In Silico Functional Networks Identified in Fish Nucleated Red Blood Cells by Means of Transcriptomic and Proteomic Profiling

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
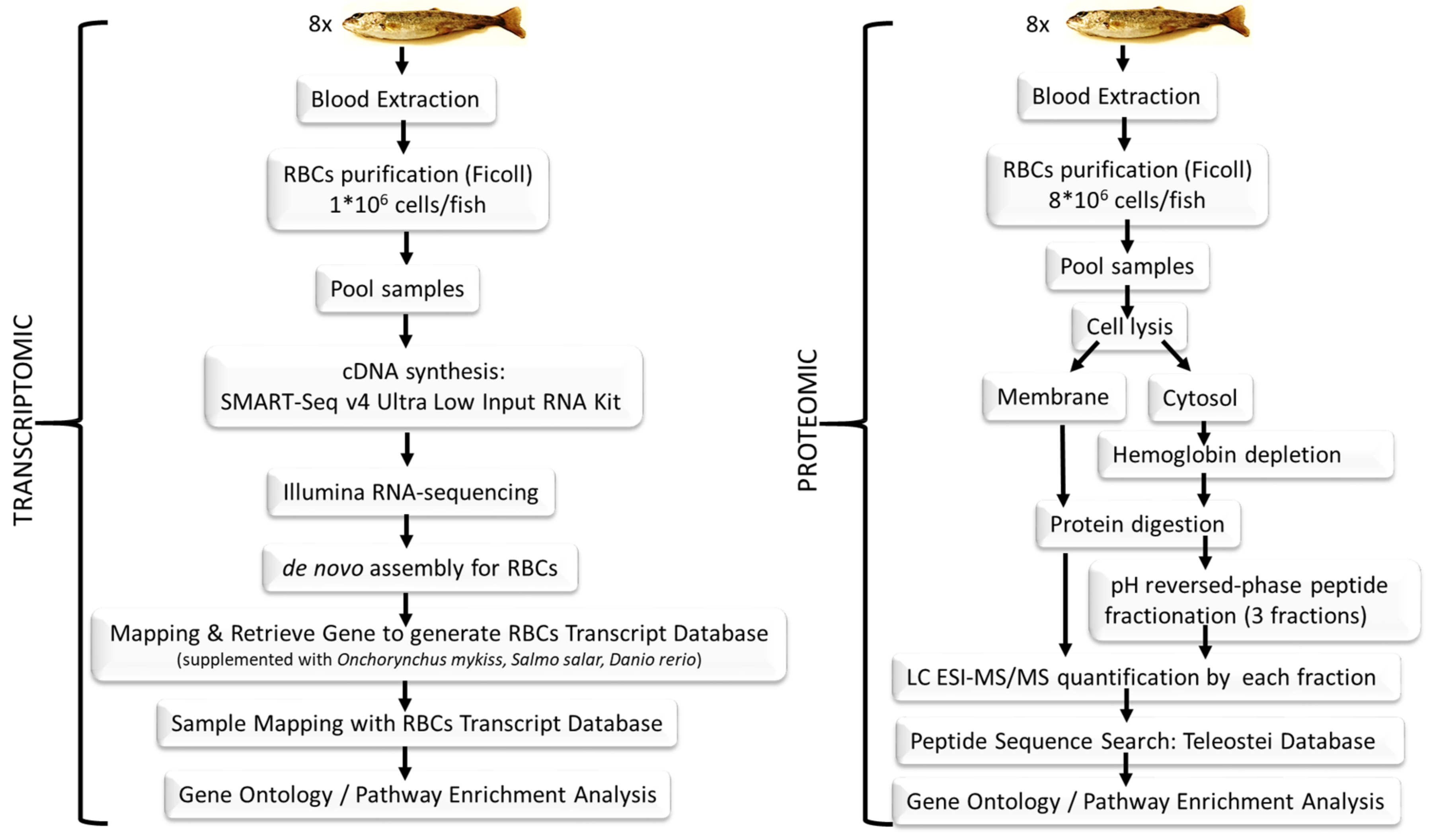
2.1. Fish
2.2. Blood Sampling and Red Blood Cells Purification
2.3. Transcriptome Sequencing
2.3.1. Complementary DNA Library Preparation and Illumina Sequencing
2.3.2. De Novo Assembly Bioinformatics Procedure
2.3.3. BLASTing of Assembled Contigs, Gene Sequence Retrieval, Red Blood Cells Transcript Database Construction and Functional Annotation
2.3.4. Red Blood Cells Transcriptome Mapping and Gene Expression Profiling
2.4. Proteome Sequencing
2.4.1. Protein Digestion
2.4.2. pH Reversed-Phase Peptide Fractionation
2.4.3. Liquid Chromatography and Mass Spectrometry Analysis
2.4.4. Proteomics Data Analysis and Sequence Search
2.5. Pathway Enrichment Analysis
2.6. RNA Extraction and Reverse Transcription Real Time Polymerase Chain Reaction Analysis
2.7. RBCs Single-Cell Sorting
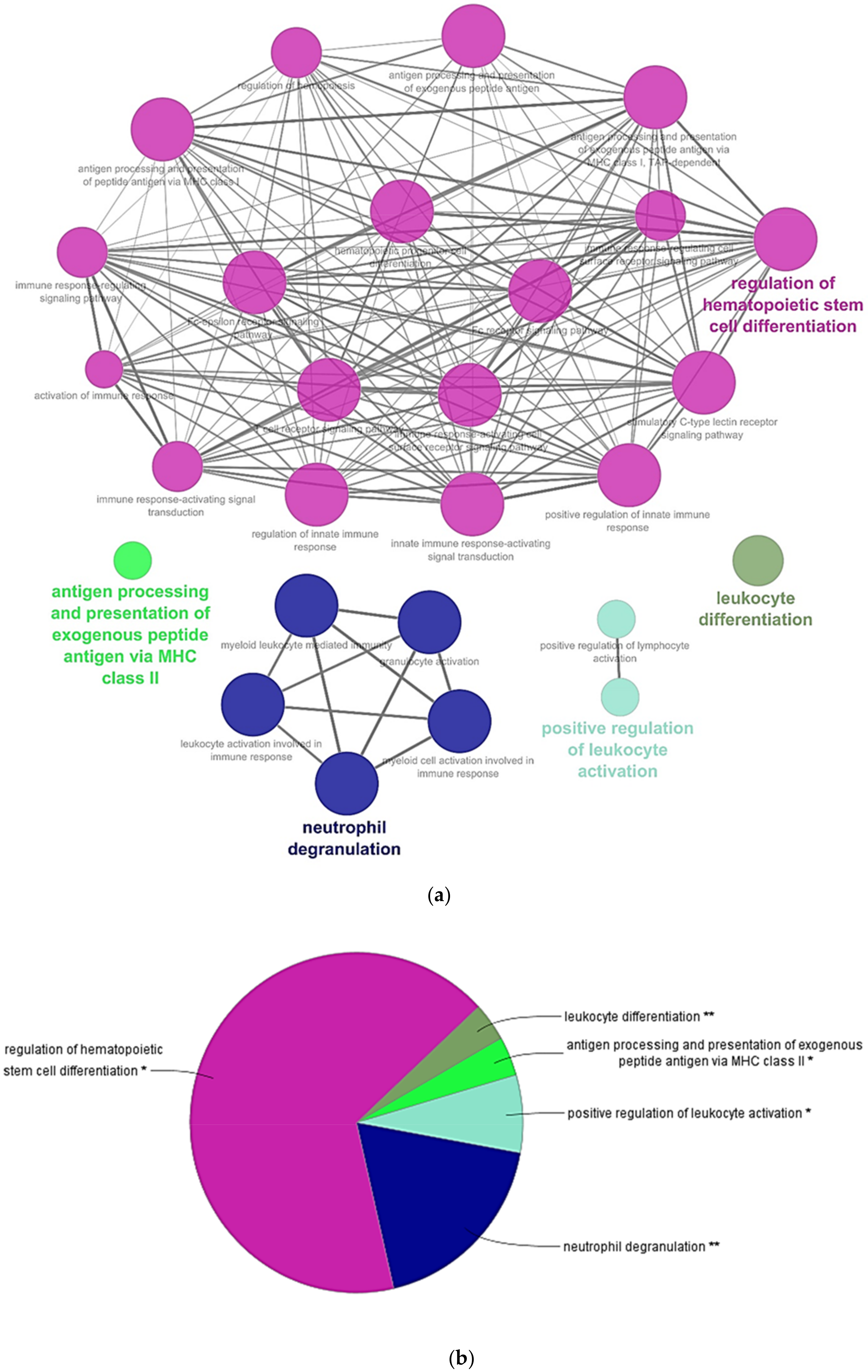
3. Results and Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Akbari, A. The Role of Red Blood Cells in Wound Healing. Master’s Thesis, University of British Columbia, Vancouver, BC, Canada, 2011. [Google Scholar]
- Moras, M.; Lefevre, S.D.; Ostuni, M.A. From erythroblasts to mature Red Blood Cells: Organelle clearance in mammals. Front. Physiol. 2017, 8, 1076. [Google Scholar] [CrossRef] [PubMed]
- Glomski, C.A.; Tamburlin, J.; Chainani, M. The phylogenetic odyssey of the erythrocyte. III. Fish, the lower vertebrate experience. Histol. Histopathol. 1992, 7, 501–528. [Google Scholar] [PubMed]
- Nombela, I.; Ortega-Villaizan, M.D.M. Nucleated red blood cells: Immune cell mediators of the antiviral response. PLoS Pathog. 2018, in press. [Google Scholar]
- Passantino, L.; Altamura, M.; Cianciotta, A.; Patruno, R.; Tafaro, A.; Jirillo, E.; Passantino, G.F. Fish immunology. I. Binding and engulfment of Candida albicans by erythrocytes of rainbow trout (Salmo gairdneri Richardson). Immunopharmacol. Immunotoxicol. 2002, 24, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Dahle, M.K.; Wessel, O.; Timmerhaus, G.; Nyman, I.B.; Jorgensen, S.M.; Rimstad, E.; Krasnov, A. Transcriptome analyses of Atlantic salmon (Salmo salar L.) erythrocytes infected with piscine orthoreovirus (PRV). Fish Shellfish Immunol. 2015, 45, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Nombela, I.; Carrion, A.; Puente-Marin, S.; Chico, V.; Mercado, L.; Perez, L.; Coll, J.; Ortega-Villaizan, M.D.M. Infectious pancreatic necrosis virus triggers antiviral immune response in rainbow trout red blood cells, despite not being infective. F1000Research 2017, 6, 1968. [Google Scholar] [CrossRef] [PubMed]
- Nombela, I.; Puente-Marin, S.; Chico, V.; Villena, A.J.; Carracedo, B.; Ciordia, S.; Mena, M.C.; Mercado, L.; Perez, L.; Coll, J.; et al. Identification of diverse defense mechanisms in rainbow trout red blood cells in response to halted replication of VHS virus. F1000Research 2017, 6, 1958. [Google Scholar] [CrossRef] [PubMed]
- Workenhe, S.T.; Kibenge, M.J.; Wright, G.M.; Wadowska, D.W.; Groman, D.B.; Kibenge, F.S. Infectious salmon anaemia virus replication and induction of alpha interferon in Atlantic salmon erythrocytes. Virol. J. 2008, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Passantino, L.; Massaro, M.A.; Jirillo, F.; Di Modugno, D.; Ribaud, M.R.; Modugno, G.D.; Passantino, G.F.; Jirillo, E. Antigenically activated avian erythrocytes release cytokine-like factors: A conserved phylogenetic function discovered in fish. Immunopharmacol. Immunotoxicol. 2007, 29, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Morera, D.; Roher, N.; Ribas, L.; Balasch, J.C.; Donate, C.; Callol, A.; Boltana, S.; Roberts, S.; Goetz, G.; Goetz, F.W.; et al. RNA-Seq reveals an integrated immune response in nucleated erythrocytes. PLoS ONE 2011, 6, e26998. [Google Scholar] [CrossRef] [PubMed]
- Bryk, A.H.; Wisniewski, J.R. Quantitative analysis of human Red Blood Cell proteome. J. Proteome Res. 2017, 16, 2752–2761. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Righetti, P.G.; Zolla, L. The red blood cell proteome and interactome: An update. J. Proteome Res. 2010, 9, 144–163. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Perez, D.A.; Garcia-Valiente, R.; Ibarrola, N.; Muro, A.; Patarroyo, M.A. The Aotus nancymaae erythrocyte proteome and its importance for biomedical research. J. Proteom. 2017, 152, 131–137. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Nemkov, T.; Reisz, J.; Dzieciatkowska, M.; Wither, M.J.; Hansen, K.C. Omics markers of the red cell storage lesion and metabolic linkage. Blood Trans. 2017, 15, 137–144. [Google Scholar]
- Gautier, E.F.; Ducamp, S.; Leduc, M.; Salnot, V.; Guillonneau, F.; Dussiot, M.; Hale, J.; Giarratana, M.C.; Raimbault, A.; Douay, L.; et al. Comprehensive proteomic analysis of human erythropoiesis. Cell Rep. 2016, 16, 1470–1484. [Google Scholar] [CrossRef] [PubMed]
- CLC Genomics Workbench; Version 9.5.4; Qiagen: Aarhus, Denmark, 2017.
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. CD-HIT: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Villaizan, M.D.M. Oncorhynchus mykiss Red Blood Cells_De Novo Transcriptome Assembly. 2018. Available online: http://osf.io/gv2w9 (accessed on 8 March 2018).
- CLC Genomics Workbench; Version 10.1.1; Qiagen: Aarhus, Denmark, 2017.
- Wessel, D.; Flugge, U.I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Walpurgis, K.; Kohler, M.; Thomas, A.; Wenzel, F.; Geyer, H.; Schanzer, W.; Thevis, M. Validated hemoglobin-depletion approach for red blood cell lysate proteome analysis by means of 2D PAGE and Orbitrap MS. Electrophoresis 2012, 33, 2537–2545. [Google Scholar] [CrossRef] [PubMed]
- PeakView; Version 2.2; SCIEX: Foster City, CA, USA, 2014.
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, T.R.; Raida, M.K.; Kania, P.W.; Buchmann, K. Response of rainbow trout (Oncorhynchus mykiss) in skin and fin tissue during infection with a variant of Gyrodactylus salaris (Monogenea: Gyrodactylidae). Folia Parasitol. 2009, 56, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Raida, M.K.; Buchmann, K. Temperature-dependent expression of immune-relevant genes in rainbow trout following Yersinia ruckeri vaccination. Dis. Aquat. Org. 2007, 77, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Ganassin, R.C.; Barlow, J.; Bols, N.C. Influence of glutamine on phytohemagglutinin stimulated mitogenesis of leucocytes from the rainbow trout head kidney. Fish Shellfish Immunol. 1998, 8, 561–564. [Google Scholar] [CrossRef]
- Ten Broeke, T.; Wubbolts, R.; Stoorvogel, W. MHC class II antigen presentation by dendritic cells regulated through endosomal sorting. Cold Spring Harb. Perspect. Biol. 2013, 5, a016873. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, J.M.; Kollner, B.; Aoyagi, K.; Sawamoto, Y.; Kuroda, A.; Ototake, M.; Nakanishi, T.; Fischer, U. The rainbow trout classical MHC class I molecule Onmy-UBA*501 is expressed in similar cell types as mammalian classical MHC class I molecules. Fish Shellfish Immunol. 2003, 14, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Villadangos, J.A.; Schnorrer, P.; Wilson, N.S. Control of MHC class II antigen presentation in dendritic cells: A balance between creative and destructive forces. Immunol. Rev. 2005, 207, 191–205. [Google Scholar] [CrossRef] [PubMed]
- St Paul, M.; Paolucci, S.; Barjesteh, N.; Wood, R.D.; Sharif, S. Chicken erythrocytes respond to Toll-like receptor ligands by up-regulating cytokine transcripts. Res. Vet. Sci. 2013, 95, 87–91. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| De Novo Assembly | |
| Total reads | 404,825,036 |
| Number of aligned reads | 286,555,140 |
| Total contigs | 1,056,546 |
| Contigs after CD HIT EST c 0.85 | 862,667 |
| RBCs Transcript Database | |
| Genes after assembly BLAST, gene retrieval, removal of duplicates and 95% similar sequences | 106,361 |
| Genes after adding Oncorhynchus mykiss, Salmo salar, and Danio rerio NCBI sequences | 137,444 |
| Raw Data and Mapping | |
| Total reads | 93,177,954 |
| Reads after trimming | 92,391,474 |
| Mapped reads | 52,118,053 |
| Un-mapped reads | 40,273,421 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puente-Marin, S.; Nombela, I.; Ciordia, S.; Mena, M.C.; Chico, V.; Coll, J.; Ortega-Villaizan, M.D.M. In Silico Functional Networks Identified in Fish Nucleated Red Blood Cells by Means of Transcriptomic and Proteomic Profiling. Genes 2018, 9, 202. https://doi.org/10.3390/genes9040202
Puente-Marin S, Nombela I, Ciordia S, Mena MC, Chico V, Coll J, Ortega-Villaizan MDM. In Silico Functional Networks Identified in Fish Nucleated Red Blood Cells by Means of Transcriptomic and Proteomic Profiling. Genes. 2018; 9(4):202. https://doi.org/10.3390/genes9040202
Chicago/Turabian StylePuente-Marin, Sara, Iván Nombela, Sergio Ciordia, María Carmen Mena, Verónica Chico, Julio Coll, and María Del Mar Ortega-Villaizan. 2018. "In Silico Functional Networks Identified in Fish Nucleated Red Blood Cells by Means of Transcriptomic and Proteomic Profiling" Genes 9, no. 4: 202. https://doi.org/10.3390/genes9040202
APA StylePuente-Marin, S., Nombela, I., Ciordia, S., Mena, M. C., Chico, V., Coll, J., & Ortega-Villaizan, M. D. M. (2018). In Silico Functional Networks Identified in Fish Nucleated Red Blood Cells by Means of Transcriptomic and Proteomic Profiling. Genes, 9(4), 202. https://doi.org/10.3390/genes9040202