Global Warming Potential (GWP) for Methane: Monte Carlo Analysis of the Uncertainties in Global Tropospheric Model Predictions


Abstract
1. Introduction
2. Monte Carlo Uncertainty Analysis and Its Methodology
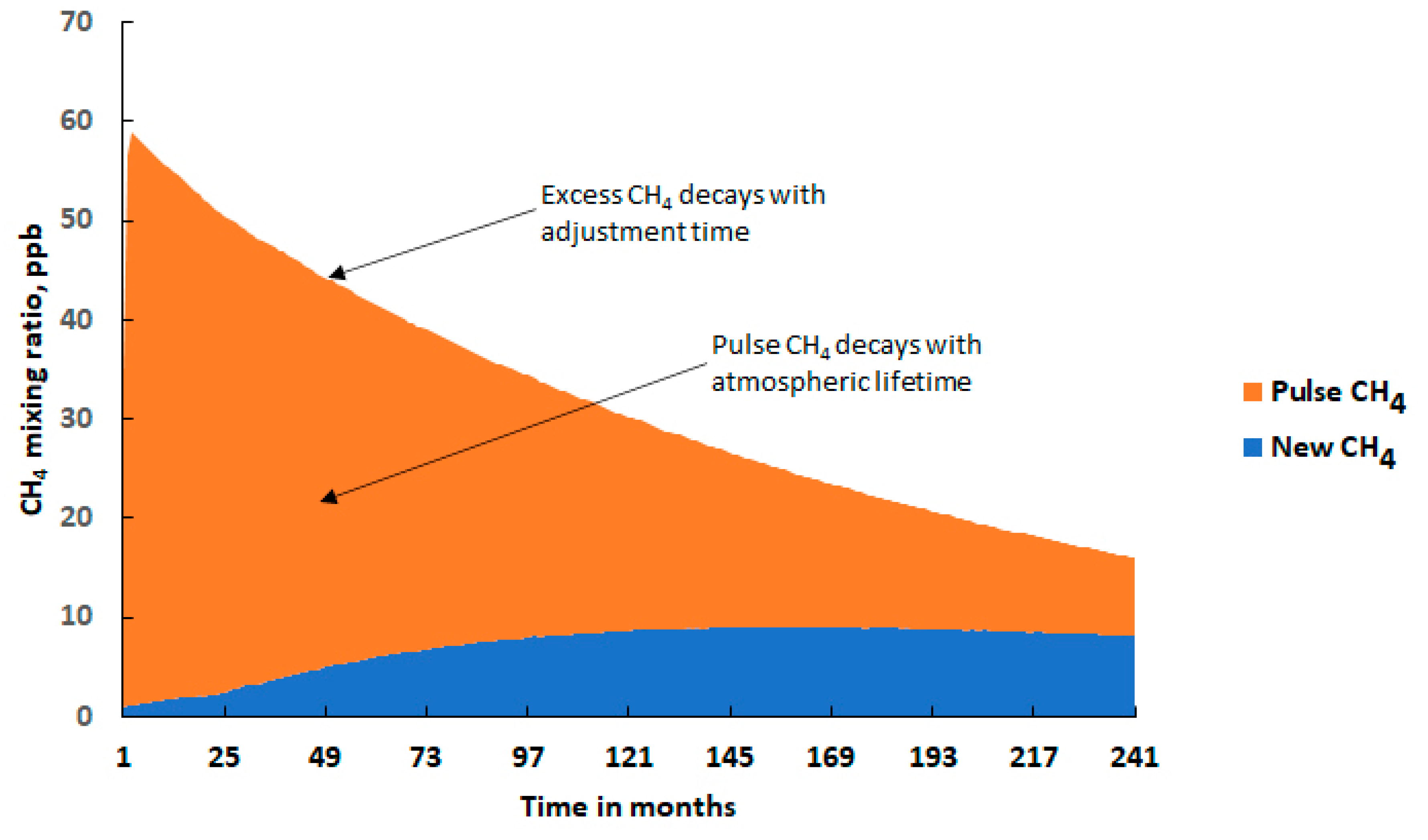
2.1. The Pulse Behaviour of Methane
2.2. TROPOS Model
2.3. Monte Carlo Uncertainty Analysis
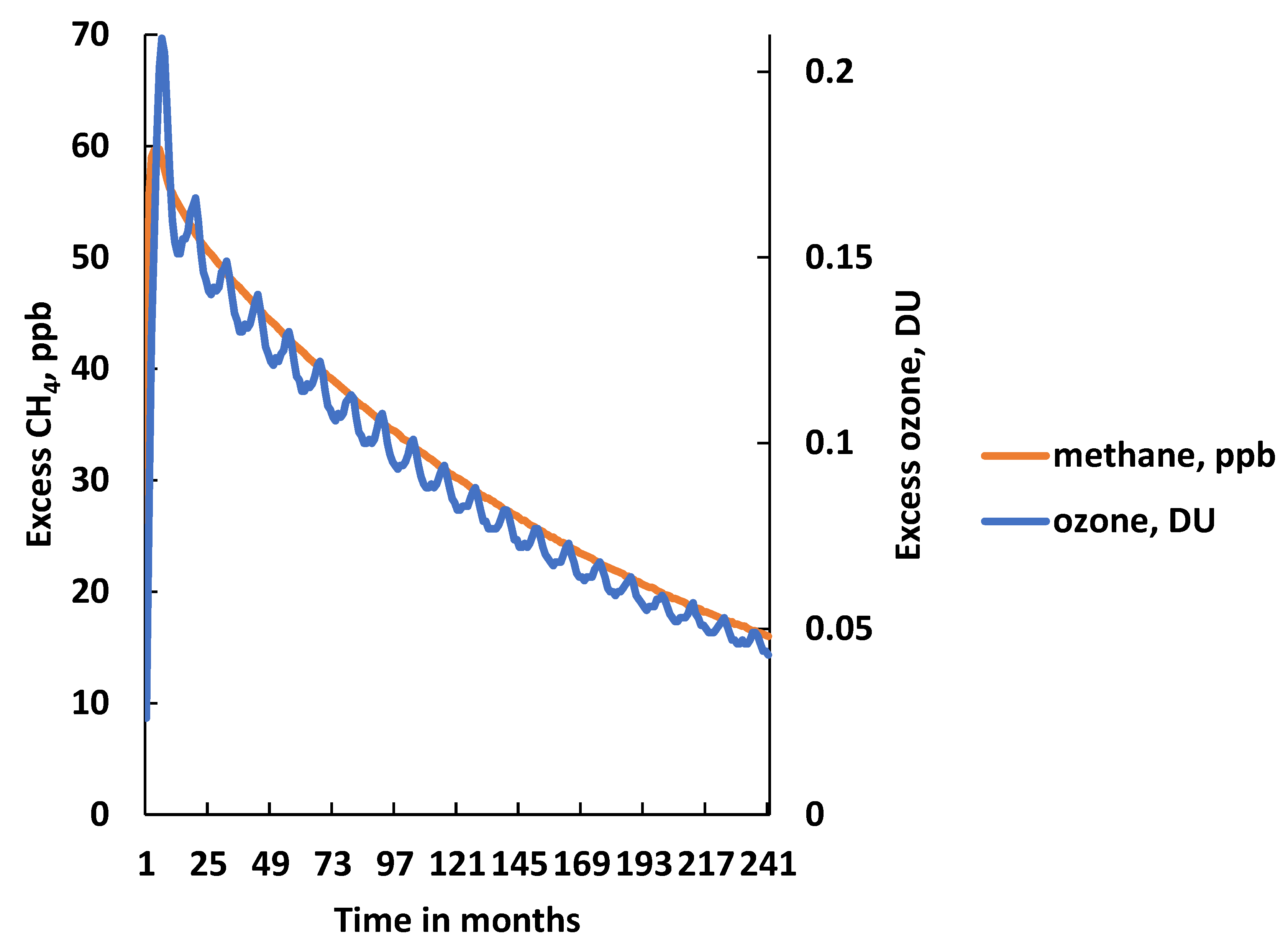
3. CH4 Pulse Experiments under Uncertainty
- The time-integrated excess CH4 mixing ratio in ppb years over the 20-year model experiment: 649 ppb year (668 ± 162 ppb year);
- The excess CH4 mixing ratio at the end of the 20-year model experiment: 16 ppb (17.6 ± 11 ppb);
- The CH4 adjustment time: 15.7 years (17.5 ± 9.7 years).
- The time-integrated excess O3 in DU years over the 20-year model experiment: 1.85 DU year in the BE run (1.90 ± 0.75 DU year in the MC replicates);
- The excess O3 in DU at the end of the 20-year model experiment: 0.043 DU in the BE run (0.045 ± 0.019 DU in the MC replicates);
- The O3 adjustment time: 15.3 year in the BE run (15.7 ± 4.6 year in the MC replicates).
4. Estimation of the Global Warming Potential for CH4 under Uncertainty
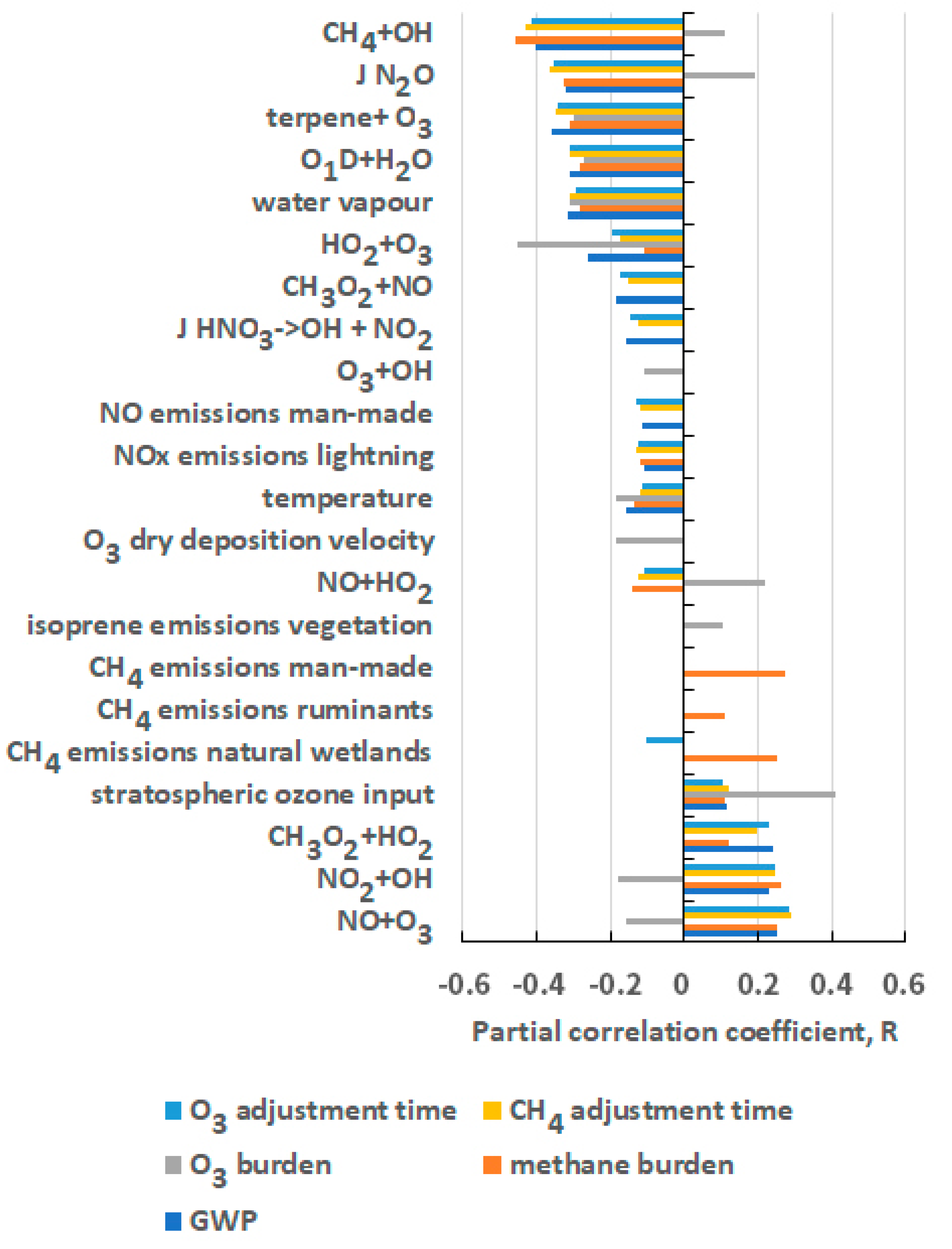
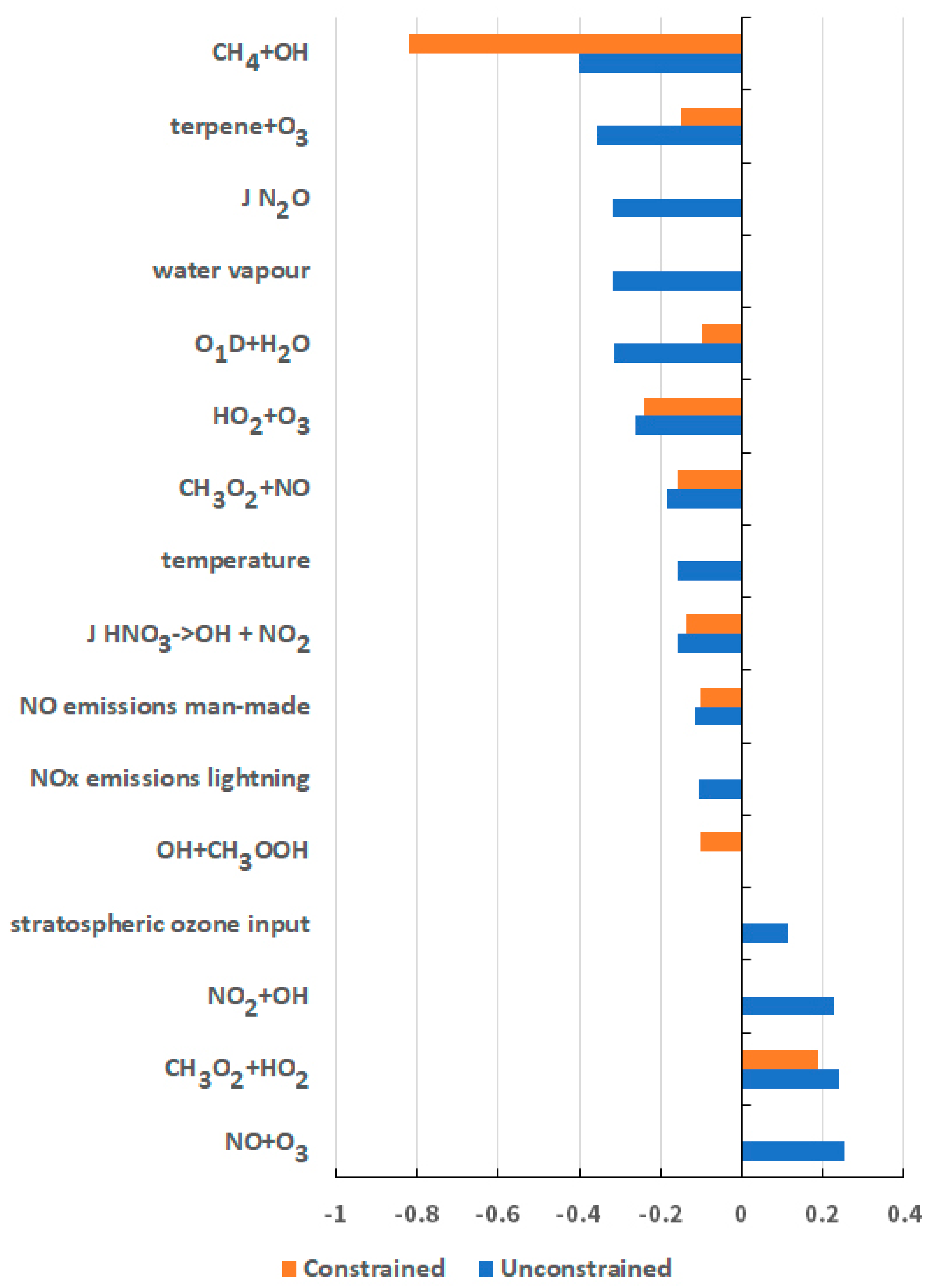
5. Origins of the Uncertainty in the GWP for CH4
6. Discussion and Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- IPCC. Climate Change 2018: The Physical Science Basis; Cambridge University Press: Cambridge, UK, 2018. [Google Scholar]
- United Nations Framework Convention on Climate Change. United Nations Framework Convention on Climate Change; UNFCCC: New York, NY, USA, 1992. [Google Scholar]
- Etheridge, D.M.; Pearman, G.I.; Fraser, P.J. Changes in tropospheric methane between 1841 and 1978 from a high accumulation-rate Antarctic ice core. Tellus B 1992, 44, 282–294. [Google Scholar] [CrossRef]
- IPCC. Climate Change 1994. In Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 1995. [Google Scholar]
- Turner, A.J.; Frankenburg, C.; Wennberg, P.O.; Jacob, D.J. Ambiguity in the causes for decadal trends in atmospheric methane and hydroxyl. Proc. Natl. Acad. Sci. USA 2017, 114, 5367–5372. [Google Scholar] [CrossRef] [PubMed]
- Rigby, M.; Montzka, S.A.; Prinn, R.G.; White, J.W.; Young, D.; O’Doherty, S.; Lunt, M.F.; Ganesan, A.L.; Manning, A.J.; Simmonds, P.G.; et al. Role of atmospheric oxidation in recent methane growth. Proc. Natl. Acad. Sci. USA 2017, 114, 5373–5377. [Google Scholar] [CrossRef] [PubMed]
- IPCC. Climate Change: The IPCC Scientific Assessment; Cambridge University Press: Cambridge, UK, 1990. [Google Scholar]
- Shine, K.P. The global warming potential—The need for an interdisciplinary retrial. Clim. Chang. 2009, 96, 467–472. [Google Scholar] [CrossRef]
- Shine, K.P.; Fuglestvedt, J.S.; Hailemariam, K.; Stuber, N. Alternatives to the global warming potential for comparing climate impacts of emissions of greenhouse gases. Clim. Chang. 2005, 68, 281–302. [Google Scholar] [CrossRef]
- Derwent, R.G.; Parrish, D.D.; Galbally, I.E.; Stevenson, D.S.; Doherty, R.M.; Naik, V.; Young, P.J. Uncertainties in models of tropospheric ozone based on Monte Carlo analysis: Tropospheric ozone burdens, atmospheric lifetimes and surface distributions. Atmos. Environ. 2018, 180, 93–102. [Google Scholar] [CrossRef]
- Derwent, R.G. Monte Carlo analyses of the uncertainties in the predictions from global tropospheric ozone models: Tropospheric burdens and seasonal cycles. Atmos. Environ. 2020, 213, 117545. [Google Scholar] [CrossRef]
- Bolin, B.; Rodhe, H. Note on concepts of age and transit-time in natural reservoirs. Tellus 1973, 25, 58–62. [Google Scholar] [CrossRef]
- Prather, M.J. Lifetimes and eigenstates in atmospheric chemistry. Geophys. Res. Lett. 1994, 21, 801–804. [Google Scholar] [CrossRef]
- Isaksen, I.S.A.; Hov, O. Calculation of trends in tropospheric O3, OH, CH4 and NOx. Tellus B 1987, 39, 271–285. [Google Scholar] [CrossRef][Green Version]
- Prather, M.J. Lifetimes and time scales in atmospheric chemistry. Phil. Trans. R. Soc. A 2007, 365, 1705–1726. [Google Scholar] [CrossRef] [PubMed]
- Wild, O.; Prather, M.J. Excitation of the primary tropospheric chemical mode in a global three-dimensional model. J. Geophys. Res. 2000, 105, 24647–24660. [Google Scholar] [CrossRef]
- Derwent, R.G.; Collins, W.J.; Johnson, C.E.; Stevenson, D.S. Transient behaviour of tropospheric ozone precursors in a global 3-D CTM and their indirect greenhouse effects. Clim. Chang. 2001, 49, 463–487. [Google Scholar] [CrossRef]
- Hough, A.M.; Derwent, R.G. Changes in the global concentration of tropospheric ozone due to human activities. Nature 1990, 344, 645–648. [Google Scholar] [CrossRef]
- Utembe, S.R.; Watson, L.A.; Shallcross, D.E.; Jenkin, M.E. A Common Representative Intermediates (CRI) mechanism for VOC degradation. Part 3: Development of a secondary organic aerosol module. Atmos. Environ. 2009, 43, 1982–1990. [Google Scholar] [CrossRef]
- Utembe, S.R.; Cooke, M.C.; Archibald, A.T.; Shallcross, D.E.; Derwent, R.G.; Jenkin, M.E. Simulating secondary organic aerosol in a 3-D Lagrangian chemistry transport model using the reduced Common Representative Intermediates mechanism (CRI v2-R5). Atmos. Environ. 2011, 45, 1604–1614. [Google Scholar] [CrossRef]
- Curtis, A.R.; Sweetenham, W.P. FACSIMILE Release H User’s Manual; AERE Report R11771; H.M. Stationery Office: London, UK, 1987. [Google Scholar]
- Cooper, O.R.; Parrish, D.D.; Ziemke, J.; Balashov, N.V.; Cupeiro, M.; Galbally, I.E.; Gilge, S.; Horowitz, L.; Jensen, N.R.; Lamarque, J.-F.; et al. Global distribution and trends of tropospheric ozone: An observation-based review. Elem. Sci. Anthr. 2014, 2, 000029. [Google Scholar] [CrossRef]
- Naik, V.; Voulgarakis, A.; Fiore, A.M.; Horowitz, L.W.; Lamarque, J.F.; Lin, M.; Prather, M.J.; Young, P.J.; Bergmann, D.; Cameron-Smith, P.J.; et al. Pre-industrial to present-day changes in tropospheric hydroxyl and methane lifetime from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP). Atmos. Chem. Phys. 2013, 13, 5277–5298. [Google Scholar] [CrossRef]
- Young, P.J.; Archibald, A.T.; Bowman, K.W.; Lamarque, J.F.; Naik, V.; Stevenson, D.S.; Tilmes, S.; Voulgarakis, A.; Wild, O.; Bergmann, D.; et al. Pre-industrial to end 21st century projections of tropospheric ozone from the Atmospheric Chemistry and Climate Model Intercomparison Project. Atmos. Chem. Phys. 2013, 13, 2063–2090. [Google Scholar] [CrossRef]
- Ziemke, J.R.; Chandra, S.; Labow, G.J.; Bhartia, P.K.; Froidevaux, L.; Witte, J.C. A global climatology of tropospheric and stratospheric ozone derived from Aura OMI and MLS measurements. Atmos. Chem. Phys. 2011, 11, 9237–9251. [Google Scholar] [CrossRef]
- Revell, L.E.; Stenke, A.; Tummon, F.; Feinberg, A.; Rozanov, E.; Peter, T.; Abraham, N.L.; Akiyoshi, H.; Archibald, A.T.; Butchart, N.; et al. Tropospheric ozone in CCMI models and Gaussian process emulation to understand biases in the SOCLv3 chemistry-climate model. Atmos. Chem. Phys. 2018, 18, 16155–16172. [Google Scholar] [CrossRef]
- Newsome, B.; Evans, M. Impact of uncertainties in inorganic chemical rate constants on tropospheric composition and ozone radiative forcing. Atmos. Chem. Phys. 2017, 17, 14333–14352. [Google Scholar] [CrossRef]
- Wild, O.; Voulgarakis, A.; O’Connor, F.; Lamarque, J.F.; Ryan, E.; Lee, L. Global sensitivity analysis of chemistry-climate model budgets of tropospheric ozone and OH: Exploring model diversity. Atmos. Chem. Phys. 2020. [Google Scholar] [CrossRef]
- Stevenson, D.S.; Young, P.J.; Naik, V.; Lamarque, J.F.; Shindell, D.T.; Voulgarakis, A.; Skeie, R.B.; Dalsoren, S.B.; Myhre, G.; Berntsen, T.K.; et al. Tropospheric ozone changes, radiative forcing and attribution to emissions in the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP). Atmos. Chem. Phys. 2013, 13, 3063–3085. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Units | This Study | TROPOS [11] | STOCHEM [10] | ACCMIP [21,24] | |
|---|---|---|---|---|---|
| O3 burden | Tg | 349 ± 101 | 324 ± 182 | 374 ± 182 | 337 ± 46 |
| O3 lifetime | days | 22.6 ± 6 | 20.5 ± 9 | 23.0 ± 8 | 22.3 ± 4 |
| CO burden | Tg | 484 ± 326 | 428 ± 253 | 374 ± 209 | 323 ± 76 |
| CO lifetime | days | 66 ± 36 | 57 ± 42 | 53 ± 34 | - |
| CH4 burden | Tg | 4842 ± 2230 | 5195 ± 756 | 4620 ±460 | 4813 ± 162 |
| CH4 lifetime | years | 11.4 ± 6.4 | 9.5 ± 4.0 | 9.0 ± 4.6 | 9.7 ± 3.0 |
| Pulse Size, Tg | Time-Integrated CH4 rf over 100 Years (mWm−2 year) | Time-Integrated O3 rf over 100 Years (mWm−2 year) | Total Time-Integrated rf over 100 Years (mWm−2 year) | Total Time-Integrated rf over 100 Years per 1 Tg Pulse (mWm−2 year) a | GWP over 100 Years Time Horizon a |
|---|---|---|---|---|---|
| 149 | 326.7 | 84.7 | 411.4 | 2.761 | 30.1 |
| 134 | 293.7 | 76.5 | 370.3 | 2.761 | 30.1 |
| 119 | 261.1 | 67.6 | 328.7 | 2.757 | 30.1 |
| 104 | 228.4 | 59.3 | 287.7 | 2.758 | 30.1 |
| 89 | 195.7 | 50.9 | 246.7 | 2.759 | 30.1 |
| 75 | 163.0 | 42.1 | 205.1 | 2.754 | 30.0 |
| 60 | 130.3 | 33.7 | 164.1 | 2.753 | 30.0 |
| 45 | 97.7 | 25.5 | 123.2 | 2.757 | 30.1 |
| 30 | 65.0 | 16.6 | 81.6 | 2.739 | 29.9 |
| 15 | 32.6 | 8.3 | 40.9 | 2.745 | 29.9 |
| Chemical Kinetic Rate Coefficients | Emission Sources and Sectors |
| CH4 + OH | NOx from man-made sources |
| O1D + H2O | NOx from lightning |
| HO2 + O3 | CH4 from man-made sources |
| O3 + OH | CH4 from natural wetlands |
| NO + HO2 | CH4 from ruminants |
| NO2 + OH | isoprene from vegetation |
| NO + O3 | - |
| CH3O2 + HO2 | - |
| Chemical Kinetic Rate Coefficients | Meteorological and Physical Parameters |
| CH3O2 + NO | Water vapour |
| terpene + O3 | Temperature |
| J HNO3 | O3 deposition velocity |
| J N2O | O3 input from stratosphere |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Derwent, R.G. Global Warming Potential (GWP) for Methane: Monte Carlo Analysis of the Uncertainties in Global Tropospheric Model Predictions. Atmosphere 2020, 11, 486. https://doi.org/10.3390/atmos11050486
Derwent RG. Global Warming Potential (GWP) for Methane: Monte Carlo Analysis of the Uncertainties in Global Tropospheric Model Predictions. Atmosphere. 2020; 11(5):486. https://doi.org/10.3390/atmos11050486
Chicago/Turabian StyleDerwent, Richard G. 2020. "Global Warming Potential (GWP) for Methane: Monte Carlo Analysis of the Uncertainties in Global Tropospheric Model Predictions" Atmosphere 11, no. 5: 486. https://doi.org/10.3390/atmos11050486
APA StyleDerwent, R. G. (2020). Global Warming Potential (GWP) for Methane: Monte Carlo Analysis of the Uncertainties in Global Tropospheric Model Predictions. Atmosphere, 11(5), 486. https://doi.org/10.3390/atmos11050486