7285–7294. [Google Scholar]
- Gustin, M.S.; Huang, J.; Miller, M.B.; Peterson, C.; Jaffe, D.A.; Ambrose, J.; Finley, B.D.; Lyman, S.N.; Call, K.; Talbot, R.; et al. Do we understand what the mercury speciation instruments are actually measuring? Results of RAMIX. Environ. Sci. Technol. 2013, 47, 7295–7306. [Google Scholar]
- Bauer, D.; Campuzano-Jost, P.; Hynes, A.J. Rapid, ultra-sensitive detection of gas phase elemental mercury under atmospheric conditions using sequential two-photon laser induced fluorescence. J. Environ. Monit. 2002, 4, 339–343. [Google Scholar] [CrossRef]
- Luke, W.T.; Kelley, P.; Lefer, B.L.; Flynn, J.; Rappenglück, B.; Leuchner, M.; Dibb, J.E.; Ziemba, L.D.; Anderson, C.H.; Buhr, M. Measurements of primary trace gases and NOy composition in Houston, Texas. Atmos. Environ. 2010, 44, 4068–4080. [Google Scholar] [CrossRef]
- Liao, J.; Huey, L.G.; Tanner, D.J.; Brough, N.; Brooks, S.; Dibb, J.E.; Stutz, J.; Thomas, J.L.; Lefer, B.; Haman, C.; et al. Observations of hydroxyl and peroxy radicals and the impact of BrO at Summit, Greenland in 2007 and 2008. Atmos. Chem. Phys. 2011, 11, 8577–8591. [Google Scholar] [CrossRef]
- Dibb, J.E.; Talbot, R.W.; Klemm, K.I.; Gregory, G.L.; Singh, H.B.; Bradshaw, J.D.; Sandholm, S.T. Asian influence over the western North Pacific during the fall season: Inferences from lead 210, soluble ionic species, and ozone. J. Geophys. Res. 1996, 101, 1779–1792. [Google Scholar] [CrossRef]
- Dibb, J.E.; Talbot, R.W.; Scheuer, E.; Seid, G.; DeBell, L.; Lefer, B.; Ridley, B. Stratospheric influence on the northern North American free troposphere during TOPSE: 7Be as a stratospheric tracer. J. Geophys. Res.: Atmos. 2003. [Google Scholar] [CrossRef]
- Luke, W.T.; Arnold, J.R.; Gunter, R.L.; Watson, T.B.; Wellman, D.L.; Dasgupta, P.K.; Li, J.; Riemer, D.; Tate, P. The NOAA Twin Otter and its role in BRACE: Platform description. Atmos. Environ. 2007, 41, 4177–4189. [Google Scholar] [CrossRef]
- Ernest, C.T.; Donohoue, D.; Bauer, D.; Ter Schure, A.; Hynes, A.J. Programmable thermal dissociation of reactive gaseous mercury—A potential approach to chemical speciation: results from a field study. Atmos. Chem. Phys. Discuss. 2012, 12, 33291–33322. [Google Scholar] [CrossRef]
- Draxler, R.R.; Rolph, G.D. HYSPLIT (HYbrid Single-Particle Lagrangian Integrated Trajectory) Model; NOAA Air Resources Laboratory: Maryland, MD, USA, 2014. Available online: http://ready.arl.noaa.gov/HYSPLIT.php (accessed on 25 April 2013).
- Ngan, F.; Cohen, M.; Luke, W.; Ren, X.; Draxler, R. Meteorological modeling using WRF-ARW model for Grand Bay Intensive studies of atmospheric mercury. Atmosphere 2014. in preparation. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ren, X.; Luke, W.T.; Kelley, P.; Cohen, M.; Ngan, F.; Artz, R.; Walker, J.; Brooks, S.; Moore, C.; Swartzendruber, P.; et al. Mercury Speciation at a Coastal Site in the Northern Gulf of Mexico: Results from the Grand Bay Intensive Studies in Summer 2010 and Spring 2011. Atmosphere 2014, 5, 230-251. https://doi.org/10.3390/atmos5020230
Ren X, Luke WT, Kelley P, Cohen M, Ngan F, Artz R, Walker J, Brooks S, Moore C, Swartzendruber P, et al. Mercury Speciation at a Coastal Site in the Northern Gulf of Mexico: Results from the Grand Bay Intensive Studies in Summer 2010 and Spring 2011. Atmosphere. 2014; 5(2):230-251. https://doi.org/10.3390/atmos5020230
Chicago/Turabian StyleRen, Xinrong, Winston T. Luke, Paul Kelley, Mark Cohen, Fong Ngan, Richard Artz, Jake Walker, Steve Brooks, Christopher Moore, Phil Swartzendruber, and et al. 2014. "Mercury Speciation at a Coastal Site in the Northern Gulf of Mexico: Results from the Grand Bay Intensive Studies in Summer 2010 and Spring 2011" Atmosphere 5, no. 2: 230-251. https://doi.org/10.3390/atmos5020230
APA StyleRen, X., Luke, W. T., Kelley, P., Cohen, M., Ngan, F., Artz, R., Walker, J., Brooks, S., Moore, C., Swartzendruber, P., Bauer, D., Remeika, J., Hynes, A., Dibb, J., Rolison, J., Krishnamurthy, N., Landing, W. M., Hecobian, A., Shook, J., & Huey, L. G. (2014). Mercury Speciation at a Coastal Site in the Northern Gulf of Mexico: Results from the Grand Bay Intensive Studies in Summer 2010 and Spring 2011. Atmosphere, 5(2), 230-251. https://doi.org/10.3390/atmos5020230