Assessing Metagenomic Signals Recovered from Lyuba, a 42,000-Year-Old Permafrost-Preserved Woolly Mammoth Calf

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. DNA Extraction
2.3. Library Preparation and Sequencing
2.4. Sequence Quality Control and Filtering
2.5. Mapping to Reference Genomes and Deamination Pattern Analysis
2.6. Mitogenome Analysis
2.7. Shotgun Metagenomic Analysis
2.8. 16S Metagenomic Analysis
2.9. MetaPhlAn2 Analysis
2.10. Authentication of Bacterial Reads
2.11. Data Availability
3. Results
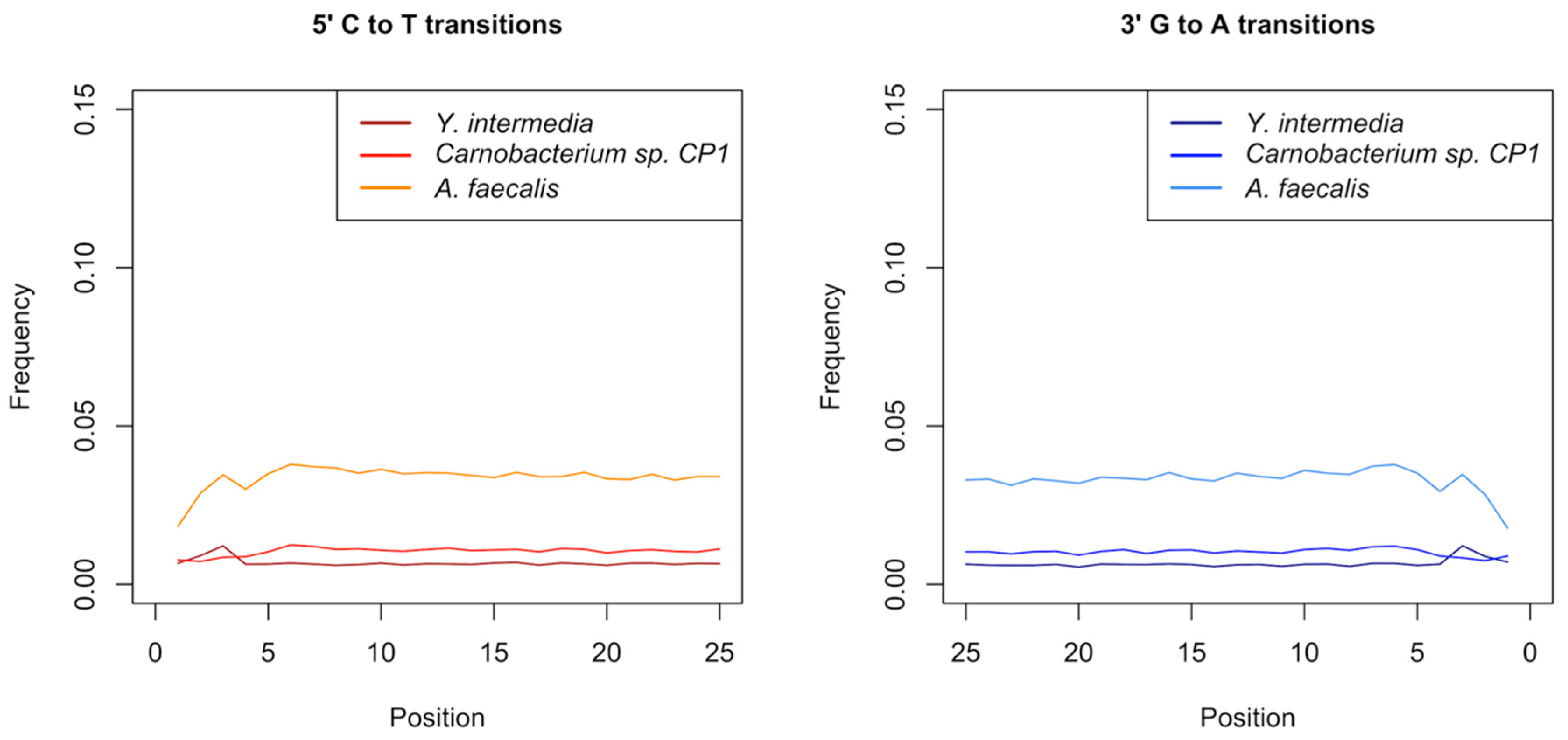
3.1. Data Quality Control and Authentication
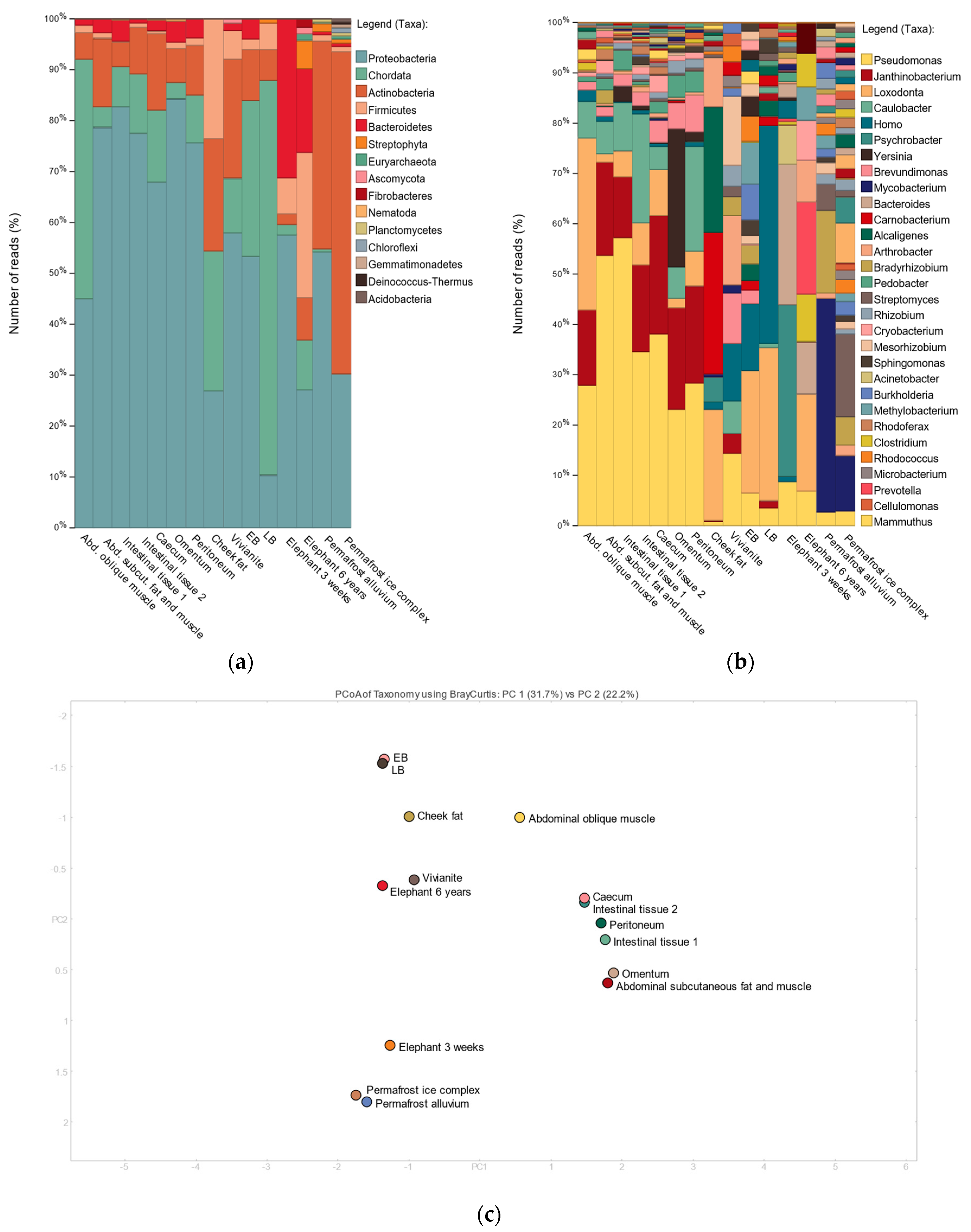
3.2. Shotgun Metagenomic Analysis
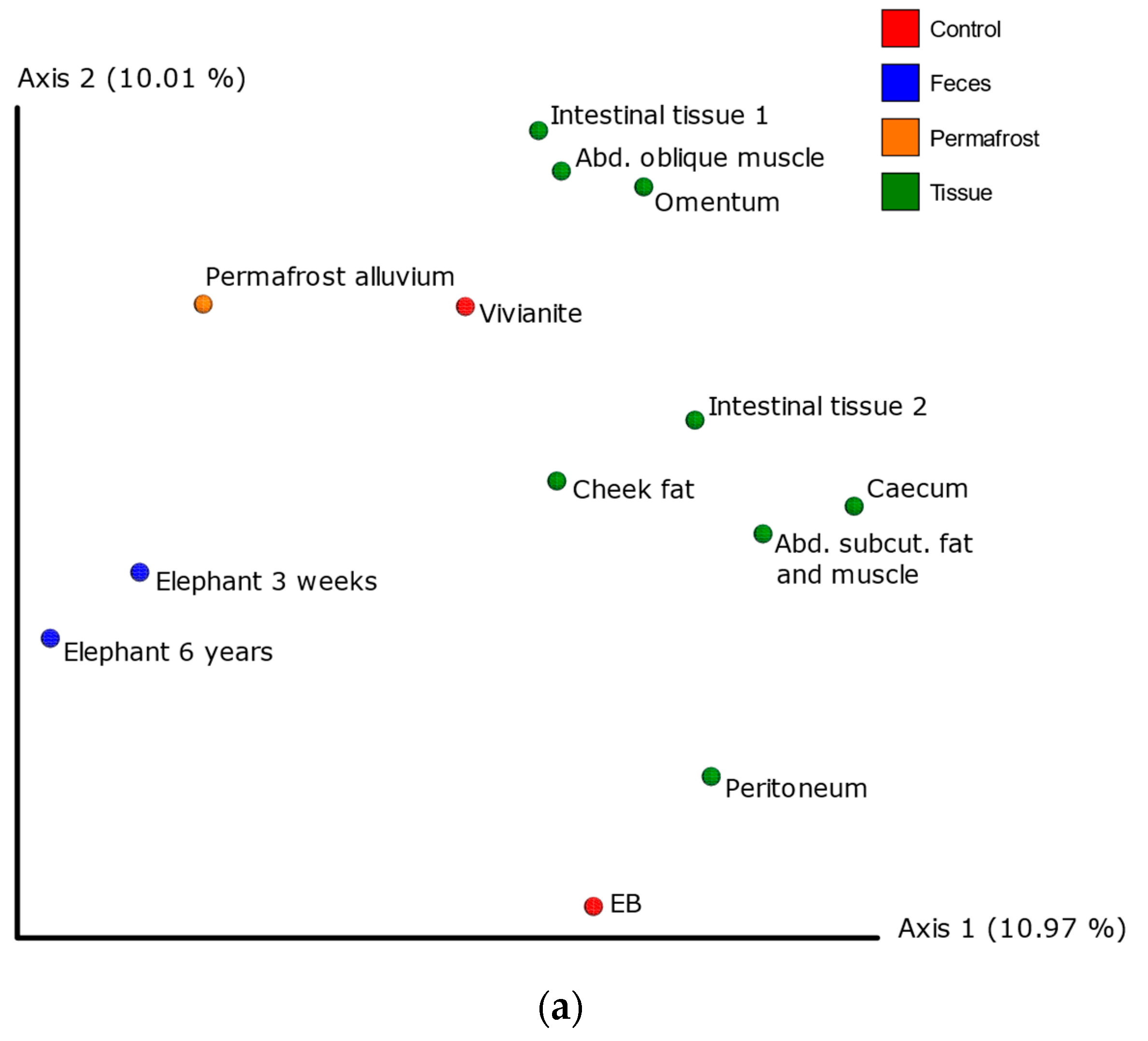
3.3. 16S Metagenomic Analysis
3.4. MetaPhlAn2 Analysis
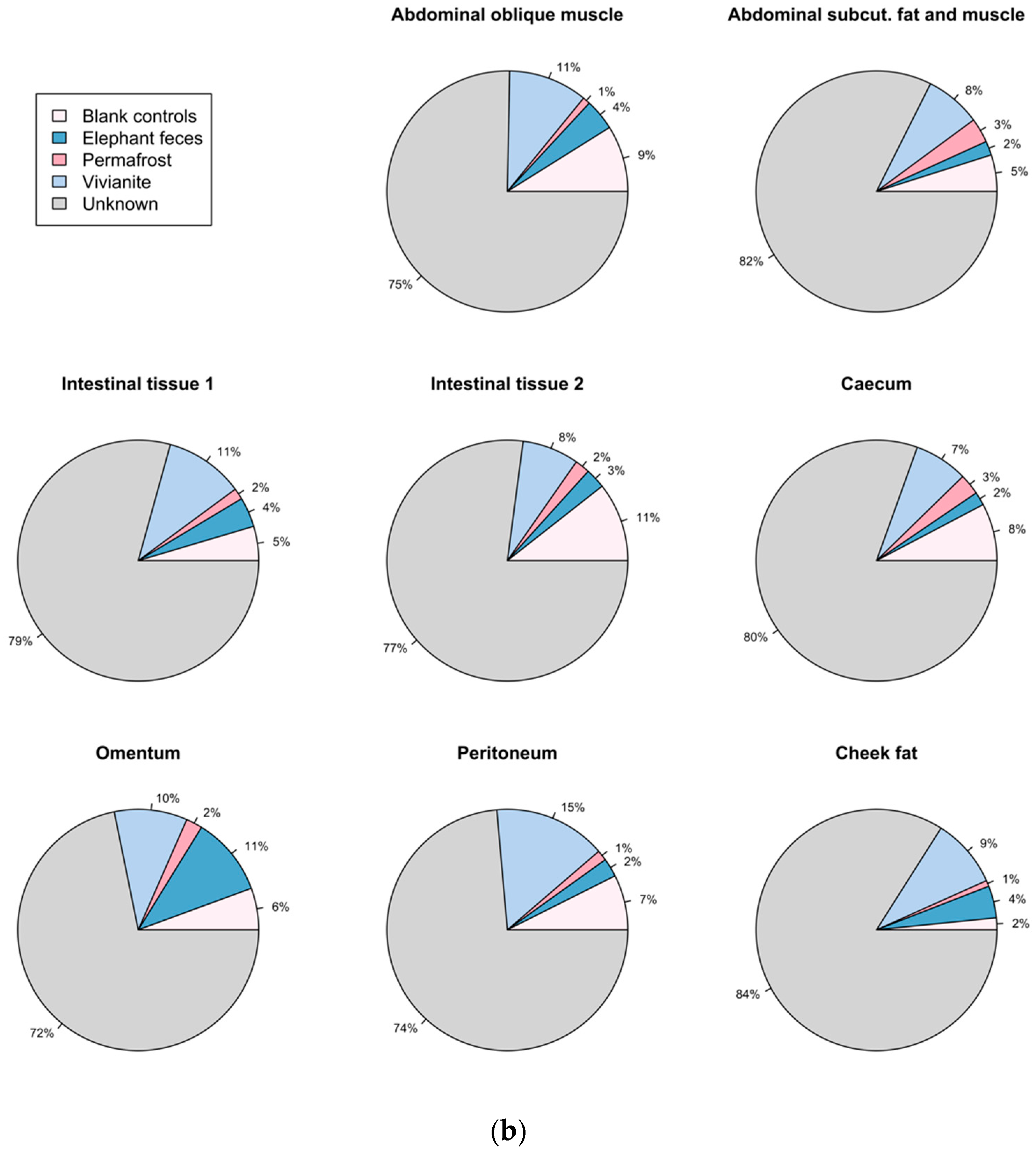
3.5. Authentication of Potential Ancient Bacterial Signals
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paabo, S. Molecular cloning of Ancient Egyptian mummy DNA. Nature 1985, 314, 644–645. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.M.; Crossey, M.; Saldanha, J.; Waldron, T. DNA from Mycobacterium tuberculosis identified in Mediaeval human skeletal remains using polymerase chain reaction. J. Archaeol. Sci. 1996, 23, 789–798. [Google Scholar] [CrossRef]
- Nerlich, A.G.; Haas, C.J.; Zink, A.; Szeimies, U.; Hagedorn, H.G. Molecular evidence for tuberculosis in an ancient Egyptian mummy. Lancet 1997, 350, 1404. [Google Scholar] [CrossRef]
- Zink, A.; Reischl, U.; Wolf, H.; Nerlich, A.G. Molecular evidence of bacteremia by gastrointestinal pathogenic bacteria in an infant mummy from ancient Egypt. Arch. Pathol. Lab. Med. 2000, 124, 1614–1618. [Google Scholar] [PubMed]
- Bos, K.I.; Schuenemann, V.J.; Golding, G.B.; Burbano, H.A.; Waglechner, N.; Coombes, B.K.; McPhee, J.B.; DeWitte, S.N.; Meyer, M.; Schmedes, S.; et al. A draft genome of Yersinia pestis from victims of the Black Death. Nature 2011, 478, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Schuenemann, V.J.; Singh, P.; Mendum, T.A.; Krause-Kyora, B.; Jager, G.; Bos, K.I.; Herbig, A.; Economou, C.; Benjak, A.; Busso, P.; et al. Genome-wide comparison of medieval and modern Mycobacterium leprae. Science 2013, 341, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Adler, C.J.; Dobney, K.; Weyrich, L.S.; Kaidonis, J.; Walker, A.W.; Haak, W.; Bradshaw, C.J.A.; Townsend, G.; Sołtysiak, A.; Alt, K.W.; et al. Sequencing ancient calcified dental plaque shows changes in oral microbiota with dietary shifts of the Neolithic and Industrial revolutions. Nat. Genet. 2013, 45, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Warinner, C.; Rodrigues, J.F.M.; Vyas, R.; Trachsel, C.; Shved, N.; Grossmann, J.; Radini, A.; Hancock, Y.; Tito, R.Y.; Fiddyment, S.; et al. Pathogens and host immunity in the ancient human oral cavity. Nat. Genet. 2014, 46, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Gordon, J.I. Commensal host-bacterial relationships in the gut. Science 2001, 292, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222. [Google Scholar] [CrossRef] [PubMed]
- Tito, R.Y.; Knights, D.; Metcalf, J.; Obregon-Tito, A.J.; Cleeland, L.; Najar, F.; Roe, B.; Reinhard, K.; Sobolik, K.; Belknap, S.; et al. Insights from characterizing extinct human gut microbiomes. PLoS ONE 2012, 7, e51146. [Google Scholar] [CrossRef] [PubMed]
- Weyrich, L.S.; Duchene, S.; Soubrier, J.; Arriola, L.; Llamas, B.; Breen, J.; Morris, A.G.; Alt, K.W.; Caramelli, D.; Dresely, V.; et al. Neanderthal behaviour, diet, and disease inferred from ancient DNA in dental calculus. Nature 2017, 544, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Campana, M.G.; Robles Garcia, N.; Ruhli, F.J.; Tuross, N. False positives complicate ancient pathogen identifications using high-throughput shotgun sequencing. BMC Res. Notes 2014, 7, 111. [Google Scholar] [CrossRef] [PubMed]
- Bos, K.I.; Jager, G.; Schuenemann, V.J.; Vagene, A.J.; Spyrou, M.A.; Herbig, A.; Nieselt, K.; Krause, J. Parallel detection of ancient pathogens via array-based DNA capture. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20130375. [Google Scholar] [CrossRef] [PubMed]
- Warinner, C.; Herbig, A.; Mann, A.; Fellows Yates, J.A.; Weiß, C.L.; Burbano, H.A.; Orlando, L.; Krause, J. A robust framework for microbial archaeology. Annu. Rev. Genom. Hum. Genet. 2017, 18, 321–356. [Google Scholar] [CrossRef] [PubMed]
- Key, F.M.; Posth, C.; Krause, J.; Herbig, A.; Bos, K.I. Mining metagenomic data sets for ancient DNA: Recommended protocols for authentication. Trends Genet. 2017, 33, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12. [Google Scholar] [CrossRef] [PubMed]
- Glassing, A.; Dowd, S.E.; Galandiuk, S.; Davis, B.; Chiodini, R.J. Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 2016, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Lauder, A.P.; Roche, A.M.; Sherrill-Mix, S.; Bailey, A.; Laughlin, A.L.; Bittinger, K.; Leite, R.; Elovitz, M.A.; Parry, S.; Bushman, F.D. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome 2016, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Rohland, N.; Malaspinas, A.-S.; Pollack, J.L.; Slatkin, M.; Matheus, P.; Hofreiter, M. Proboscidean mitogenomics: Chronology and mode of elephant evolution using mastodon as outgroup. PLoS Biol. 2007, 5, e207. [Google Scholar] [CrossRef] [PubMed]
- Long, S.Y.; Latimer, E.M.; Hayward, G.S. Review of elephant endotheliotropic Herpesviruses and acute hemorrhagic disease. ILAR J. 2015, 56, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Zlot, A.; Vines, J.; Nystrom, L.; Lane, L.; Behm, H.; Denny, J.; Finnegan, M.; Hostetler, T.; Matthews, G.; Storms, T.; et al. Diagnosis of tuberculosis in three zoo elephants and a human contact—Oregon, 2013. Centers Dis. Control Prev. Morb. Mortal. Wkly. Rep. 2016, 64, 1398–1402. [Google Scholar] [CrossRef] [PubMed]
- Kosintsev, P.A.; Lapteva, E.G.; Trofimova, S.S.; Zanina, O.G.; Tikhonov, A.N.; van der Plicht, J. The intestinal contents of a baby woolly mammoth (Mammuthus primigenius Blumenbach, 1799) from the Yuribey River (Yamal Peninsula). Dokl. Biol. Sci. 2010, 432, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.C.; Tikhonov, A.N.; Kosintsev, P.A.; Rountrey, A.N.; Buigues, B.; van der Plicht, J. Anatomy, death, and preservation of a woolly mammoth (Mammuthus primigenius) calf, Yamal Peninsula, northwest Siberia. Quat. Int. 2012, 255, 94–105. [Google Scholar] [CrossRef]
- Rountrey, A.N.; Fisher, D.C.; Tikhonov, A.N.; Kosintsev, P.A.; Lazarev, P.A.; Boeskorov, G.; Buigues, B. Early tooth development, gestation, and season of birth in mammoths. Quat. Int. 2012, 255, 196–205. [Google Scholar] [CrossRef]
- Papageorgopoulou, C.; Link, K.; Rühli, F.J. Histology of a woolly mammoth (Mammuthus primigenius) preserved in permafrost, Yamal Peninsula, Northwest Siberia. Anat. Rec. 2015, 298, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Krüttli, A.; Bouwman, A.; Akgül, G.; Della Casa, P.; Rühli, F.; Warinner, C. Ancient DNA analysis reveals high frequency of European lactase persistence allele (T-13910) in medieval central Europe. PLoS ONE 2014, 9, e86251. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Poinar, H.N. Ancient DNA: Do it right or not at all. Science 2000, 289, 1139. [Google Scholar] [CrossRef] [PubMed]
- Llamas, B.; Valverde, G.; Fehren-Schmitz, L.; Weyrich, L.S.; Cooper, A.; Haak, W. From the field to the laboratory: Controlling DNA contamination in human ancient DNA research in the high-throughput sequencing era. STAR Sci. Technol. Archaeol. Res. 2017, 3, 1–14. [Google Scholar] [CrossRef]
- Meyer, M.; Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Sawyer, S.; Meyer, M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2012, 40, e3. [Google Scholar] [CrossRef] [PubMed]
- Renaud, G.; Stenzel, U.; Maricic, T.; Wiebe, V.; Kelso, J. deML: Robust demultiplexing of Illumina sequences using a likelihood-based approach. Bioinformatics 2015, 31, 770–772. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- SeqPrep. Available online: https://github.com/jstjohn/SeqPrep (accessed on 19 February 2016).
- Maixner, F.; Krause-Kyora, B.; Turaev, D.; Herbig, A.; Hoopmann, M.R.; Hallows, J.L.; Kusebauch, U.; Vigl, E.E.; Malfertheiner, P.; Megraud, F.; et al. The 5300-year-old Helicobacter pylori genome of the Iceman. Science 2016, 351, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 8 April 2014).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Ginolhac, A.; Lindgreen, S.; Thompson, J.F.; AL-Rasheid, K.A.; Willerslev, E.; Krogh, A.; Orlando, L. Improving ancient DNA read mapping against modern reference genomes. BMC Genom. 2012, 13, 178. [Google Scholar] [CrossRef] [PubMed]
- Picard. Available online: http://broadinstitute.github.io/picard/ (accessed on 5 February 2016).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Jónsson, H.; Ginolhac, A.; Schubert, M.; Johnson, P.; Orlando, L. mapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 2013, 29, 1682–1684. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA. Ph.D. Thesis, The Pennsylvania State University, State College, PA, USA, 2007. [Google Scholar]
- Enk, J.; Devault, A.; Widga, C.; Saunders, J.; Szpak, P.; Southon, J.; Rouillard, J.-M.; Shapiro, B.; Golding, G.B.; Zazula, G.; et al. Mammuthus population dynamics in Late Pleistocene North America: Divergence, phylogeography and introgression. Front. Ecol. Evol. 2016, 4. [Google Scholar] [CrossRef]
- Ilmberger, N.; Güllert, S.; Dannenberg, J.; Rabausch, U.; Torres, J.; Wemheuer, B.; Alawi, M.; Poehlein, A.; Chow, J.; Turaev, D.; et al. A comparative metagenome survey of the fecal microbiota of a breast- and a plant-fed Asian elephant reveals an unexpectedly high diversity of glycoside hydrolase family enzymes. PLoS ONE 2014, 9, e106707. [Google Scholar] [CrossRef] [PubMed]
- Krivushin, K.; Kondrashov, F.; Shmakova, L.; Tutukina, M.; Petrovskaya, L.; Rivkina, E. Two metagenomes from late Pleistocene northeast Siberian permafrost. Genome Announc. 2015, 3, e01380-14. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Vågene, Å.J.; Herbig, A.; Campana, M.G.; Robles García, N.M.; Warinner, C.; Sabin, S.; Spyrou, M.A.; Andrades Valtueña, A.; Huson, D.; Tuross, N.; et al. Salmonella enterica genomes from victims of a major sixteenth-century epidemic in Mexico. Nat. Ecol. Evol. 2018, 2, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN Community edition—interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. EMPeror: A tool for visualizing high-throughput microbial community data. Gigascience 2013, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Baeza, Y.; Gonzalez, A.; Smarr, L.; McDonald, D.; Morton, J.T.; Navas-Molina, J.A.; Knight, R. Bringing the dynamic microbiome to life with animations. Cell Host Microbe 2017, 21, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Knights, D.; Kuczynski, J.; Charlson, E.S.; Zaneveld, J.; Mozer, M.C.; Collman, R.G.; Bushman, F.D.; Knight, R.; Kelley, S.T. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 2011, 8, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef] [PubMed]
- Dabney, J.; Meyer, M.; Pääbo, S. Ancient DNA Damage. Cold Spring Harb. Perspect. Biol. 2013, 5, a012567. [Google Scholar] [CrossRef] [PubMed]
- Sulakvelidze, A. Yersiniae other than Y. enterocolitica, Y. pseudotuberculosis, and Y. pestis: The ignored species. Microbes Infect. 2000, 2, 497–513. [Google Scholar] [CrossRef]
- Phung, L.T.; Trimble, W.L.; Meyer, F.; Gilbert, J.A.; Silver, S. Draft genome sequence of Alcaligenes faecalis subsp. faecalis NCIB 8687 (CCUG 2071). J. Bacteriol. 2012, 194, 5153. [Google Scholar] [CrossRef] [PubMed]
- Leisner, J.J.; Laursen, B.G.; Prévost, H.; Drider, D.; Dalgaard, P. Carnobacterium: Positive and negative effects in the environment and in foods. FEMS Microbiol. Rev. 2007, 31, 592–613. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raw Read Pairs | Merged Reads (% of Total) | Unique Reads (Clonality) | Unique Mapped Reads (% of Unique Reads) | |||
|---|---|---|---|---|---|---|
| Loxodonta africana | Mammuthus primigenius | Homo sapiens | ||||
| Nuclear Genome | Mitogenome | |||||
| Abdominal | 35,511,605 | 31,176,532 | 102,34,165 (3.05×) | 4,371,482 | 55532 | 78047 |
| oblique muscle | −87.80% | −42.71% | −0.54% | −0.76% | ||
| Abd. subcut. | 22,977,668 | 21,176,646 | 6,834,930 (3.10×) | 143221 | 16216 | 20252 |
| fat and muscle | −92.20% | −2.10% | −0.24% | −0.29% | ||
| Intestinal | 25,053,246 | 23,305,474 | 7,496,017 (3.11×) | 605598 | 32548 | 16223 |
| tissue 1 | −93.00% | −8.08% | −0.43% | −0.22% | ||
| Intestinal | 21,130,716 | 17,066,995 | 2,829,012 (6.03×) | 270976 | 12162 | 6882 |
| tissue 2 | −80.80% | −9.58% | −0.43% | −0.24% | ||
| Caecum | 17,386,802 | 16,168,853 | 5,819,269 (2.78×) | 634528 | 46103 | 12498 |
| −93.00% | −10.90% | −0.79% | −0.21% | |||
| Omentum | 22,924,111 | 21,390,754 | 6,909,388 (3.10×) | 186132 | 9453 | 7581 |
| −93.30% | −2.69% | −0.14% | −0.11% | |||
| Peritoneum | 18,925,818 | 17,416,113 | 5,994,320 (2.91×) | 405067 | 6093 | 14650 |
| −92.00% | −6.76% | −0.10% | −0.24% | |||
| Cheek | 47,716,128 | 42,628,440 | 11,029,081 (3.87×) | 1947669 | 8582 | 37331 |
| fat | −89.30% | −42.08% | −0.19% | −0.81% | ||
| Vivianite | 21,888,799 | 8,481,750 | 824,368 (10.29×) | 2334 | 194 | 6298 |
| −38.80% | −0.28% | −0.02% | −0.76% | |||
| Extraction | 7,036,101 | 3,224,382 | 711,189 (4.53×) | 22774 | 227 | 5038 |
| blank | −45.80% | −3.20% | −0.03% | −0.71% | ||
| Library | 4,820,063 | 285,265 | 33387 (8.54×) | 8417 | 151 | 4479 |
| blank | −5.90% | −25.21% | −0.45% | −13.42% | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrari, G.; Lischer, H.E.L.; Neukamm, J.; Rayo, E.; Borel, N.; Pospischil, A.; Rühli, F.; Bouwman, A.S.; Campana, M.G. Assessing Metagenomic Signals Recovered from Lyuba, a 42,000-Year-Old Permafrost-Preserved Woolly Mammoth Calf. Genes 2018, 9, 436. https://doi.org/10.3390/genes9090436
Ferrari G, Lischer HEL, Neukamm J, Rayo E, Borel N, Pospischil A, Rühli F, Bouwman AS, Campana MG. Assessing Metagenomic Signals Recovered from Lyuba, a 42,000-Year-Old Permafrost-Preserved Woolly Mammoth Calf. Genes. 2018; 9(9):436. https://doi.org/10.3390/genes9090436
Chicago/Turabian StyleFerrari, Giada, Heidi E. L. Lischer, Judith Neukamm, Enrique Rayo, Nicole Borel, Andreas Pospischil, Frank Rühli, Abigail S. Bouwman, and Michael G. Campana. 2018. "Assessing Metagenomic Signals Recovered from Lyuba, a 42,000-Year-Old Permafrost-Preserved Woolly Mammoth Calf" Genes 9, no. 9: 436. https://doi.org/10.3390/genes9090436
APA StyleFerrari, G., Lischer, H. E. L., Neukamm, J., Rayo, E., Borel, N., Pospischil, A., Rühli, F., Bouwman, A. S., & Campana, M. G. (2018). Assessing Metagenomic Signals Recovered from Lyuba, a 42,000-Year-Old Permafrost-Preserved Woolly Mammoth Calf. Genes, 9(9), 436. https://doi.org/10.3390/genes9090436