Metagenomic Composition Analysis of an Ancient Sequenced Polar Bear Jawbone from Svalbard

 , ,
, ,  , , ,
, , ,  and
and 
Abstract
1. Introduction
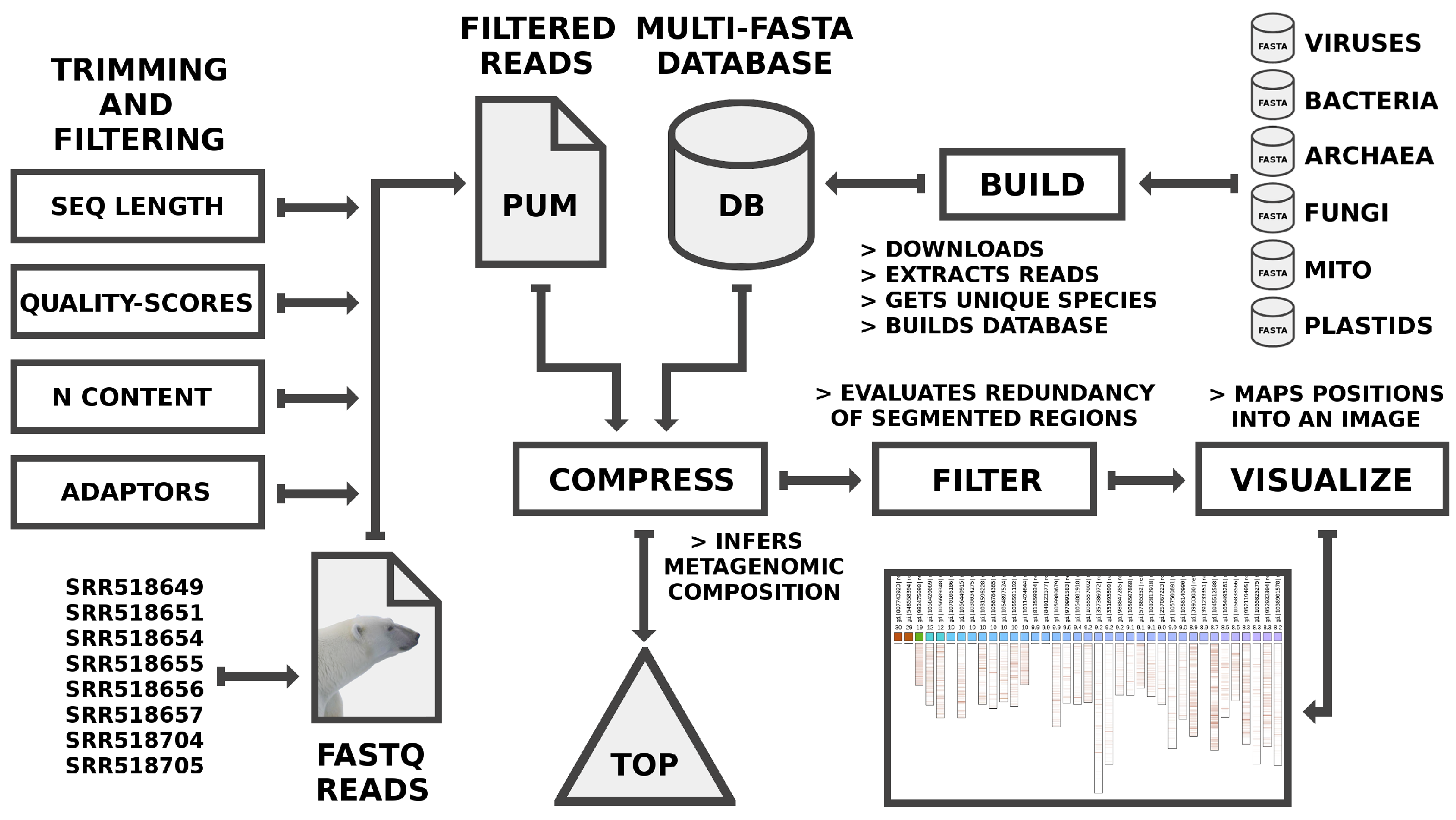
2. Methods
2.1. Filtering and Trimming Reads
2.2. Building the Database
2.3. Running FALCON-Meta
- Tolerant CM: depth: 20, alpha: 0.1, tolerance: 5;
- CM: depth: 20, alpha: 0.005, inverted repeats: yes;
- CM: depth: 14, alpha: 0.01, inverted repeats: yes;
- CM: depth: 11, alpha: 0.1, inverted repeats: no; and
- CM: depth: 6, alpha: 1, inverted repeats: no.
- ./FALCON -v -n 1 -t 800 -l 45 -F -Z -c 250 -y complexity.com PUM.fq DB.fa
- ./FALCON-FILTER -v -F -sl 0.001 -du 20000000 -t 0.5 -o positions.csv complexity.com
- ./FALCON-EYE -v -e 500 -s 4 -o top.svg positions.csv
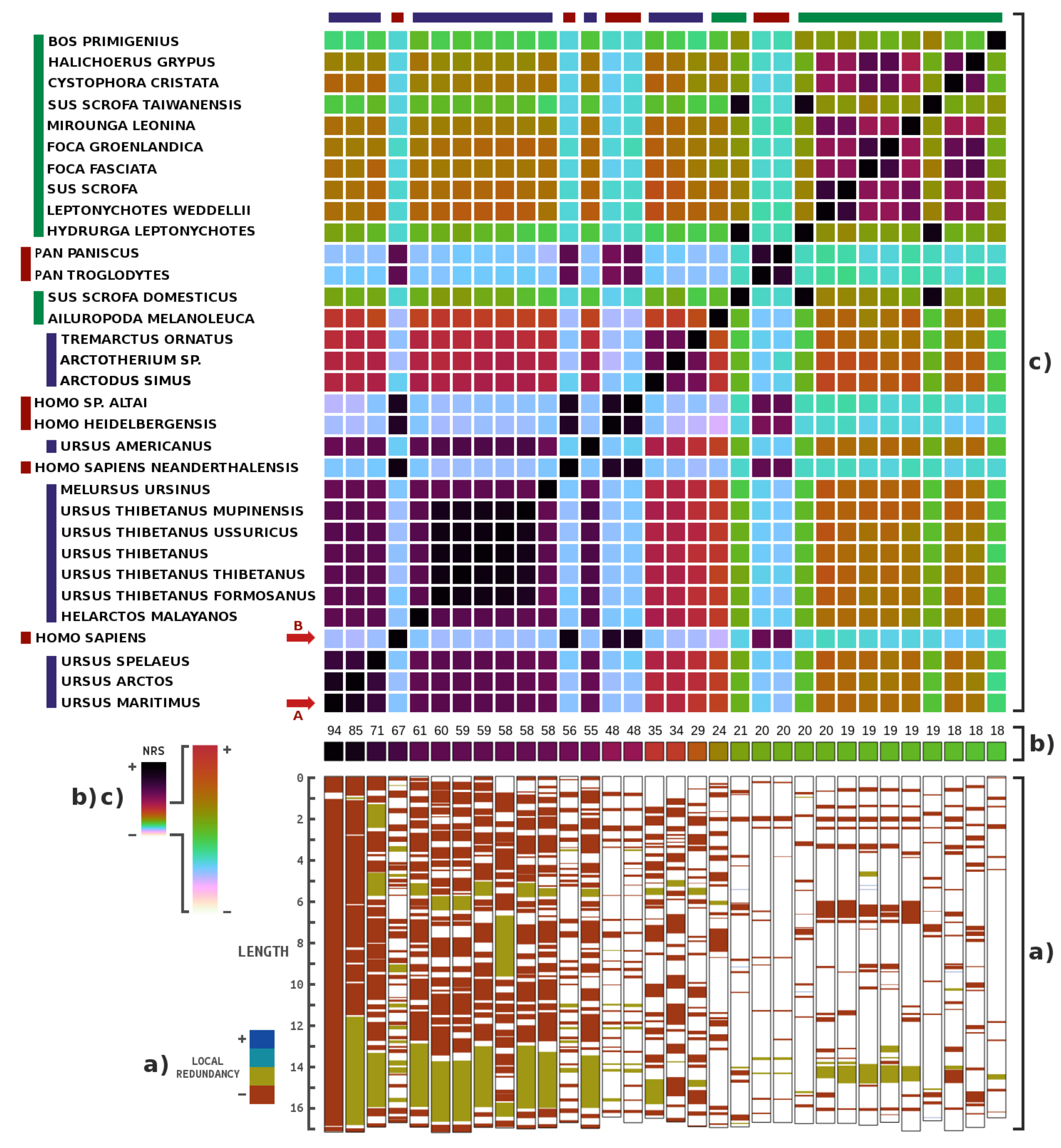
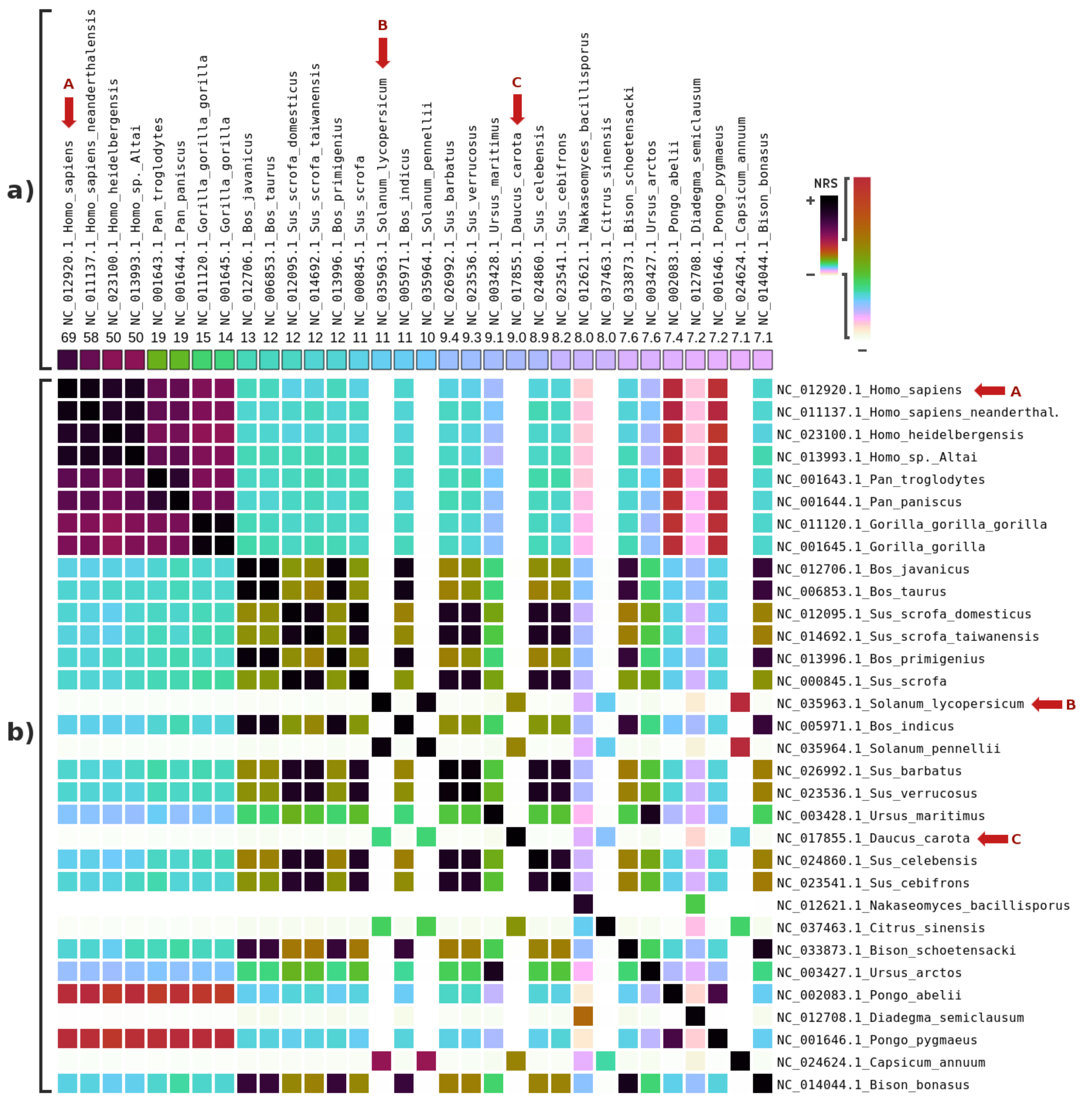
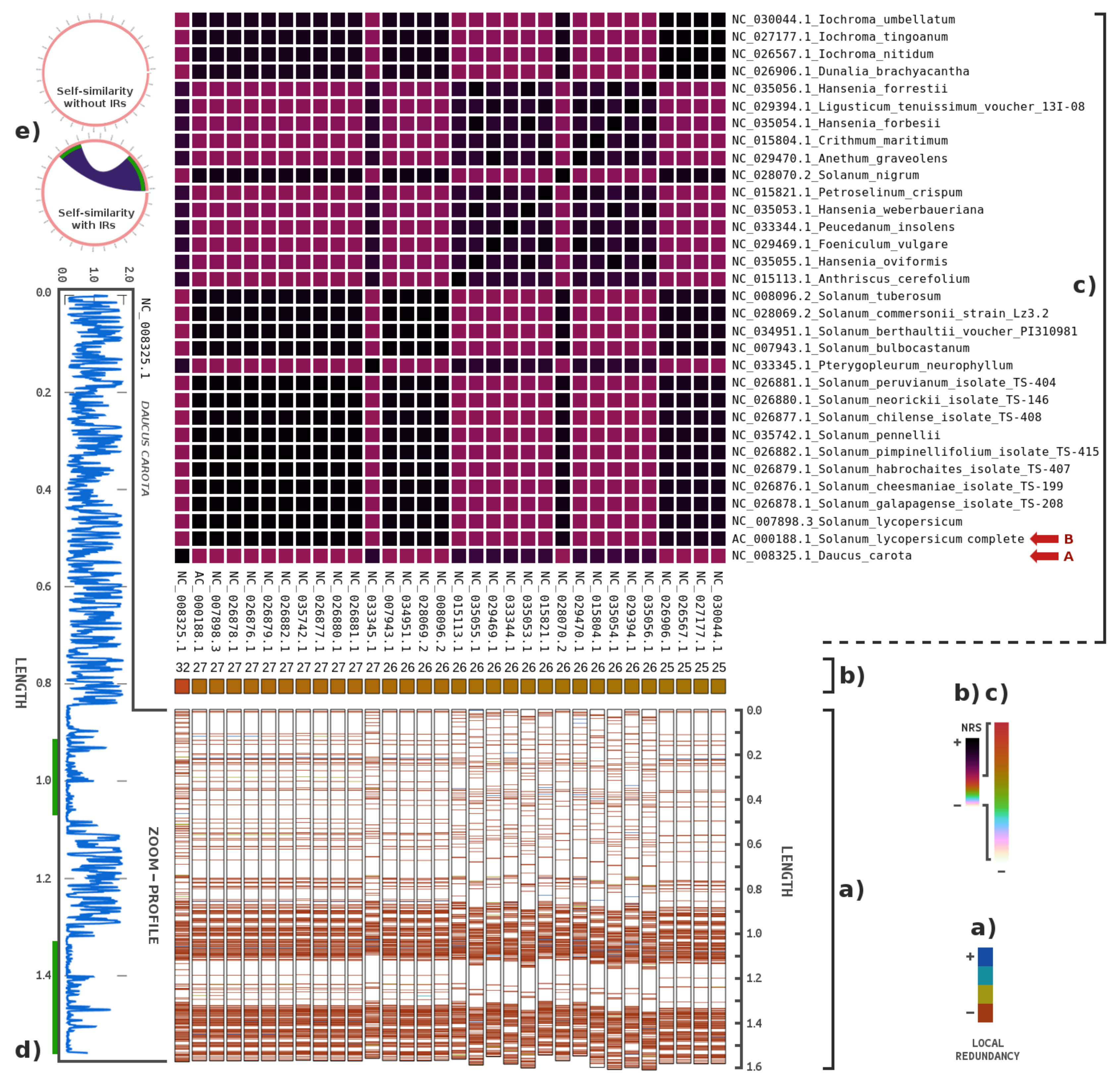
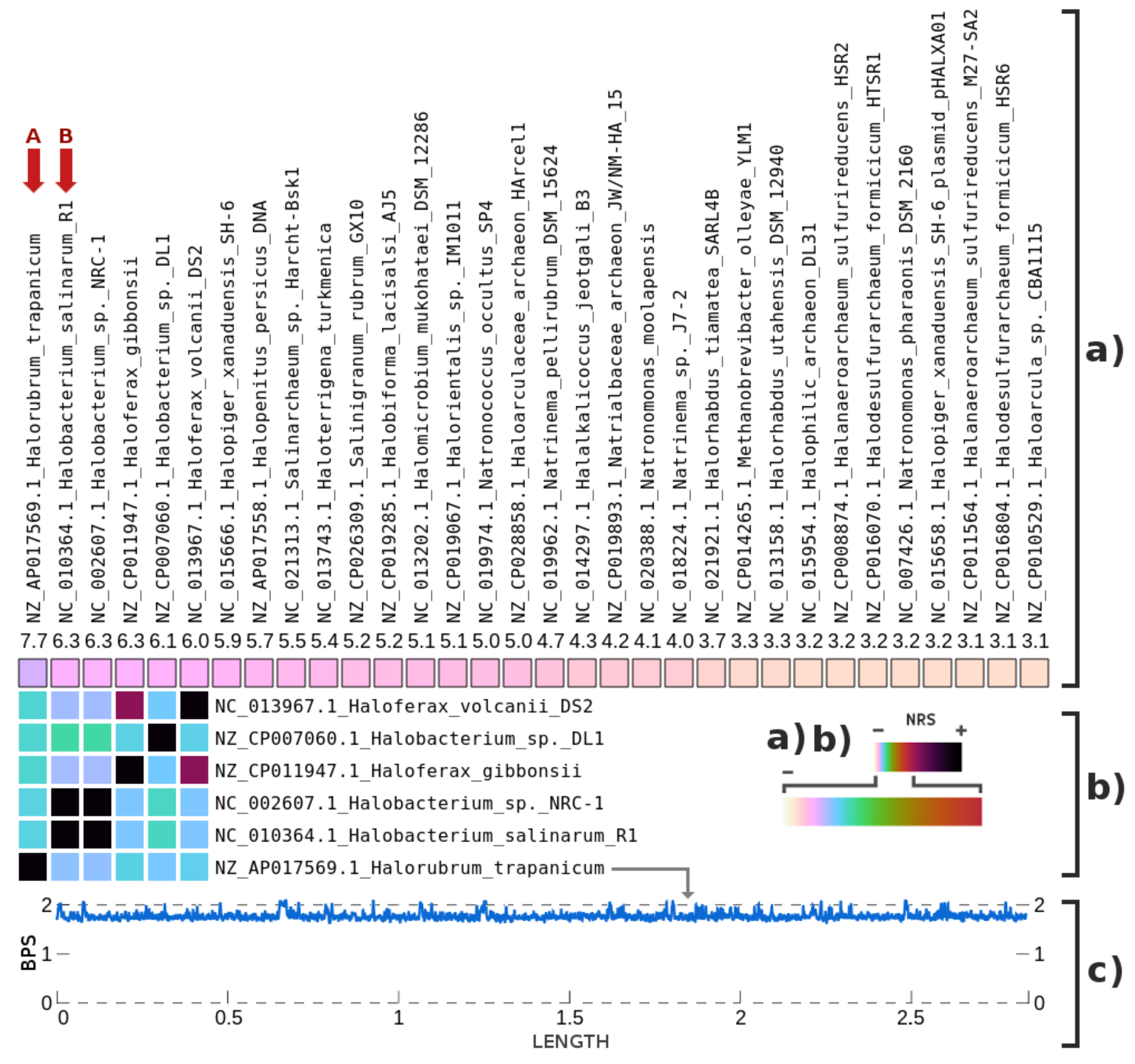
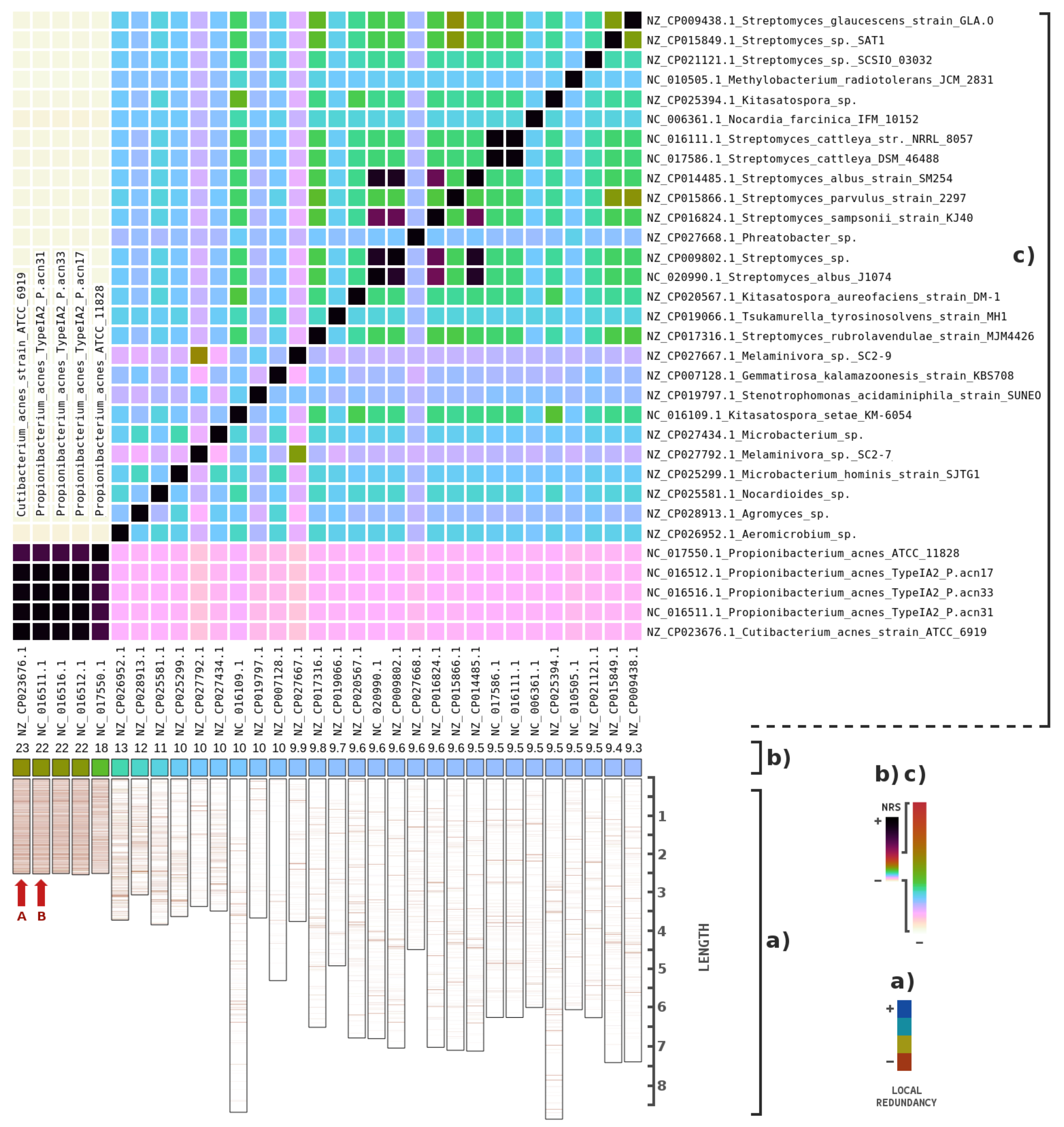
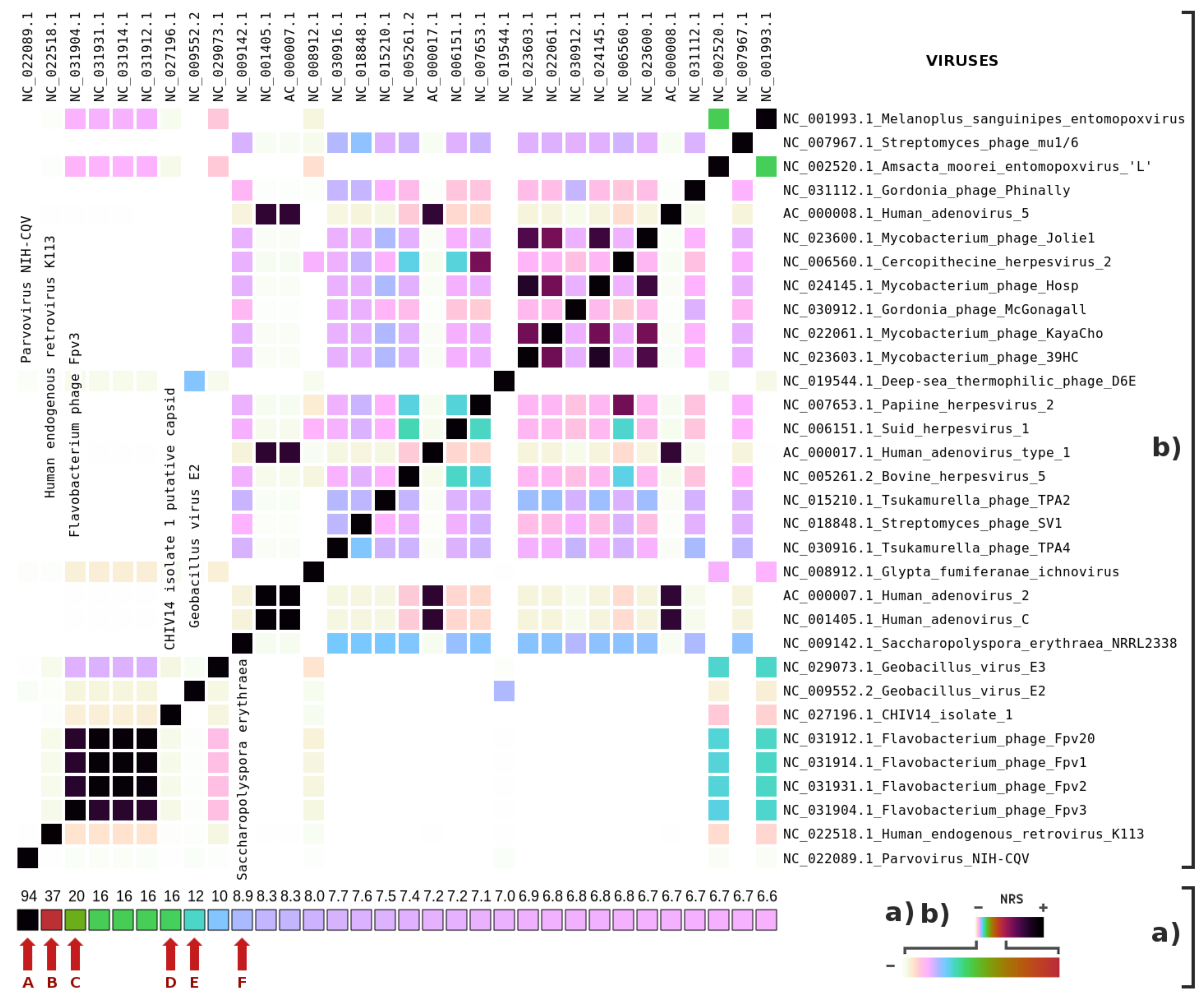
3. Results
4. Discussions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| aDNA | ancient DNA |
| CM | Context Model |
| DB | Database |
| DNA | Deoxyribonucleic acid |
| NRC | Normalized Relative Compression |
| NRS | Normalized Relative Similarity |
| PB | Polar Bear |
| PE | Paired Ends |
| PUM | Poolepynten Ursus maritimus (ancient Polar Bear) |
| RAM | Random Access Memory |
References
- Pääbo, S.; Poinar, H.; Serre, D.; Jaenicke-Després, V.; Hebler, J.; Rohland, N.; Kuch, M.; Krause, J.; Vigilant, L.; Hofreiter, M. Genetic analyses from ancient DNA. Annu. Rev. Genet. 2004, 38, 645–679. [Google Scholar] [CrossRef] [PubMed]
- Willerslev, E.; Hansen, A.J.; Binladen, J.; Brand, T.B.; Gilbert, M.T.P.; Shapiro, B.; Bunce, M.; Wiuf, C.; Gilichinsky, D.A.; Cooper, A. Diverse plant and animal genetic records from Holocene and Pleistocene sediments. Science 2003, 300, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Willerslev, E.; Hansen, A.J.; Poinar, H.N. Isolation of nucleic acids and cultures from fossil ice and permafrost. Trends Ecol. Evolut. 2004, 19, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Hofreiter, M.; Paijmans, J.L.; Goodchild, H.; Speller, C.F.; Barlow, A.; Fortes, G.G.; Thomas, J.A.; Ludwig, A.; Collins, M.J. The future of ancient DNA: Technical advances and conceptual shifts. BioEssays 2015, 37, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Ingólfsson, Ó.; Wiig, ∅. Late Pleistocene fossil find in Svalbard: The oldest remains of a polar bear (Ursus maritimus Phipps, 1744) ever discovered. Polar Res. 2009, 28, 455–462. [Google Scholar] [CrossRef]
- Lindqvist, C.; Schuster, S.C.; Sun, Y.; Talbot, S.L.; Qi, J.; Ratan, A.; Tomsho, L.P.; Kasson, L.; Zeyl, E.; Aars, J.; et al. Complete mitochondrial genome of a Pleistocene jawbone unveils the origin of polar bear. Proc. Natl. Acad. Sci. USA 2010, 107, 5053–5057. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.; Schuster, S.C.; Welch, A.J.; Ratan, A.; Bedoya-Reina, O.C.; Zhao, F.; Kim, H.L.; Burhans, R.C.; Drautz, D.I.; Wittekindt, N.E.; et al. Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proc. Natl. Acad. Sci. USA 2012, 109, E2382–E2390. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Lammers, F.; Bidon, T.; Pfenninger, M.; Kolter, L.; Nilsson, M.A.; Janke, A. The evolutionary history of bears is characterized by gene flow across species. Sci. Rep. 2017, 7, 46487. [Google Scholar] [CrossRef] [PubMed]
- Tsangaras, K.; Mayer, J.; Alquezar-Planas, D.E.; Greenwood, A.D. An evolutionarily young polar bear (Ursus maritimus) endogenous retrovirus identified from next generation sequence data. Viruses 2015, 7, 6089–6107. [Google Scholar] [CrossRef] [PubMed]
- Houldcroft, C.J.; Beale, M.A.; Breuer, J. Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 2017, 15, 183. [Google Scholar] [CrossRef] [PubMed]
- Duggan, A.T.; Perdomo, M.F.; Piombino-Mascali, D.; Marciniak, S.; Poinar, D.; Emery, M.V.; Buchmann, J.P.; Duchêne, S.; Jankauskas, R.; Humphreys, M.; et al. 17th century variola virus reveals the recent history of smallpox. Curr. Biol. 2016, 26, 3407–3412. [Google Scholar] [CrossRef] [PubMed]
- Weyrich, L.S.; Duchene, S.; Soubrier, J.; Arriola, L.; Llamas, B.; Breen, J.; Morris, A.G.; Alt, K.W.; Caramelli, D.; Dresely, V.; et al. Neanderthal behaviour, diet, and disease inferred from ancient DNA in dental calculus. Nature 2017, 544, 357. [Google Scholar] [CrossRef] [PubMed]
- Sajantila, A. Editors’ Pick: Contamination has always been the issue! Investig. Genet. 2014, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Louvel, G.; Der Sarkissian, C.; Hanghøj, K.; Orlando, L. metaBIT, an integrative and automated metagenomic pipeline for analysing microbial profiles from high-throughput sequencing shotgun data. Mol. Ecol. Res. 2016, 16, 1415–1427. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Herbig, A.; Maixner, F.; Bos, K.I.; Zink, A.; Krause, J.; Huson, D.H. MALT: Fast alignment and analysis of metagenomic DNA sequence data applied to the Tyrolean Iceman. bioRxiv 2017. [Google Scholar] [CrossRef]
- Wandelt, S.; Leser, U. MRCSI: Compressing and searching string collections with multiple references. Proc. VLDB Endow. 2015, 8, 461–472. [Google Scholar] [CrossRef]
- Jaenicke, S.; Albaum, S.P.; Blumenkamp, P.; Linke, B.; Stoye, J.; Goesmann, A. Flexible metagenome analysis using the MGX framework. Microbiome 2018, 6, 76. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yao, H.; Thompson, E.J.; Tannir, N.M.; Weinstein, J.N.; Su, X. VirusSeq: Software to identify viruses and their integration sites using next-generation sequencing of human cancer tissue. Bioinformatics 2013, 29, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Naccache, S.N.; Federman, S.; Veeraraghavan, N.; Zaharia, M.; Lee, D.; Samayoa, E.; Bouquet, J.; Greninger, A.L.; Luk, K.C.; Enge, B.; et al. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res. 2014, 24, 1180–1192. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Nie, K.; Zhang, C.; Zhang, Y.; Wang, J.; Niu, P.; Ma, X. VIP: An integrated pipeline for metagenomics of virus identification and discovery. Sci. Rep. 2016, 6, 23774. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Jun, S.R.; Leuze, M.; Ussery, D.; Nookaew, I. Viral phylogenomics using an alignment-free method: A three-step approach to determine optimal length of k-mer. Sci. Rep. 2017, 7, 40712. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Rampelli, S.; Soverini, M.; Turroni, S.; Quercia, S.; Biagi, E.; Brigidi, P.; Candela, M. ViromeScan: A new tool for metagenomic viral community profiling. BMC Genom. 2016, 17, 165. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 2017, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Costea, P.I.; Munch, R.; Coelho, L.P.; Paoli, L.; Sunagawa, S.; Bork, P. metaSNV: A tool for metagenomic strain level analysis. PLoS ONE 2017, 12, e0182392. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.Y.; Chen, T.; Fuhrman, J.A.; Sun, F. COCACOLA: Binning metagenomic contigs using sequence COmposition, read CoverAge, CO-alignment and paired-end read LinkAge. Bioinformatics 2017, 33, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.G.Z.; Green, K.T.; Dutilh, B.E.; Edwards, R.A. SUPER-FOCUS: A tool for agile functional analysis of shotgun metagenomic data. Bioinformatics 2015, 32, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Ramazzotti, M.; Berná, L.; Donati, C.; Cavalieri, D. riboFrame: An improved method for microbial taxonomy profiling from non-targeted metagenomics. Front. Genet. 2015, 6, 329. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Zhang, X.; Ligo, J.; Farnoud, F.; Veeravalli, V.; Milenkovic, O. MetaCRAM: An integrated pipeline for metagenomic taxonomy identification and compression. BMC Bioinform. 2016, 17, 94. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- Zielezinski, A.; Vinga, S.; Almeida, J.; Karlowski, W.M. Alignment-free sequence comparison: Benefits, applications, and tools. Genome Biol. 2017, 18, 186. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Bai, X.; Lu, Y.Y.; Tang, K.; Wang, Y.; Reinert, G.; Sun, F. Alignment-Free Sequence Analysis and Applications. Annu. Rev. Biomed. Data Sci. 2018, 1, 93–114. [Google Scholar] [CrossRef]
- Harbert, R.S. Algorithms and strategies in short-read shotgun metagenomic reconstruction of plant communities. Appl. Plant Sci. 2018, 6, e1034. [Google Scholar] [CrossRef] [PubMed]
- Pratas, D.; Pinho, A.J.; Silva, R.M.; Rodrigues, J.M.O.S.; Hosseini, M.; Caetano, T.; Ferreira, P.J.S.G. FALCON-meta: A method to infer metagenomic composition of ancient DNA. bioRxiv 2018. [Google Scholar] [CrossRef]
- Dabney, J.; Knapp, M.; Glocke, I.; Gansauge, M.T.; Weihmann, A.; Nickel, B.; Valdiosera, C.; García, N.; Pääbo, S.; Arsuaga, J.L.; et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. USA 2013, 110, 15758–15763. [Google Scholar] [CrossRef] [PubMed]
- Pratas, D.; Silva, R.M.; Pinho, A.J. Comparison of Compression-Based Measures with Application to the Evolution of Primate Genomes. Entropy 2018, 20, 393. [Google Scholar] [CrossRef]
- Pinho, A.J.; Pratas, D.; Ferreira, P.J.S.G. Authorship attribution using relative compression. In Proceedings of the 2016 Data Compression Conference, Snowbird, UT, USA, 30 March–1 April 2016. [Google Scholar]
- Budowle, B.; Connell, N.D.; Bielecka-Oder, A.; Colwell, R.R.; Corbett, C.R.; Fletcher, J.; Forsman, M.; Kadavy, D.R.; Markotic, A.; Morse, S.A.; et al. Validation of high throughput sequencing and microbial forensics applications. Investig. Genet. 2014, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Skoglund, P.; Northoff, B.H.; Shunkov, M.V.; Derevianko, A.P.; Pääbo, S.; Krause, J.; Jakobsson, M. Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proc. Natl. Acad. Sci. USA 2014, 111, 2229–2234. [Google Scholar] [CrossRef] [PubMed]
- Jónsson, H.; Ginolhac, A.; Schubert, M.; Johnson, P.L.; Orlando, L. mapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 2013, 23, 1682–1684. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Ginolhac, A.; Lindgreen, S.; Thompson, J.F.; Al-Rasheid, K.A.; Willerslev, E.; Krogh, A.; Orlando, L. Improving ancient DNA read mapping against modern reference genomes. BMC Genom. 2012, 13, 178. [Google Scholar] [CrossRef] [PubMed]
- Taron, U.H.; Lell, M.; Barlow, A.; Paijmans, J.L. Testing of Alignment Parameters for Ancient Samples: Evaluating and Optimizing Mapping Parameters for Ancient Samples Using the TAPAS Tool. Genes 2018, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Grüning, B.; Dale, R.; Sjödin, A.; Chapman, B.; Rowe, J.; Tomkins-Tinch, C.; Valieris, R.; Köster, J.; Bioconda, T. Bioconda: Sustainable and comprehensive software distribution for the life sciences. Nat. Methods 2018, 15, 475. [Google Scholar] [CrossRef] [PubMed]
- Pratas, D.; Pinho, A.J.; Ferreira, P.J.S.G. Efficient compression of genomic sequences. In Proceedings of the 2016 Data Compression Conference (DCC), Snowbird, UT, USA, 30 March–1 April 2016; pp. 231–240. [Google Scholar]
- Bell, T.C.; Cleary, J.G.; Witten, I.H. Text Compression; Prentice Hall: Upper Saddle River, NJ, USA, 1990. [Google Scholar]
- Pinho, A.J.; Pratas, D.; Ferreira, P.J.S.G. Bacteria DNA sequence compression using a mixture of finite-context models. In Proceedings of the 2011 IEEE Statistical Signal Processing Workshop (SSP), Nice, France, 28–30 June 2011. [Google Scholar]
- Pratas, D.; Pinho, A.J. Exploring deep Markov models in genomic data compression using sequence pre-analysis. In Proceedings of the 2014 22nd European Signal Processing Conference (EUSIPCO), Lisbon, Portugal, 1–5 September 2014; pp. 2395–2399. [Google Scholar]
- Pratas, D.; Hosseini, M.; Pinho, A.J. Substitutional Tolerant Markov Models for Relative Compression of DNA Sequences. In Proceedings of the International Conference on Practical Applications of Computational Biology & Bioinformatics, Porto, Portugal, 21–23 June 2017; Springer: Cham, Switzerland, 2017; pp. 265–272. [Google Scholar]
- Ferreira, P.J.S.G.; Pinho, A.J. Compression-based normal similarity measures for DNA sequences. In Proceedings of the 2014 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP-2014), Florence, Italy, 4–9 May 2014; pp. 419–423. [Google Scholar]
- Pratas, D. Compression and Analysis of Genomic Data. Ph.D. Thesis, University of Aveiro, Aveiro, Portugal, 2016. [Google Scholar]
- Posada, D. Phylogenomics for Systematic Biology. Syst. Biol. 2016, 65, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Pinho, A.J.; Garcia, S.P.; Pratas, D.; Ferreira, P.J. DNA sequences at a glance. PLoS ONE 2013, 8, e79922. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.; Pratas, D.; Pinho, A.J. On the role of inverted repeats in DNA sequence similarity. In Proceedings of the International Conference on Practical Applications of Computational Biology & Bioinformatics, Porto, Portugal, 21–23 June 2017; Springer: Cham, Switzerland, 2017; pp. 228–236. [Google Scholar]
- Scholz, C.F.; Kilian, M. The natural history of cutaneous propionibacteria, and reclassification of selected species within the genus Propionibacterium to the proposed novel genera Acidipropionibacterium gen. nov., Cutibacterium gen. nov. and Pseudopropionibacterium gen. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 4422–4432. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.A.; Korobeynikov, A.; Lapidus, A.; Prjibelski, A.D.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 2013, 20, 714–737. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Naccache, S.N.; Hackett, J.; Delwart, E.L.; Chiu, C.Y. Concerns over the origin of NIH-CQV, a novel virus discovered in Chinese patients with seronegative hepatitis. Proc. Natl. Acad. Sci. USA 2014, 111, E976. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; Xu, G.; Morici, L.; Bon-Durant, S.S.; Baddoo, M.; Lin, Z.; Fewell, C.; Taylor, C.M.; Flemington, E.K. Microbial contamination in next generation sequencing: Implications for sequence-based analysis of clinical samples. PLoS Pathog. 2014, 10, e1004437. [Google Scholar] [CrossRef] [PubMed]
- Stenholm, A.R.; Dalsgaard, I.; Middelboe, M. Isolation and characterization of bacteriophages infecting the fish pathogen Flavobacterium psychrophilum. Appl. Environ. Microbiol. 2008, 74, 4070–4078. [Google Scholar] [CrossRef] [PubMed]
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.; Green, R.E.; Kelso, J.; Prüfer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M.; et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef] [PubMed]
- Green, R.E.; Briggs, A.W.; Krause, J.; Prüfer, K.; Burbano, H.A.; Siebauer, M.; Lachmann, M.; Pääbo, S. The Neandertal genome and ancient DNA authenticity. EMBO J. 2009, 28, 2494–2502. [Google Scholar] [CrossRef] [PubMed]
- Dabney, J.; Meyer, M.; Pääbo, S. Ancient DNA damage. Cold Spring Harb. Perspect. Biol. 2013, 5, a012567. [Google Scholar] [CrossRef] [PubMed]
- Key, F.M.; Posth, C.; Krause, J.; Herbig, A.; Bos, K.I. Mining Metagenomic Data Sets for Ancient DNA: Recommended Protocols for Authentication. Trends Genet. 2017, 33, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Firtina, C.; Alkan, C. On genomic repeats and reproducibility. Bioinformatics 2016, 32, 2243–2247. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Kidd, J.M.; Marques-Bonet, T.; Aksay, G.; Antonacci, F.; Hormozdiari, F.; Kitzman, J.O.; Baker, C.; Malig, M.; Mutlu, O.; et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 2009, 41, 1061. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Domain/Kingdom/Type | Number of Sequences | Length | Script |
|---|---|---|---|
| Viruses | 9626 | 338 MB | DownloadViruses.pl |
| Archaea | 40,322 | 3.4 GB | DownloadArchaea.pl |
| Bacteria | 2,245,000 | 130 GB | DownloadBacteria.pl |
| Fungi | 2,205,000 | 11 GB | DownloadFungi.pl |
| Mitochondrion v2 | 8670 | 212 MB | DownloadMTV2.sh |
| Plastid v2 | 2938 | 308 MB | DownloadPlastidV2.sh |
| Total (DB) | 4,511,556 | 145.2 GB |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pratas, D.; Hosseini, M.; Grilo, G.; Pinho, A.J.; Silva, R.M.; Caetano, T.; Carneiro, J.; Pereira, F. Metagenomic Composition Analysis of an Ancient Sequenced Polar Bear Jawbone from Svalbard. Genes 2018, 9, 445. https://doi.org/10.3390/genes9090445
Pratas D, Hosseini M, Grilo G, Pinho AJ, Silva RM, Caetano T, Carneiro J, Pereira F. Metagenomic Composition Analysis of an Ancient Sequenced Polar Bear Jawbone from Svalbard. Genes. 2018; 9(9):445. https://doi.org/10.3390/genes9090445
Chicago/Turabian StylePratas, Diogo, Morteza Hosseini, Gonçalo Grilo, Armando J. Pinho, Raquel M. Silva, Tânia Caetano, João Carneiro, and Filipe Pereira. 2018. "Metagenomic Composition Analysis of an Ancient Sequenced Polar Bear Jawbone from Svalbard" Genes 9, no. 9: 445. https://doi.org/10.3390/genes9090445
APA StylePratas, D., Hosseini, M., Grilo, G., Pinho, A. J., Silva, R. M., Caetano, T., Carneiro, J., & Pereira, F. (2018). Metagenomic Composition Analysis of an Ancient Sequenced Polar Bear Jawbone from Svalbard. Genes, 9(9), 445. https://doi.org/10.3390/genes9090445