
Experimental Study of the Reaction of OH Radicals with Carbonyl Sulfide between 365 and 960 K: Kinetics and Products


Abstract
1. Introduction
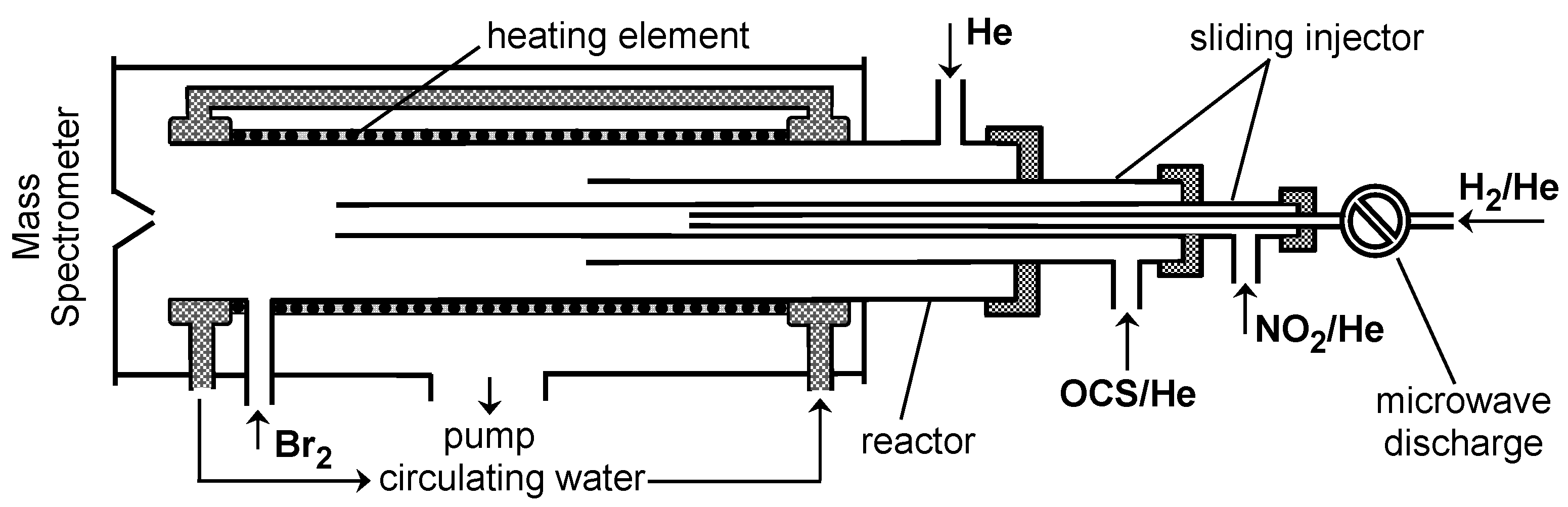
2. Materials and Methods
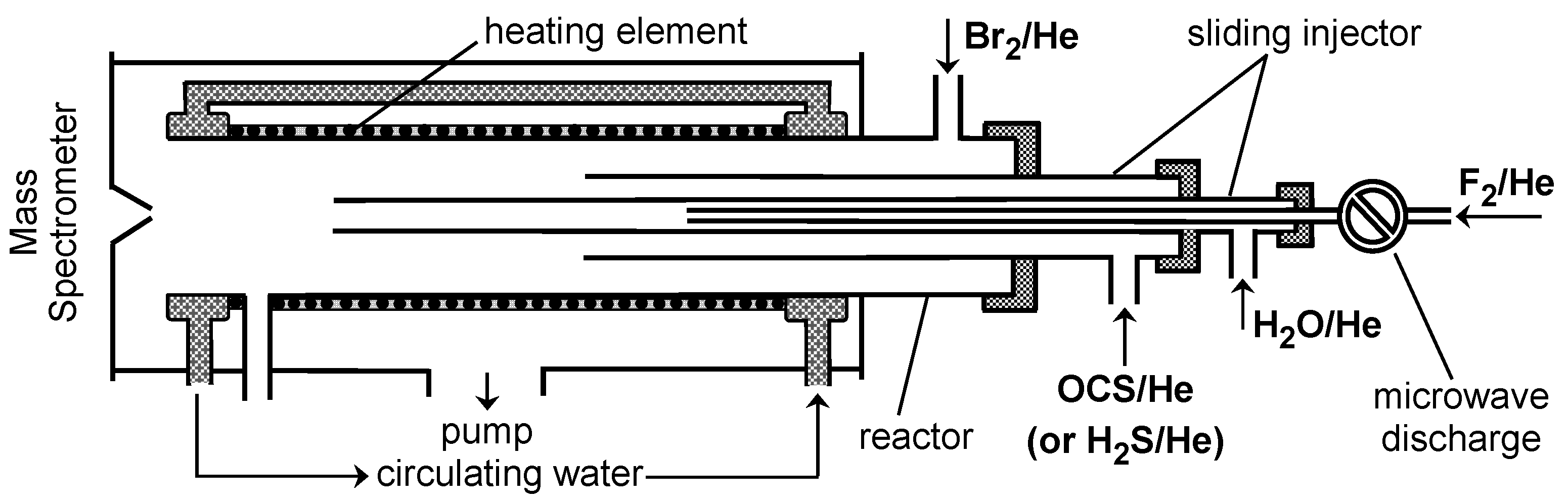
3. Results
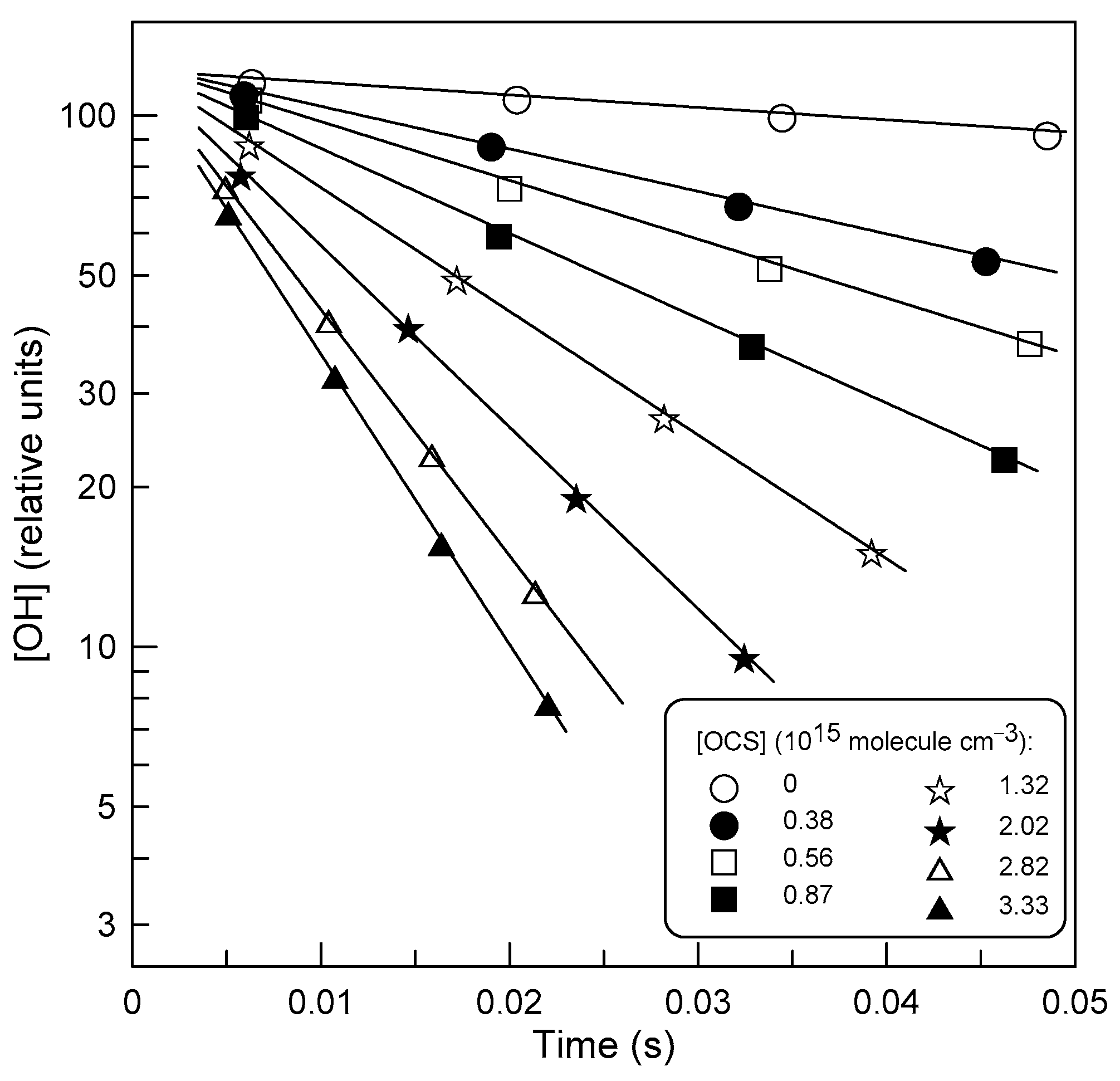
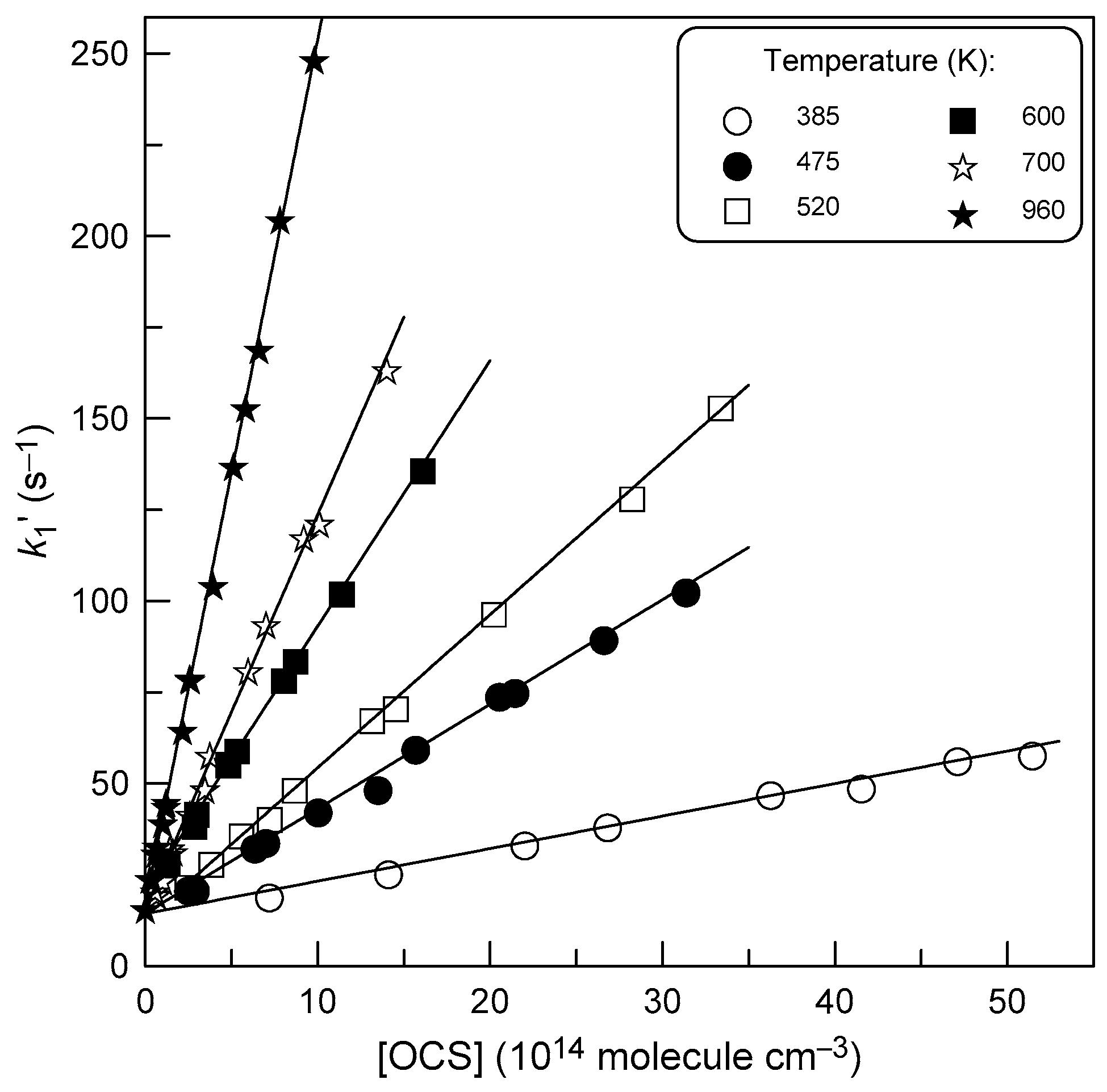
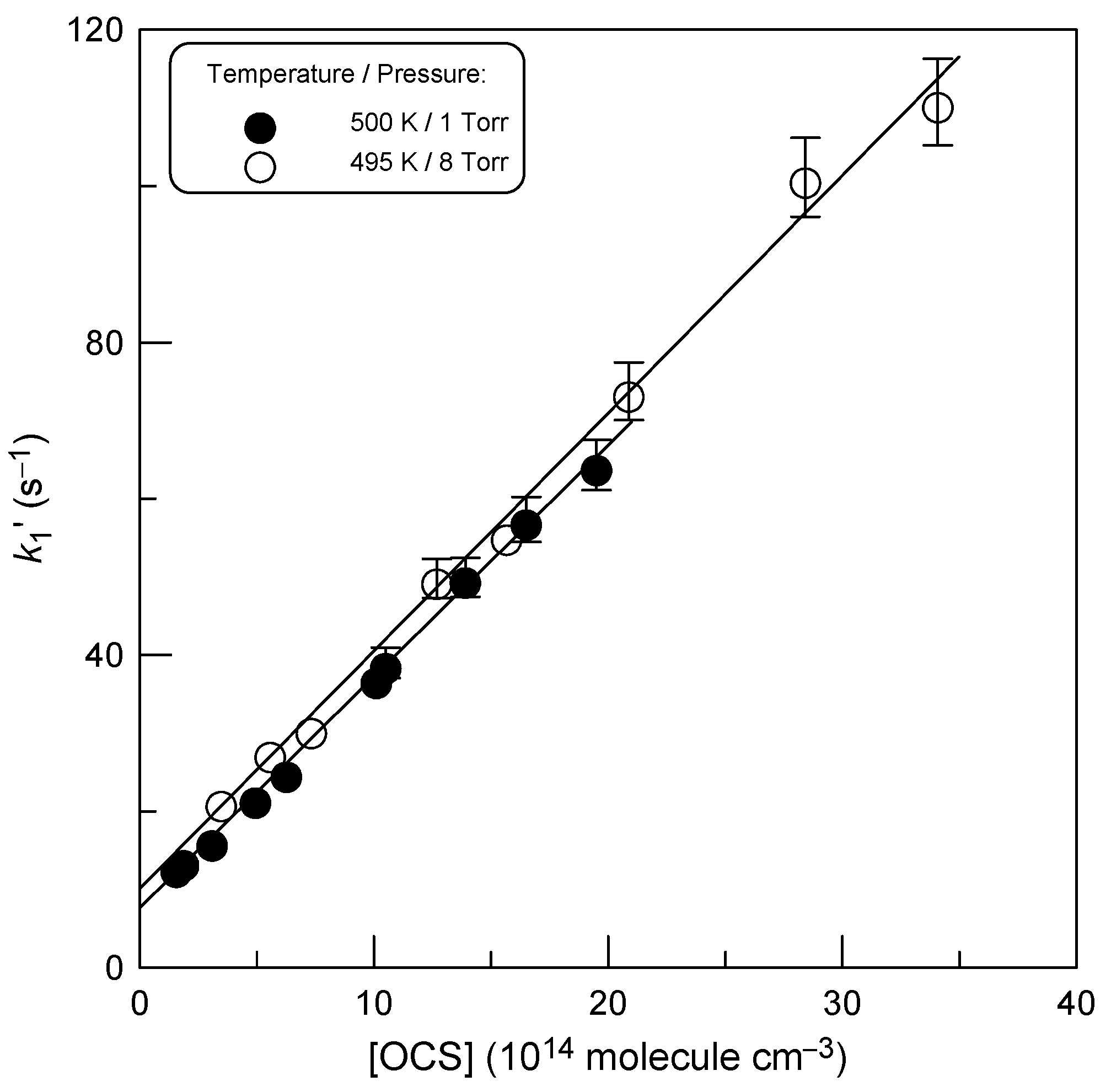
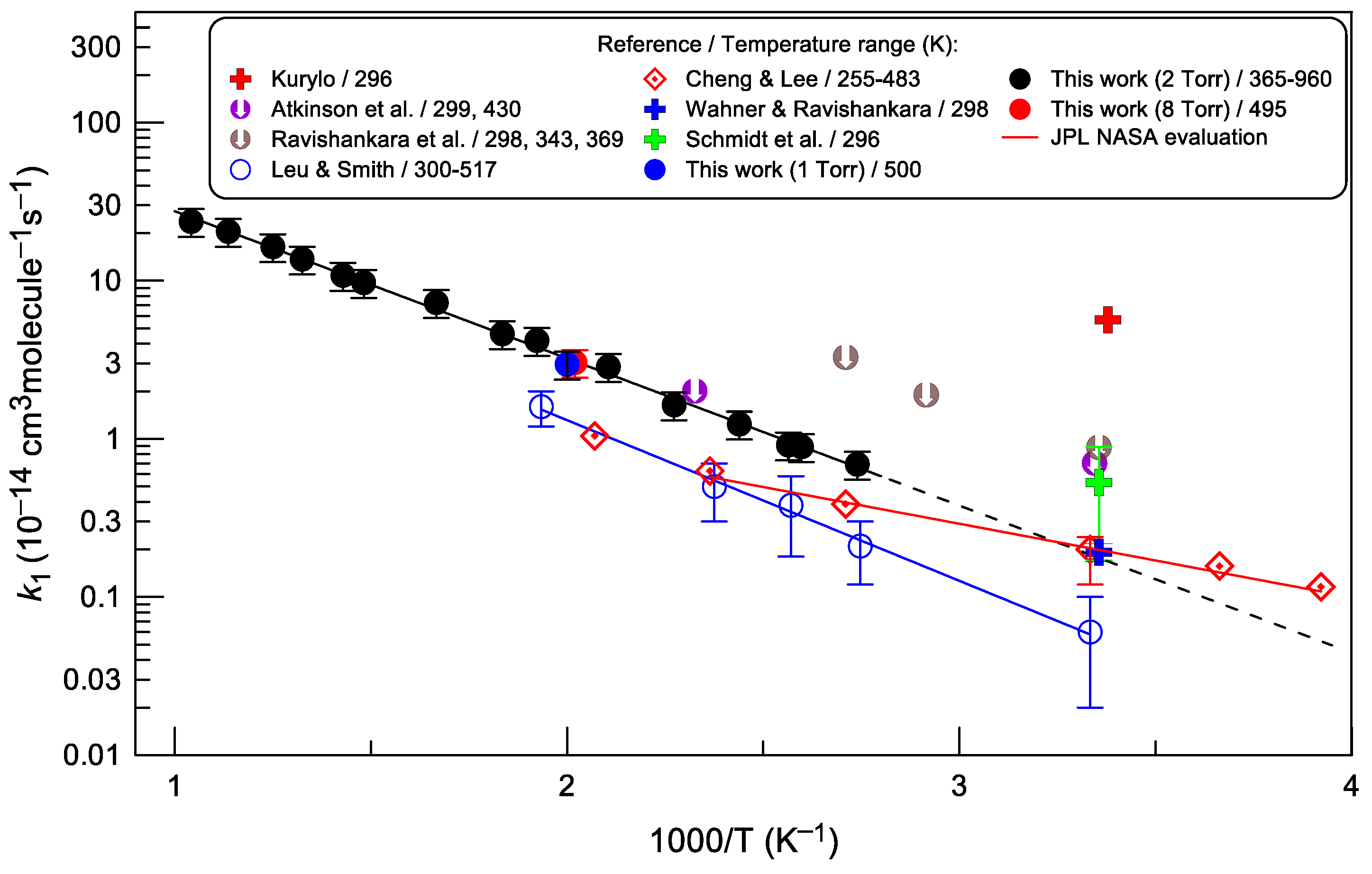
3.1. Rate Constant of Reaction (1)
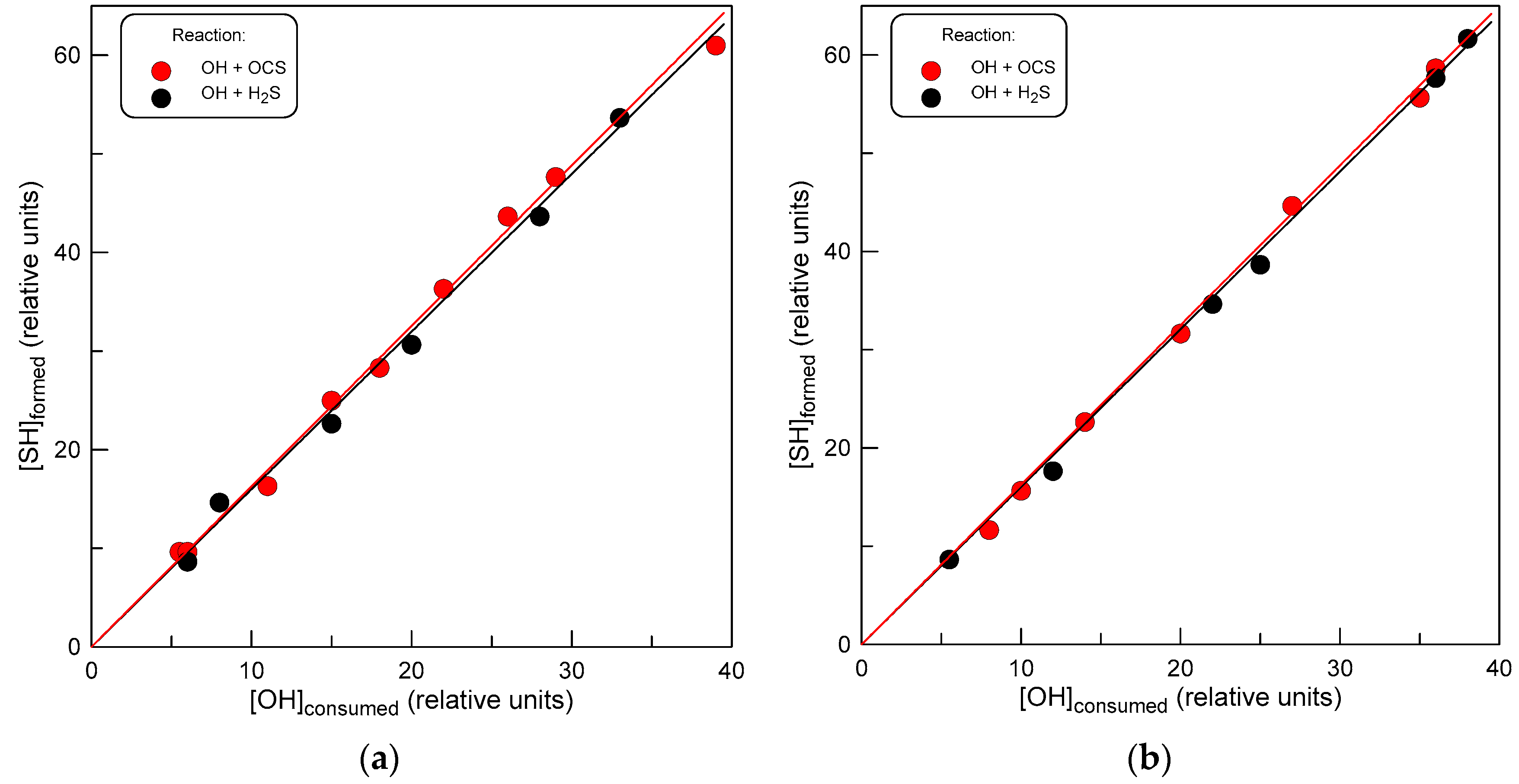
3.2. Products of Reaction (1)
4. Discussion
4.1. Temperature Dependence of k1
4.2. Pressure Dependence of k1
4.3. Products of Reaction (1)
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kettle, A.J.; Kuhn, U.; von Hobe, M.; Kesselmeier, J.; Andreae, M.O. Global budget of atmospheric carbonyl sulfide: Temporal and spatial variations of the dominant sources and sinks. J. Geophys. Res. Atmos. 2002, 107, 4658. [Google Scholar] [CrossRef]
- Atkinson, R.; Perry, R.A.; Pitts, J.N. Rate constants for the reaction of OH radicals with COS, CS2 and CH3SCH3 over the temperature range 299–430 K. Chem. Phys. Lett. 1978, 54, 14–18. [Google Scholar] [CrossRef]
- Kurylo, M.J. Flash photolysis resonance fluorescence investigation of the reactions of OH radicals with OCS and CS2. Chem. Phys. Lett. 1978, 58, 238–242. [Google Scholar] [CrossRef]
- Ravishankara, A.R.; Kreutter, N.M.; Shah, R.C.; Wine, P.H. Rate of reaction of OH with COS. Geophys. Res. Lett. 1980, 7, 861–864. [Google Scholar] [CrossRef]
- Leu, M.-T.; Smith, R.H. Kinetics of the gas-phase reaction between hydroxyl and carbonyl sulfide over the temperature range 300–517 K. J. Phys. Chem. 1981, 85, 2570–2575. [Google Scholar] [CrossRef]
- Cheng, B.-M.; Lee, Y.-P. Rate constant of OH + OCS reaction over the temperature range 255–483 K. Int. J. Chem. Kinet. 1986, 18, 1303–1314. [Google Scholar] [CrossRef]
- Wahner, A.; Ravishankara, A.R. The kinetics of the reaction of OH With COS. J. Geophys. Res. Atmos. 1987, 92, 2189–2194. [Google Scholar] [CrossRef]
- Schmidt, J.A.; Kyte, M.; Østerstrøm, F.F.; Joelsson, L.M.T.; Knap, H.C.; Jørgensen, S.; Nielsen, O.J.; Murakami, T.; Johnson, M.S. On adduct formation and reactivity in the OCS+OH reaction: A combined theoretical and experimental study. Chem. Phys. Lett. 2017, 675, 111–117. [Google Scholar] [CrossRef]
- Danielache, S.O.; Johnson, M.S.; Nanbu, S.; Grage, M.M.L.; McLinden, C.; Yoshida, N. Ab initio study of sulfur isotope fractionation in the reaction of OCS with OH. Chem. Phys. Lett. 2008, 450, 214–220. [Google Scholar] [CrossRef]
- Saheb, V.; Alizadeh, M.; Rezaei, F.; Shahidi, S. Quantum chemical and theoretical kinetics studies on the reaction of carbonyl sulfide with H, OH and O(3P). Comput. Theor. Chem. 2012, 994, 25–33. [Google Scholar] [CrossRef]
- Glarborg, P.; Marshall, P. Oxidation of Reduced Sulfur Species: Carbonyl Sulfide. Int. J. Chem. Kinet. 2013, 45, 429–439. [Google Scholar] [CrossRef]
- Roberts, T.; Dayma, G.; Oppenheimer, C. Reaction rates control high-temperature chemistry of volcanic gases in air. Front. Earth Sci. 2019, 7, 154. [Google Scholar] [CrossRef]
- Bedjanian, Y. Temperature dependent rate constant for the reaction of H-atoms with carbonyl sulfide. Int. J. Chem. Kinet. 2024, 56, 162–167. [Google Scholar] [CrossRef]
- Bedjanian, Y. Experimental Study of the Reaction of O(3P) with Carbonyl Sulfide between 220 and 960 K. J. Phys. Chem. A 2022, 126, 4080–4086. [Google Scholar] [CrossRef] [PubMed]
- Su, M.C.; Kumaran, S.S.; Lim, K.P.; Michael, J.V.; Wagner, A.F.; Harding, L.B.; Fang, D.C. Rate Constants, 1100 ≤ T ≤ 2000 K, for H + NO2 → OH + NO Using Two Shock Tube Techniques: Comparison of Theory to Experiment. J. Phys. Chem. A 2002, 106, 8261–8270. [Google Scholar] [CrossRef]
- Atkinson, R.; Baulch, D.L.; Cox, R.A.; Crowley, J.N.; Hampson, R.F.; Hynes, R.G.; Jenkin, M.E.; Rossi, M.J.; Troe, J. Evaluated kinetic and photochemical data for atmospheric chemistry: Volume III—Gas phase reactions of inorganic halogens. Atmos. Chem. Phys. 2007, 7, 981–1191. [Google Scholar] [CrossRef]
- Bedjanian, Y. Temperature-Dependent Rate Constant for the Reaction of Hydroxyl Radical with 3-Hydroxy-3-methyl-2-butanone. J. Phys. Chem. A 2019, 123, 10446–10453. [Google Scholar] [CrossRef] [PubMed]
- Burkholder, J.B.; Sander, S.P.; Abbatt, J.; Barker, J.R.; Cappa, C.; Crounse, J.D.; Dibble, T.S.; Huie, R.E.; Kolb, C.E.; Kurylo, M.J.; et al. Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 19, JPL Publication 19-5, Jet Propulsion Laboratory. Available online: http://jpldataeval.jpl.nasa.gov (accessed on 20 April 2024).
- Wang, N.S.; Lovejoy, E.R.; Howard, C.J. Temperature dependence of the rate constant for the reaction mercapto + nitrogen dioxide. J. Phys. Chem. 1987, 91, 5743–5749. [Google Scholar] [CrossRef]
- Lovejoy, E.R.; Wang, N.S.; Howard, C.J. Kinetic Studies of the Reactions of HSO with NO2, NO, and O2. J. Phys. Chem. 1987, 91, 5749–5755. [Google Scholar] [CrossRef]
- Kaufman, F. Kinetics of elementary radical reactions in the gas phase. J. Phys. Chem. 1984, 88, 4909–4917. [Google Scholar] [CrossRef]
- Goumri, A.; Rocha, J.-D.R.; Laakso, D.; Smith, C.E.; Marshall, P. Characterization of Reaction Pathways on the Potential Energy Surfaces for H + SO2 and HS + O2. J. Phys. Chem. A 1999, 103, 11328–11335. [Google Scholar] [CrossRef]
- Nicovich, J.M.; Kreutter, K.D.; Van Dijk, C.A.; Wine, P.H. Temperature-dependent kinetics studies of the reactions Br(2P3/2) + H2S ↔ SH + HBr and Br(2P3/2) + CH3SH ↔ CH3S + HBr. Heats of formation of SH and CH3S radicals. J. Phys. Chem. 1992, 96, 2518–2528. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T (K) | [OCS] a | k1 b | OH Source | Pressure (Torr) |
|---|---|---|---|---|
| 365 | 3.50–51.5 | 0.69 ± 0.04 | H + NO2 | 2 |
| 385 | 7.19–51.5 | 0.89 ± 0.03 | H + NO2 | 2 |
| 390 | 3.73–50.7 | 0.91 ± 0.02 | H + NO2 | 2 |
| 410 | 2.29–33.6 | 1.24 ± 0.05 | H + NO2 | 2 |
| 440 | 1.45–28.3 | 1.64 ± 0.04 | H + NO2 | 2 |
| 475 | 2.50–31.4 | 2.86 ± 0.09 | H + NO2 | 2 |
| 495 | 3.48–34.1 | 3.04 ± 0.09 | H + NO2 | 8 |
| 500 | 1.96–19.5 | 2.96 ± 0.09 | H + NO2 | 1 |
| 520 | 2.44–33.9 | 4.19 ± 0.08 | H + NO2 | 2 |
| 545 | 2.00–27.7 | 4.62 ± 0.26 | F + H2O | 2 |
| 600 | 1.29–16.1 | 7.27 ± 0.14 | H + NO2 | 2 |
| 675 | 1.16–17.2 | 9.73 ± 0.30 | H + NO2 | 2 |
| 700 | 0.55–14.0 | 10.8 ± 0.34 | H + NO2 | 2 |
| 755 | 3.58–18.1 | 13.7 ± 0.65 | F + H2O | 2 |
| 800 | 0.356–7.12 | 16.4 ± 0.44 | H + NO2 | 2 |
| 880 | 1.42–15.6 | 20.5 ± 0.45 | H + NO2 | 2 |
| 960 | 0.26–9.79 | 23.7 ± 0.34 | H + NO2 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bedjanian, Y. Experimental Study of the Reaction of OH Radicals with Carbonyl Sulfide between 365 and 960 K: Kinetics and Products. Atmosphere 2024, 15, 576. https://doi.org/10.3390/atmos15050576
Bedjanian Y. Experimental Study of the Reaction of OH Radicals with Carbonyl Sulfide between 365 and 960 K: Kinetics and Products. Atmosphere. 2024; 15(5):576. https://doi.org/10.3390/atmos15050576
Chicago/Turabian StyleBedjanian, Yuri. 2024. "Experimental Study of the Reaction of OH Radicals with Carbonyl Sulfide between 365 and 960 K: Kinetics and Products" Atmosphere 15, no. 5: 576. https://doi.org/10.3390/atmos15050576
APA StyleBedjanian, Y. (2024). Experimental Study of the Reaction of OH Radicals with Carbonyl Sulfide between 365 and 960 K: Kinetics and Products. Atmosphere, 15(5), 576. https://doi.org/10.3390/atmos15050576